

Abstract
An increasing number of cell types, including peripheral blood mononuclear cells (PBMCs), have been demonstrated to release heat shock proteins (Hsps). In this paper we investigate further the hypothesis that Hsps are danger signals. PBMCs and Jurkat cells released Hsp70 (0·22 and 0·7 ng/106 cells, respectively) into medium over 24 h at 37°C. Release of Hsp70 was stimulated 10-fold by GroEL (P < 0·001) and more than threefold by lipopolysaccharide (LPS) (P < 0·001). Although Hsp60 could be detected in the medium of cells cultured at 37°C for 24 h, the low rates of release were due probably to cell damage. Significant release of Hsp60 was observed when Jurkat cells were exposed to GroEL (2·88 ng/106 cells) or LPS (1·40 ng/106 cells). The data are consistent with the hypothesis that Hsp70 and Hsp60 are part of a danger signalling cascade in response to bacterial infection.
Keywords: danger signals, GroEL, Hsp60 release, Hsp70 release, LPS, lymphocytes, peripheral blood mononuclear cells
Introduction
Responses to bacterial infection involve recognition of foreign antigens and a signalling cascade alerting, or priming, the immune system. Recently heat shock proteins (Hsps) have been implicated as members of a danger signalling cascade [1–3]. This paper examines whether Hsp70 and Hsp60 could form part of the response to the bacterial antigens LPS and GroEL.
Bacterial-derived LPS and Hsps are stimulators of the innate immune system [4–7] and have been demonstrated to initiate activation and cytokine release from monocytes, macrophages and dendritic cells [8–14]. Innate immune responses to bacterial LPS and Hsps have been demonstrated to occur through CD14/TLR2/TLR4 receptors on antigen-presenting cells [15–18]. Responses to mammalian Hsp60 and Hsp70 have been reported to occur through both CD14-dependent and CD14-independent pathways [7,19–21].
Hsps are normally intracellular proteins that have functions involved in protein folding and maintenance of protein integrity under both normal and stress conditions, yet if they are to act as signals in response to infection they would need to be present in the extracellular environment. A number of bacterial Hsps (such as the chaperonin 60 protein of Escherichia coli, GroEL) have been found in extracellular locations and implicated in immune reactions to infection [22–25]. Human Hsps have also been shown to be present on cell surfaces [26–28], in serum [29–32] and to be released from a variety of cell types [33–38]. We have reported recently the release of Hsp70 from lymphocytes [36].
In order for Hsps to act as danger signals there is a requirement to demonstrate that their release can be up-regulated in response to stresses that would occur in vivo, such as bacterial infection; for Hsp60 and/or Hsp70 to have a role as a danger signal we would predict that bacterial antigens, such as LPS and GroEL, would stimulate their release. This paper provides evidence for this stimulation in both PBMCs and cultured Jurkat cells.
Materials and methods
The Biological Sciences Ethics Committee (University of Chester) approved the study. Whole blood was taken from healthy volunteers and informed written consent was obtained from all subjects.
Isolation of cells from whole blood
Whole blood was diluted 1:1 in phosphate buffered saline (PBS; 0·14 M, pH 7·4) and subjected to density centrifugation using Histopaque® (Sigma 1077–1, Poole, UK). Peripheral blood mononuclear cell (PBMC) pellets were resuspended (∼2 × 107 cells/ml) in RPMI medium (Sigma, R-8758) supplemented with 10% fetal calf serum, 2 mM l-glutamine and 1% antibiotic/anti-mycotic solution (Sigma, A-5955).
Jurkat cell culture
Jurkat E6·1 human T cell leukaemia cells were cultured in RPMI-1640 medium (Sigma, R-8758) supplemented with 10% fetal calf serum (Fetalclone I; Perbio Science, Cramlington, UK) 2 mM l-glutamine (Sigma, G-7513) and 1% antibiotic/anti-mycotic solution (Sigma, A-5955). Cells were grown at 37°C, 5% CO2. Experiments were performed on cells that had achieved a density of ∼ 4 × 105 cells/ml (mid-log expansion), and contained < 1% of dead cells as determined by Trypan blue staining.
Giant cell tumour cell (GCT) culture
GCT stromal cells were cultured in α-Minimum Essential Medium (MEM) (Sigma, M-8042) supplemented with 10% fetal calf serum (Fetalclone I; Perbio) 2 mM l-glutamine (Sigma, G-7513), and 1% antibiotic/anti-mycotic solution (Sigma, A-5955). In preparation for experiments 1 ml cells were seeded into 12-well plates at a density of ∼2 × 105 cells/ml, and were incubated at 37°C in 5% CO2 for 3 days or until confluent. Medium was removed from the confluent cells and replaced with 1 ml of supplemented α-MEM medium.
Cell treatments
Prior to treatment PBMCs and Jurkat cells were resuspended in 10 ml supplemented RPMI and washed by centrifugation (500 g, 3 min at room temperature). Medium was removed from the pellet and the cells were resuspended in supplemented RPMI to give a density of ∼ 3 × 106 cells/ml (Jurkat) and ∼ 2 × 107 cells/ml (PBMCs). Cell suspensions were transferred into 12-well plates (1 ml/well) and t0 (time zero) samples were harvested immediately. Cell treatments consisting of Hsp60 (10 µg/ml) (NSP-540; Stressgen, Victoria, Canada), Hsp70 (10 µg/ml) (Stressgen, NSP-555), GroEL (10 µg/ml) (Stressgen, SPP-610-G) and LPS (10 µg/ml) (Sigma, L-4516), diluted in the appropriate medium, were added to the remaining wells in triplicate and the cells were incubated at 37°C, 5% CO2 for 24 h.
Measurement of released Hsp60 and Hsp70
Following treatments Jurkat and PBMCs were harvested by centrifugation (500 g, 3 min at room temperature). Cell lysates were prepared from the pellet as below for the measurement of intracellular Hsp60, Hsp70 and lactate dehydrogenase (LDH). The culture medium was removed from each cell pellet and transferred to clean centrifuge tubes. The media obtained were centrifuged (500 g, 3 min at room temperature), decanted and transferred to clean tubes. This was repeated twice, and the media were checked to ensure that no cells remained in the samples. Medium from GCT cells was removed from each well by pipette, and cells were resuspended in 1 ml PBS. All samples were stored at −80°C prior to measurement. Hsp60 and Hsp70 were quantified in medium and cell extracts using Hsp60 and Hsp70 enzyme-linked immunosorbent assay (ELISA) kits (Stressgen Biotechnologies, EKS-600 and EKS-700).
Western blotting
Cell lysates and media were suspended in 1 ml of extraction buffer [0·1% Triton X-100, 100 mM KCl, 8 mM MgCl2, 150 mM NaCl, 20 mM Tris-HCl pH 7·4 and 1 mM phenylmethylsulphonyl fluoride (PMSF)].
The cellular and media samples (10 µl per sample) and a control sample (5 µl), recombinant human Hsp60 control (Stressgen, ESP-540E) or recombinant human Hsp70 (Stressgen, ESP-555E), were diluted 1:1 in sodium dodecyl sulphate (SDS) sample buffer, boiled for 3 min, then separated on 8% SDS polyacrylamide gels and transferred to a nitrocellulose membrane. The blots were blocked for 1 h in 1% b ovine serum albumin (BSA) in Tris-buffered saline (2·85 g Tris, 29·24 g NaCl, in 1 dm3, pH 7·5) and then probed with the relevant primary antibody, murine anti-Hsp60 monoclonal antibody (Stressgen, SPA-806) or murine anti-Hsp70 monoclonal antibody (Stressgen, SPA-810) at a concentration of 1 µg/ml for 1 h at room temperature. The primary antibody was detected using anti-mouse IgG peroxidase (Sigma, A-5278) at a concentration of 1:1000 for 1 h at room temperature. The blot was developed using metal enhanced 3, 3′-diaminobenzidine (DAB) substrate (Pierce, 34065, Illinois, IL, USA).
Lactate dehydrogenase (LDH) assay
A reaction mixture was prepared consisting of: 2·6 ml glycine buffer, 0·6 M, pH 9·2 (Sigma, 826–3), 3 mg sodium lactate (Sigma, L-7022), 10 mg nicotinamide adenine dinucleotide (NAD) (Sigma, 260–110) and 3·4 ml distilled water. Media samples (50 µl/well) and cell extracts (50 µl/well) were added to a 96-well microplate alongside LDH standards (Sigma, 826–6). The reaction mixture was added to the plate (100 µl/well) and the plate read at 340 nm at 2-min intervals up to 30 min.
Cell counting and viability staining
Total cell counts and cell viability counts were carried out on all samples before and after treatments using a haemocytometer. Counts to determine cell viability were performed by diluting the suspended cell sample 1:1 with Trypan blue (Sigma, T-8154); cells that took up the blue stain were counted as non-viable.
Statistical analysis
All data are presented as the means ± standard errors of three separate experiments. Difference between sample means was evaluated through use of Student’s t-test or Dunnett’s post hoc multiple comparison test.
Results
PBMCs incubated at 37°C for 24 h released Hsp70 (Fig. 1a). LPS caused a significant increase (P < 0·001) in Hsp70 release (Fig. 1a). Incubation with GroEL also resulted in a significant increase (P < 0·001) in Hsp70 release (> 10-fold) (Fig. 1A). Incubation with Hsp60 for 24 h caused a small reduction (P < 0·05) in Hsp70 release from PBMCs (Fig. 1a). Jurkat cells incubated at 37°C for 24 h also released Hsp70 (Fig. 1b). Jurkat cells showed a similar pattern of Hsp70 release to PBMCs in response to Hsp60, GroEL and LPS treatments (Fig. 1b): LPS caused Hsp70 release to increase (P < 0·01); GroEL caused a 10-fold increase (P < 0·001); Hsp60 had no significant effect on Hsp70 release.
Fig. 1.
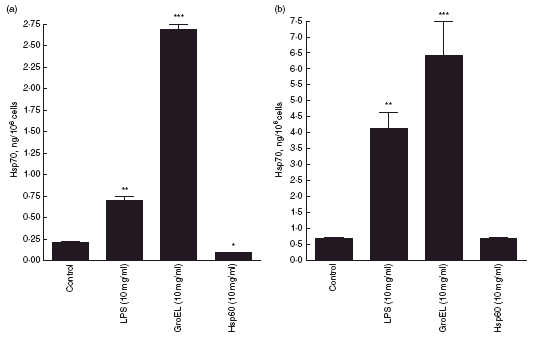
Heat shock protein (Hsp)70 release into culture media from peripheral blood mononuclear cells (a) and Jurkat cells (b) following incubation with lipopolysaccharide (LPS) (10 µg/ml), GroEL (10 µg/ml) and Hsp60 (10 µg/ml) for 24 h. Data presented as mean ± s.e.m., n = 3. *Indicates significant difference from 37°C control; *P < 0·05; **P < 0·01; ***P < 0·001.
PBMCs incubated at 37°C for 24 h released low amounts of Hsp60 (Fig. 2a). Following 24 h incubation of PBMCs with LPS, Hsp60 release was significantly increased (P < 0·01) (Fig. 2a). Incubation with GroEL or Hsp70 caused significant increases (P < 0·001) in Hsp60 release from PBMCs (Fig. 2a). Jurkat cells incubated at 37°C for 24 h released Hsp60 (Fig. 2b). Incubation of Jurkat cells with LPS resulted in a rate of Hsp60 release which was not significantly different from the control (Fig. 2b). However, incubation with GroEL caused a significant increase (P < 0·001) in Hsp60 (Fig. 2b). Treatment with Hsp70 resulted in a >10-fold increase (P < 0·001) in Hsp60 release (Fig. 2b).
Fig. 2.
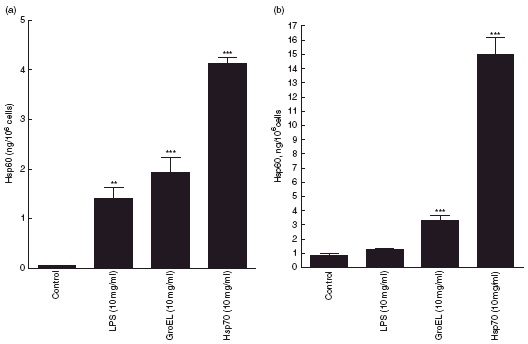
Heat shock protein (Hsp)60 release into culture media from peripheral blood mononuclear cells (a) and Jurkat cells (b) following incubation with lipopolysaccharide (LPS) (10 µg/ml), GroEL (10 µg/ml) and Hsp70 (10 µg/ml) for 24 h. Data presented as mean ± s.e.m., n = 3. *Indicates significant difference from 37°C control; **P < 0·01; ***P < 0·001).
Western blots confirmed that the Hsp60 and Hsp70 release responses observed from PBMCs (Fig. 3) and Jurkat (data not shown) were due to the release of complete 60 kDa and 70 kDa Hsps. The intracellular enzyme LDH was used as a control for damage related protein release. In control cells (PBMCs and Jurkat cells) the release of LDH was 0·95% and 1·03%, respectively (Table 1). The release of Hsp70 from cells under normal conditions was significantly greater in terms of the total cell contents (P < 0·001) than these rates (12·72% and 3·40%, respectively). The release of Hsp60 from these control cells was not significantly different from the percentage LDH release (0·85% and 1·66%, respectively) (Table 1). Only LPS caused a small increase in percentage LDH release in both cell types (Table 1). However, in all cases when Hsp70 and Hsp60 release was increased (to 20% or more) the LDH release remained below 3% (Table 1), indicating that cell damage was not responsible for the effects observed. Percentage cell viability as determined by Trypan blue showed no significant difference between controls and treated samples following 24 h incubation (data not shown). MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay results for PBMCs and Jurkat indicated that 24 h treatment with Hsp60, Hsp70 and LPS had no effect on cell number/metabolic activity. GroEL caused a small, but significant, increase in cell number/metabolic activity (data not shown).
Fig. 3.
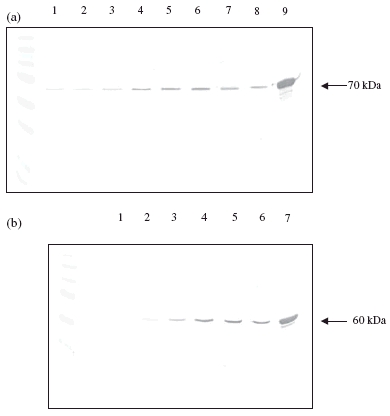
Western blots showing the presence of heat shock protein (Hsp)60 and Hsp70 in peripheral blood mononuclear cells media and cell extracts following 24 h treatment with Hsp60, Hsp70, GroEL and lipopolysaccharide (LPS). (a) Hsp70 in media: lane 1, control; lane 2, Hsp60 (10 µg/ml); lane 3, LPS (10 µg/ml); lane 4, GroEL (10 µg/ml). Hsp70 in cells: lane 5, control; lane 6, Hsp60 (10 µg/ml); lane 7, LPS (10 µg/ml); lane 8, GroEL (10 µg/ml); lane 9, Hsp70 standard. (b) Hsp60 in media: lane 1, control; lane 2, LPS (10 µg/ml); lane 3, Hsp70 (10 µg/ml). Hsp60 in cells: lane 4, control; lane 5, LPS (10 µg/ml); lane 6, Hsp70 (10 µg/ml); lane 7, Hsp60 standard.
Table 1.
Percentage release of heat shock protein (Hsp)70, Hsp60 and lactate dehydrogenase (LDH). Hsp70, Hsp60 and LDH were measured in culture media following incubation with lipopolysaccharide (LPS) (10 µg/ml), GroEL (10 µg/ml), Hsp60 (10 µg/ml) and Hsp70 (10 µg/ml) for 24 h. Percentage release was calculated from measurements of protein released and that present in cell extracts.
| % Release | |||
|---|---|---|---|
| Treatment | PBMC | Jurkat | |
| Hsp70 release | Control | 12·72 ± 0·82 | 3·40 ± 0·18 |
| % total cell Hsp70 | LPS | 19·16 ± 0·74 | 29·14 ± 1·47 |
| n = 3 | GroEL | 71·35 ± 1·98 | 24·18 ± 5·11 |
| Hsp60 | 8·78 ± 0·15 | 3·97 ± 0·27 | |
| Hsp60 release | Control | 0·85 ± 0·13 | 1·66 ± 0·24 |
| % total cell Hsp60 | LPS | 20·27 ± 1·96 | 2·91 ± 0·13 |
| n = 3 | GroEL | 33·66 ± 2·04 | 5·99 ± 0·48 |
| Hsp70 | 59·22 ± 0·49 | 26·93 ± 1·39 | |
| LDH release | Control | 0·95 ± 0·19 | 1·03 ± 0·28 |
| % of total cell LDH | LPS | 1·87 ± 0·38 | 2·74 ± 0·83 |
| n = 3 | GroEL | 1·22 ± 0·27 | 1·81 ± 0·40 |
| Hsp60 | 1·67 ± 0·79 | 0·87 ± 0·08 | |
| Hsp70 | 2·06 ± 0·18 | 1·87 ± 0·39 | |
PBMC: peripheral blood mononuclear cells.
Following the observed release from PBMCs and Jurkat cells it was decided that a non-lymphocyte cell line would be investigated for effects of the same treatments. Hsp70 was released from GCT cells under normal cell culture conditions during 24 h cell culture at 37°C. Both LPS and Hsp60 inhibited (P < 0·01) Hsp70 release from GCT cells, while GroEL caused a large increase (P < 0·001) (Fig. 4). GCT osteoblast-like cells did not release any Hsp60 under control conditions or following treatment with LPS for 24 h (Fig. 4). Hsp60 release increased significantly (P < 0·001) following treatment with both GroEL and Hsp70.
Fig. 4.
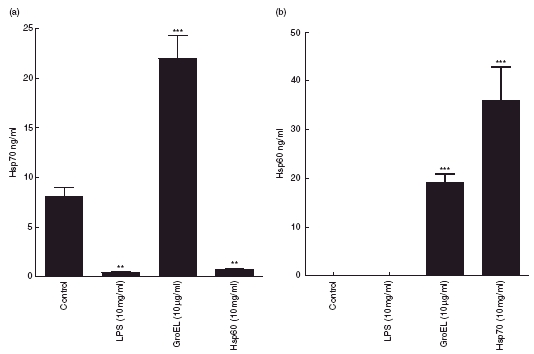
Heat shock protein (Hsp)70 (a) and Hsp60 (b) release into culture media from giant cell tumour (GCT) cells following 24 h treatment with lipopolysaccharide (LPS) (10 µg/ml), GroEL (10 µg/ml), Hsp60 (10 µg/ml) and Hsp70 (10 µg/ml). Data presented as mean ± s.e.m., n = 3. Indicates significant difference from 37°C control; **P = < 0·01; ***P = < 0·001.
Intracellular concentrations of Hsp60 and Hsp70 were measured following all treatments (Table 2). Hsp60 treatment resulted in a decrease in intracellular Hsp70 in PBMCs (P < 0·05) and GCTs (P < 0·05) (Table 2). GroEL resulted in a decrease in intracellular Hsp70 in all cells (P < 0·05) and intracellular Hsp60 in PBMCs (P < 0·05) (Table 2). The effect of LPS on intracellular Hsp70 concentration was an increase in PBMCs (P < 0·05) and a decrease in GCTs (P < 0·05) (Table 2). The effect of LPS on intracellular Hsp60 was to cause a decrease in PBMCs and Jurkat cells (P < 0·05) and an increase in GCTs (P < 0·05) (Table 2). Hsp70 resulted in an increase in intracellular Hsp60 in PBMCs (P < 0·05) and a decrease in Jurkat cells (P < 0·01) (Table 2).
Table 2.
Intracellular heat shock protein (Hsp)70 and Hsp60 from peripheral blood mononuclear cells (PBMCs), Jurkat cells and giant cell tumour cells (GCTs) following incubation with lipopolysaccharide (LPS) (10 µg/ml), GroEL (10 µg/ml), Hsp70 (10 µg/ml) and Hsp60 (10 µg/ml) for 24 h.
| PBMC ng/106 cells | Jurkat ng/106 cells | GCT ng/ml | |
|---|---|---|---|
| Hsp70 | |||
| Control | 1·50 ± 0·08 | 19·61 ± 0·30 | 21·40 ± 0·78 |
| Hsp60 | 1·03 ± 0·04* | 16·45 ± 0·18 | 15·65 ± 0·06* |
| GroEL | 1·08 ± 0·07* | 13·00 ± 0·35* | 11·71 ± 1·06* |
| LPS | 2·93 ± 0·04 | 15·57 ± 1·69 | 10·21 ± 1·30* |
| Hsp60 | |||
| Control | 3·66 ± 0·08 | 58·32 ± 3·77 | 22·69 ± 2·09 |
| Hsp70 | 5·25 ± 0·09* | 40·42 ± 1·19** | 29·36 ± 1·34 |
| GroEL | 2·03 ± 0·19* | 51·64 ± 3·01 | 33·90 ± 1·94* |
| LPS | 3·53 ± 0·02* | 43·59 ± 2·14* | 35·35 ± 0·27* |
Data presented as mean ± s.e.m., n = 3. *Indicates significant difference from 37°C control
P < 0·05
P < 0·01.
Discussion
There is an increasing amount of evidence that Hsps have cytokine-like functions and may act as danger signals to the innate immune system [1–3,39,40]. If the role of Hsp release from cells is to act as a danger signal then it would seem reasonable that their release would be increased following exposure to bacterial infection. We have previously demonstrated release of Hsp70 from PBMCs [36]. The aim of this work was to investigate whether Hsp70 and/or Hsp60 release from cells could be up-regulated following exposure to LPS, GroEL or human Hsps.
Hsp70 was released by PBMCs, a T cell line (Jurkat) and a primary GCT when cultured at 37°C for 24 h. Under these conditions the release of Hsp60 was not significantly greater than the release of LDH from cells in percentage terms. These data suggest that under normal conditions the cells are releasing Hsp70, but not Hsp60. In general, Hsp60 and Hsp70 release from PBMCs and Jurkat cells was found to be increased following exposure to LPS or GroEL. Human Hsp70 also stimulated Hsp60 release from these cells. Cell viability counts and LDH measurements established that the responses observed were not due to cellular damage and Western blots confirmed the release of complete Hsp60 and Hsp70 proteins. The intracellular concentrations are presented in Table 2.
LPS caused a significant increase in Hsp70 and Hsp60 release from PBMCs and Hsp70 release from Jurkat cells. However, LPS did not affect Hsp60 release from Jurkat or GCT cells and inhibited Hsp70 release from GCT cells. One pathway through which LPS is known to affect cells is via CD14/TLR4 or TLR2 receptors [41]. CD14 receptors are not present on T cells and are not normally present on osteoblasts [42]; the response of Hsp60 release to LPS follows this CD14 pattern of expression. The differences between the effects of LPS on Jurkat and GCT cells suggest that there are also differences in the signalling pathways leading to Hsp70 or Hsp60 release. Although GroEL decreased the intracellular concentrations in several treatments, these decreases are not sufficient to account for the increases in release. The suggestion that Hsp60 and Hsp70 release are regulated processes, involving gene expression and membrane transport rather than simply the result of mass flow, is supported by the intracellular data.
A response identified in all cell lines (immune and non-immune) was that of elevated Hsp70 release in response to GroEL, but not to human Hsp60. These patterns of Hsp release suggested that there are different receptors and/or responses to human and bacterial Hsp60. This is supported further by the difference between the stimulation of Hsp60 release by Hsp70 and GroEL. Responses to GroEL have been shown to be species specific within bacteria [43] and microbial and mammalian Hsp60 compete for different binding sites [44]. These differences are consistent with the Hsp danger signal hypothesis.
We have therefore demonstrated that Hsp70 and Hsp60 release can be up-regulated by exposure to bacterial antigens. We have shown previously that Hsp70 is released from both B and T lymphocytes [36] and we suggest that both these cell types are probably involved in the responses to bacterial antigens observed in this study. Confirmation of the role of each cell type in this response is worthy of further study. Hsp70 and Hsp60 have been demonstrated to be capable of activating monocytes, macrophages and dendritic cells [11,45], and also of inducing secretion of a wide range of cytokines [11,13,18,20,39]. Hsp70 has also been shown here to stimulate Hsp60 release in all cells tested. Conversely, Hsp60 had no effect on Hsp70 release in Jurkat cells, but inhibited release in PBMCs and GCT cells. Overall, these results, combined with those on the bacterial antigens, indicate different receptors and pathways are involved in the responses [20,21].
Although Hsps and LPS have been demonstrated to act through a common pathway involving CD14 [13,15,18,39] there are reports that some responses of cells to recombinant Hsps are due to LPS contamination [46,47]. The methods adopted in this paper have taken the possibility of LPS contamination into consideration. Hsp preparations were treated with polymyxin B or boiled at 100°C for 10 min. The effects on Hsp release from Jurkat cells were unaffected by the former and completely removed by the latter (data not shown). The Hsp70 preparations used in these experiments have been shown to contain LPS [47]. We have repeated experiments on Jurkat cells with the low LPS Hsp preparations (Hsp60, ESP-540E; and Hsp70, ESP-555E, Stressgen). It is possible that the treatments described did not remove all LPS activity. However, we observed differential responses to LPS and the recombinant Hsps, and the responses to GroEL and Hsp70 were significantly greater than responses to LPS. Therefore, despite the fact that Hsp60 strongly binds LPS [48], we suggest that either LPS contamination cannot be totally responsible for the results obtained, or that the biological activity of LPS is altered when bound to the Hsp.
Bacterial infection causes an increase in serum Hsp70 [2]. We have presented evidence that bacterial antigens stimulate Hsp70 and Hsp60 release from cells of the immune system. The danger model requires a signalling system in response to pathogens [49,50]. The data presented here support the hypothesis that Hsp70 and Hsp60 form part of a danger-signalling cascade. HSPs have a dual role in such a system; first, as bacterial antigens molecules such as GroEL (as well as LPS) are recognized as non-self and the immune system reacts. One of these reactions, described in this paper, is the release of Hsp60 and Hsp70. These proteins have differential effects on the cells of the immune system and can therefore be considered immunoregulatory. There are potential consequences of this signalling role of Hsps in other situations where Hsps are elevated in serum, such as exercise [29] and psychological stress [2].
Many questions remain regarding the signalling pathways and receptors involved in the observed responses, although it is clear that the release observed to this point could have important implications for demonstrating the role of Hsps as chemokines and for their causative and protective roles in disease processes.
Acknowledgments
The work was funded by a University of Chester Gladstone Bursaries awarded to E. D. and M. M. F. V. G. B.
References
- 1.Prohaszka Z, Singh M, Nagy K, et al. Heat shock protein 70 is a potent activator of the human complement system. Cell Stress Chaperones. 2002;7:17–22. doi: 10.1379/1466-1268(2002)007<0017:hspiap>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campisi J, Leem TH, Fleshner M. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones. 2003;8:272–86. doi: 10.1379/1466-1268(2003)008<0272:sehiaf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W, Syldath U, Bellmann K, Burkart V, Kolb W. Human 60-kDa heat-shock protein: a danger signal to the innate immune system. J Immunol. 1999;162:3212–9. [PubMed] [Google Scholar]
- 4.Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–11. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- 5.Muller MR, Wiesmiuller KH, Jung G, et al. Lipopeptide adjuvants. monitoring and comparison of P3CSK4- and LPS-induced gene transcription. Int Immunopharmacol. 2002;2:1065–77. doi: 10.1016/s1567-5769(02)00030-9. [DOI] [PubMed] [Google Scholar]
- 6.Multhoff G. Activation of natural killer cells by heat shock protein 70. Int J Hyperthermia. 2002;18:576–85. doi: 10.1080/0265673021000017109. [DOI] [PubMed] [Google Scholar]
- 7.Gross C, Hansch D, Gastpar R, Multhoff G. Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol Chem. 2003;384:267–79. doi: 10.1515/BC.2003.030. [DOI] [PubMed] [Google Scholar]
- 8.Friedland JS, Shattock R, Remick DG, Griffin GE. Mycobacterial 65-kD heat shock protein induces release of proinflammatory cytokines from human monocytic cells. Clin Exp Immunol. 1993;91:58–62. doi: 10.1111/j.1365-2249.1993.tb03354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Retzlaff C, Yamamoto Y, Hoffman PS, Friedman H, Klein TW. Bacterial heat shock proteins directly induce cytokine mRNA and interleukin-1 secretion in macrophage cultures. Infect Immun. 1994;62:5689–93. doi: 10.1128/iai.62.12.5689-5693.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peetermans WE, Raats CJI, Langermans JAM, van Furth R. Mycobacterial heat-shock protein 65 induces proinflammatory cytokines but does not activate human mononuclear phagocytes. Scand J Immunol. 1994;39:613–17. doi: 10.1111/j.1365-3083.1994.tb03421.x. [DOI] [PubMed] [Google Scholar]
- 11.Kol A, Bourcier T, Lichman AH, Libby P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest. 1999;103:571–7. doi: 10.1172/JCI5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galdiero M, Cipollaro G, Marcatilli A. Cytokine and adhesion molecule expression in human monocytes and endothelial cells stimulated with bacterial heat shock proteins. Infect Immun. 1997;65:699–707. doi: 10.1128/iai.65.2.699-707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 14.Tabona P, Reddi K, Khan S, et al. Homogeneous Escherichia coli chaperonin 60 induces IL-1 and IL-6 gene expression in human monocytes by a mechanism independent of protein conformation. J Immunol. 1998;161:1414–21. [PubMed] [Google Scholar]
- 15.Bulut Y, Faure E, Thomas L, et al. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J Immunol. 2002;168:1435–40. doi: 10.4049/jimmunol.168.3.1435. [DOI] [PubMed] [Google Scholar]
- 16.Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol. 2000;164:13–17. doi: 10.4049/jimmunol.164.1.13. [DOI] [PubMed] [Google Scholar]
- 17.Cohen-Sfady M, Nussbaum G, Pevsner-Fischer M, et al. Heat shock protein 60 activates B cells via the TLR4-MyD88 pathway. J Immunol. 2005;175:3594–602. doi: 10.4049/jimmunol.175.6.3594. [DOI] [PubMed] [Google Scholar]
- 18.Vabulas RM, Ahmad-Nejad P, da Costa C, et al. Endocytosed HSP60s use Toll-like receptor 2 (TLR2) and TLR4 to activate the Toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–9. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 19.Habich C, Baumgart K, Kolb H, Burkart V. The receptor for heat shock protein 60 on macrophages is saturable, specific, and distinct from receptors for other heat shock proteins. J Immunol. 2002;168:569–76. doi: 10.4049/jimmunol.168.2.569. [DOI] [PubMed] [Google Scholar]
- 20.Basu S, Binder RJ, Ramalingham T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–13. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 21.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70 − role of Toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 22.Tsuzuki T, Ina K, Ohta M, et al. Clarithromycin increases the release of heat shock protein B from Helicobacter pylori. Aliment Pharmacol Ther. 2002;16:217–28. doi: 10.1046/j.1365-2036.16.s2.23.x. [DOI] [PubMed] [Google Scholar]
- 23.Hennequin C, Collignon A, Karjalainen T. Analysis of expression of GroEL (Hsp60) of Clostridium difficile in response to stress. Microb Pathog. 2001;31:255–60. doi: 10.1006/mpat.2001.0468. [DOI] [PubMed] [Google Scholar]
- 24.Frisk A, Ison CA, Lagergard T. GroEL heat shock protein of Haemophilus ducreyi: association with cell surface and capacity to bind to eukaryotic cells. Infect Immun. 1998;66:1252–7. doi: 10.1128/iai.66.3.1252-1257.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holoshitz J, Kosek J, Sibley R, Brown DA, Strober S. Lymphocyte-T–synovial fibroblast interactions induced by mycobacterial proteins in rheumatoid-arthritis. Arthritis Rheum. 1991;34:679–86. doi: 10.1002/art.1780340608. [DOI] [PubMed] [Google Scholar]
- 26.Botzler C, Li G, Issels RD, Multhoff G. Definition of extracellular localized epitopes of Hsp70 involved in an NK immune response. Cell Stress Chaperones. 1998;3:6–11. doi: 10.1379/1466-1268(1998)003<0006:doeleo>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzhova IV, Arnholdt ACV, Darieva ZA, et al. Effects of exogenous stress protein 70 on the functional properties of human promonocytes through binding to cell surface and internalization. Cell Stress Chaperones. :67–77. doi: 10.1379/1466-1268(1998)003<0067:eoespo>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gastpar R, Gross C, Rossbacher L, Ellwart J, Riegger J, Multhoff G. The cell surface-localized heat shock protein 70 epitope TKD induces migration and cytolytic activity selectively in human NK cells. J Immunol. 2004;172:972–80. doi: 10.4049/jimmunol.172.2.972. [DOI] [PubMed] [Google Scholar]
- 29.Walsh RC, Koukoulas I, Garnham A, Moseley PL, Hargreaves M, Febbraio MA. Exercise increases serum Hsp72 in humans. Cell Stress Chaperones. 2001;6:386–93. doi: 10.1379/1466-1268(2001)006<0386:eishih>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pockley AG, de Faire U, Kiessling R, Lemne C, Thulin T, Frostegard J. Circulating heat shock protein and heat shock protein antibody levels in established hypertension. J Hypertens. 2002;20:1815–20. doi: 10.1097/00004872-200209000-00027. [DOI] [PubMed] [Google Scholar]
- 31.Pockley AG, Shepherd J, Corton JM. Detection of heat shock protein 70 (Hsp70) and anti-Hsp70 antibodies in the serum of normal individuals. Immunol Invest. 1998;27:367–77. doi: 10.3109/08820139809022710. [DOI] [PubMed] [Google Scholar]
- 32.Hunter-Lavin C, Hudson PR, Mukherjee S, et al. Folate supplementation reduces serum Hsp70 levels in patients with type 2 diabetes. Cell Stress Chaperones. 2004;9:344–9. doi: 10.1379/CSC-28R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barreto A, Gonzalez JM, Kabingu E, Asea A, Fiorentino S. Stress-induced release of HSC70 from human tumors. Cell Immunol. 2003;222:97–104. doi: 10.1016/s0008-8749(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 34.Guzhova I, Kislyakova K, Moskaliova O, et al. In vitro studies show that Hsp70 can be released by glia and exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res. 2001;914:66–73. doi: 10.1016/s0006-8993(01)02774-3. [DOI] [PubMed] [Google Scholar]
- 35.Broquet AH, Thomas G, Masliah J, Trugnan G, Bachelet M. Expression of the molecular chaperone Hsp70 in detergent- resistant microdomains correlates with its membrane delivery and release. J Biol Chem. 2003;278:21601–6. doi: 10.1074/jbc.M302326200. [DOI] [PubMed] [Google Scholar]
- 36.Hunter-Lavin C, Davies EL, Bacelar MMFVG, Marshall MJ, Andrew SM, Williams JHH. Hsp70 release from periperal blood mononuclear cells. Biochem Biophys Res Commun. 2004;324:511–17. doi: 10.1016/j.bbrc.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 37.Evdonin AL, Guzhova IV, Margulis BA, Medvedeva ND. Phospholipase c inhibitor, u73122, stimulates release of hsp-70 stress protein from A431 human carcinoma cells. Cancer Cell Int. 2004 doi: 10.1186/1475-2867-4-2. 4/1/2. http://www.cancerci.com/content/4/1/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao DF, Jin ZG, Baas AS, et al. Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J Biol Chem. 2000;275:189–96. doi: 10.1074/jbc.275.1.189. [DOI] [PubMed] [Google Scholar]
- 39.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–12. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 40.Zanin-Zhorov A, Bruck R, Tal G, et al. Heat shock protein 60 inhibits Th1-mediated hepatitis model via innate regulation of Th1/Th2 transcription factors and cytokines. J Immunol. 2005;174:3227–36. doi: 10.4049/jimmunol.174.6.3227. [DOI] [PubMed] [Google Scholar]
- 41.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 42.Reyes-Botella C, Montes MJ, Vallecillo-Capilla MFV, Olivares EG, Ruiz C. Antigenic phenotype of cultured human osteoblast-like cells. Cell Physiol Biochem. 2002;12:359–64. doi: 10.1159/000067906. [DOI] [PubMed] [Google Scholar]
- 43.Maguire M, Coates ARM, Henderson B. Chaperonin 60 unfolds its secrets of cellular communication. Cell Stress Chaperones. 2002;7:317–29. doi: 10.1379/1466-1268(2002)007<0317:cuisoc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habich C, Kempe K, van der Zee R, Burkart V, Kolb H. Different heat shock protein 60 species share pro-inflammatory activity but not binding sites on macrophages. FEBS Lett. 2003;533:105–9. doi: 10.1016/s0014-5793(02)03772-9. [DOI] [PubMed] [Google Scholar]
- 45.Bethke K, Staib F, Distler M, et al. Different efficiency of heat shock proteins (HSP) to activate human monocytes and dendritic cells: superiority of HSP60. J Immunol. 2002;169:6141–8. doi: 10.4049/jimmunol.169.11.6141. [DOI] [PubMed] [Google Scholar]
- 46.Gao BC, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor a release by murine macrophages. J Biol Chem. 2003;278:174–9. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- 47.Gao BC, Tsan MF. Induction of cytokines by heat shock proteins and endotoxin in murine macrophages. Biochem Biophys Res Commun. 2004;317:1149–54. doi: 10.1016/j.bbrc.2004.03.160. [DOI] [PubMed] [Google Scholar]
- 48.Habich C, Kempe K, van der Zee R, et al. Heat shock protein 60. Specific binding of lipopolysaccharide. J Immunol. 2005;174:1298–305. doi: 10.4049/jimmunol.174.3.1298. [DOI] [PubMed] [Google Scholar]
- 49.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 50.Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. doi: 10.1006/smim.1998.0143. [DOI] [PubMed] [Google Scholar]