

Abstract
Immunologically mediated thrombocytopenia is a frequent clinical manifestation in patients with systemic lupus erythematosus (SLE). Autoantibodies targeting platelet membrane glucoproteins have a central role in peripheral platelet destruction. Autoantibodies against thrombopoietin are also present in about one-third of patients, but their pathogenetic role is obscure. Thirty-eight serum samples from SLE patients were tested for anti-platelet antibodies, anti-thrombopoietin antibodies and levels of circulating thrombopoietin. Bone marrow histology was also assessed. Thirty-nine per cent of sera displayed anti-thrombopoietin antibodies and 29% had circulating anti-platelet antibodies. Anti-thrombopoietin antibodies were associated with lower thrombopoietin concentrations, and lower mean platelet values in long-term follow-up. Anti-platelet antibodies were present in about 40% of thrombocytopenic and non-thrombocytopenic individuals but were absent in patients who had recovered from thrombocytopenia, supporting their pathogenetic role. Both autoantibodies were absent in control sera from patients with rheumatoid arthritis and primary Sjögren’s syndrome. Decreased bone marrow cellularity, normal or low number of hypolobulated, pyknotic megakaryocytes and stromal alterations were prominent findings in thrombocytopenic SLE patients, suggesting a defect in megakaryopoiesis. These findings were not evident in specimens from patients with idiopathic thrombocytopenic purpura who had increased megakaryocytes, normal cellularity and absence of stromal alterations. In conclusion, peripheral destruction due to platelet autoantibodies, anti-thrombopoetin antibodies, lower effective circulating thrombopoetin and impaired compensatory response due to bone marrow damage interact in SLE and thrombocytopenia ensues.
Keywords: autoantibodies, bone marrow, pathogenesis, systemic lupus erythematosus, thrombocytopenia, thrombopoietin
Introduction
Systemic lupus erythematosus (SLE) is the prototype of systemic autoimmune disorders, primarily affecting women of childbearing age. Despite progress made in the last decades, the disease remains a cause of irreversible organ damage and increased morbidity. Previous studies have reported that thrombocytopenia due to peripheral consumption of platelets is a common clinical manifestation, ranging from 7 to 30%[1–4]. Anti-platelet antibody formation is by far the most common pathogenetic mechanism [5,6]. Anti-phospholipid antibodies [7,8], thrombotic microangiopathies [9,10] and haemophagocytic syndrome [11] have also been implicated. The common denominator is the presence of normal or increased megakaryocytes (MKs) in the bone marrow. Recently, a few cases of thrombocytopenia have been identified, with amegakaryocytic hypoplasia due to antibodies against the c-mpl receptor [12]. Thrombopoietin (TPO), the c-mpl ligand, is the key regulator of platelet production. Serum levels of TPO are higher in SLE compared to normal controls. Autoantibodies against TPO have also been detected in 23% of the sera of lupus patients [13]. Their exact role (if any) in the pathogenesis of lupus thrombocytopenia is still undefined. In addition, despite the well-defined role of anti-platelet autoantibodies in increased platelet turnover, it is widely known that only a proportion of patients develop true thrombocytopenia, rendering their predictive value insignificant. It has been demonstrated recently that patients with active disease and complement activation are more susceptible to thrombocytopenia than their matched controls for age, sex and disease duration [14]. However, other distinctive clinical or serological profiles were not detected.
In this study we evaluated the role of autoantibodies against platelet antigens and antibodies against TPO in adult patients with SLE.
Patients and methods
Patients
Thirty-eight consecutive SLE patients according to the revised ACR criteria [15] were referred for consultation to the haematology laboratory of the department of pathophysiology. Serum samples were taken from these patients and stored. At the time of specimen collection, 11 patients were thrombocytopenic, 13 had a past history of thrombocytopenia (post-thrombocytopenic) and the remaining 14 had no history of thrombocytopenia (non-thrombocytopenic) during their disease course. Thrombocytopenia was defined as a platelet level of less than 100 000/µl, in compliance with American College of Rheumatology criteria (normal range for healthy subjects 150 000–450 000/µl). In these patients, blood smears were examined to assess morphology and verify platelet counts. At the time of collection, disease activity and complement components were assessed. Disease activity was measured using the European Consensus Lupus Activity Measurement (ECLAM score). Serum C3 and C4 were measured by nephelometry. Each sample was tested for the presence of anti-platelet antibodies (anti-PLTs), anti-TPO antibodies and levels of circulating TPO. Twenty randomly selected sera from patients with rheumatoid arthritis (RA) and primary Sjögren’s syndrome (pSS) without thrombocytopenia served as controls. The records of post-thrombocytopenic individuals were evaluated retrospectively for 10 consecutive months after the occurrence of thrombocytopenia. Platelet number, therapies and anti-cardiolipin antibodies were also recorded for the same period and individual profiles were constructed.
All patients who participated in our study were fully informed of the aim of this study. They provided written consent for their participation and agreed that the results of this study may be presented or published, solely in the interests of science, provided that their anonymity is maintained. We also confirm that this study was approved and authorized by the Scientific Board of Athens University Medical School, Clinical–Pathological Division (President Professor D. Kelekis) and conforms to standards defined by our university authorities.
Measurement of TPO levels
TPO levels were quantified by a commercial sandwich enzyme immunoassay technique (Quantikine; R&D Systems, Mineapolis, USA) according to the manufacturer’s instructions. Briefly, samples were pipetted into enzyme-linked immunosorbent assay (ELISA) plates precoated with an anti-TPO monoclonal antibody. After washing away any unbound substances, an anti-TPO polyclonal antibody conjugated with alkanine phosphatase was added. Following a washing step, a substrate solution was added and the colour was developed (in proportion to the amount of TPO bound in the initial step) and measured at 450 nm.
Detection of antibodies against TPO
For the detection of anti-TPO IgG antibodies in serum samples we utilized a modified commercial assay, designed originally for the measurement of TPO (Quantikine; R&D Systems). Preliminary experiments using recombinant human TPO (recTPO; R&D Systems) demonstrated that the anti-TPO precoated ELISA plates can be saturated with antigen by 1 h incubation at 37°C with 80 ng/ml recTPO in 0·1% bovine serum albumin (BSA)/phosphate-bufferd saline (PBS), pH = 7·2 (200 µl/well). The saturated with recTPO ELISA plates were then washed three times with the kit’s wash buffer, and incubated for 90 min with 200 µl/well of diluted sera (1:100 in 0·1% BSA/PBS, pH = 7·2). After washing away any unbound substances (seven times), anti-human IgG conjugated with alkanine phosphatase (Jackson Immunoresearch, West Grove, PA, USA) was added at a dilution of 1:1000 in 0·1% BSA/PBS, pH = 7·2. Following nine washes with the kit’s wash buffer, Para-Nitro-Phenyl-Phosphate (PNPP) substrate solution (Sigma-Aldrich, St Louis, MO, USA) was added and the colour was measured at 405 nm. Positive cut-off was set as the mean OD of 30 sera from healthy donors plus 3 standard deviations (s.d.).
Detection of circulating antibodies against platelet surface antigens
The detection of circulating antibodies against platelet surface antigens was performed using a commercially available assay (PAK12, GTI Diagnostics, Wauksesha, WI, USA), according to the manufacturer’s instructions. This assay allows the simultaneous detection of antibodies against platelet glycoproteins IIb/IIIa, Ia/IIa, Ib/IX and HLA class I antigens.
Bone marrow findings
Representative trephine biopsies of the bone marrow (BM) were taken from the posterior iliac crest, fixed in 10% buffer formalin for 12–24 h, rapidly decalcified in ethylenediamine tetraacetic acid (EDTA) for 3 h [16,17] and after dehydration embedded to paraffin according to standard protocols. BM cellularity was assessed by visual examination according to Harstock et al. [18] and graded into three groups, as follows: normocellular (30–50% of intertrabecular space occupied by haematopoietic tissue), hypercellular (> 50% of intertrabecular space occupied by haematopoietic tissue) and hypocellular ( < 30% of intertrabecular space occupied by haematopoietic tissue). BM cellularity was further corrected by age according to Tuzuner et al. [19] to avoid misclassification. The amount of erythroid, myeloid and megakaryocytic precursors was assessed as decreased, normal or increased by simultaneously taking into account BM cellularity and the myeloid–erythroid ratio [20]. BM necrosis was assessed according to Norgard et al. [21]. Fibrosis was assessed semiquantitatively by reticulin content and graded from 0 to 4, as described previously by Bauermeister et al. [22]. To ensure the accuracy of all the semiquantitative grading methods, histological slides were reviewed independently by two observers, without prior knowledge of specific clinical data, in blinded fashion. BM findings in all thrombocytopenic patients were compared with 10 specimens from patients with idiopathic thrombocytopenic purpura (ITP) also referred to our department. Bone marrow biopsies were not taken from SLE patients without thrombocytopenia, or from control (RA and pSS) populations.
Statistical analysis
Continuous variables were compared using the Mann–Whitney U-test or t-test if normality assumed. For categorical variables Pearson’s χ2 was implemented. Post-thrombocytopenic individuals were analysed using methods for longitudinal data analysis. Platelet number was the dependent variable. Panel corrected standard error (PCSE) estimates for linear time-series were used. Unstructured covariance matrix was used and patient-specific first-order autocorrelation AR(1) assumed within each patient data. Predicted profiles were plotted for anti-TPO-positive and anti-TPO-negative individuals. Level of significance was set to 0·05. The stata version 8 package was used for analysis.
Results
SLE patients were 38·6 ± 15·5 years old and had a mean disease duration of 11·4 ± 6·7 years. Median ECLAM score was 1·5 (range 0–6), C3 levels were 71·9 ± 30·9 mg/dl and C4 levels were 17·1 ± 11·1 mg/dl. RA and pSS controls were matched for sex and age with SLE patients. A distinct clinical profile for thrombocytopenic SLE patients was not evident. Four of 11 thrombocytopenic patients had received no medication at blood sampling. The remaining seven samples were taken from patients who had received low-dose corticosteroids ± azathioprine (four patients) or combination therapy with high-dose corticosteroids + cyclophosphamide pulses (two patients due to concomitant renal disease and one due to refractory thrombocytopenia with PLTs < 10 000/µl).
Anti-platelet antibodies
Autoantibodies against platelet membrane antigens were detected in the sera of 11 patients (29%). Eighty-eight per cent (eight of 11) of them targeted the Gp IIb/IIIa complex and at a lower frequencies the Gp Ia/IIa, HLA I and Gp Ib/Ix complex. Almost 16% of patients had different anti-platelet antibodies with variable antigenic specificity (Table 1a). The presence of anti-platelet antibodies was unrelated to serum C3 levels and disease activity.
Table 1.
(a) Prevalence of anti-platelet (anti-PLTs) and anti-thrombopoietin (anti-TPO) autoantibodies in the sera of lupus patients; (b) post-thrombocytopenic sera do not display autoantibody activity against platelet antigens.
| Antibody | Current | Non-thrombocytopenic | Post-thrombocytopenic | Total |
|---|---|---|---|---|
| (a) | ||||
| Anti-PLTs | 5/11 (45%) | 6/14 (43%) | 0/13 (0%) | 11/38 (29%) |
| Anti-GpIIb/IIIa | 3/11 (27%) | 5/14 (36%) | 0/13 (0%) | 8/38 (21%) |
| Anti-GpIa/IIa | 3/11 (27%) | 4/14 (29%) | 0/13 (0%) | 7/38 (18%) |
| Anti-GpIb/IX | 0/11 (0%) | 2/14 (14%) | 0/13 (0%) | 2/38 (5%) |
| Two or more | 1/11 (9%) | 5/14 (36%) | 0/13 (0%) | 6/38 (16%) |
| Anti-HLA I | 1/11 (9%) | 2/14 (14%) | 0/13 (23%) | 3/38 (8%) |
| Anti-TPO | 4/11 (36%) | 6/14 (43%) | 5/13 (38%) | 15/38 (39%) |
| (b) | Frequency (%) | P-value | ||
| Post-thrombocytopenic versus thrombocytopenic | 0/13 (0%) versus 5/11 (45%) | 0·006 | ||
| Post-thrombocytopenic versus non-thrombocytopenic | 0/13 (0%) versus 6/14 (43%) | 0·007 | ||
The prevalence of anti-platelet antibodies in the thrombocytopenic and non-thrombocytopenic groups did not differ, accounting for about 40% in each group. Notably, none of the post-thrombocytopenic individuals exhibited autoantibody activity against platelets (see differences in Table 1b). This group had received more intense immunosuppression in comparison to non-thrombocytopenic, consisting of pulse cyclophosphamide treatment (7/13 versus 2/14, P = 0·03) and restored normal platelet counts.
Prevalence of anti-TPO antibodies and TPO-levels
Anti-TPO IgG antibodies were measured using a sandwich ELISA, resulted by modification of a commercial assay, designed originally for the measurement of TPO. This assay maintains the TPO in its natural conformation, avoiding the denaturation effects that usually occur after the absorption of the antigen by the solid surface. By using this ‘natural TPO’ recognition assay, anti-TPO antibodies were detected in the sera of 15 patients (39%). The presence of these antibodies was associated with lower serum TPO concentrations (74 pg/ml versus 147 pg/ml, P = 0·04), although unrelated to platelet number at the time of specimen collection.
Overall, SLE patients had higher TPO concentrations compared to both RA (117·3 versus 45·7 pg/ml, P = 0·02) and pSS (117·3 versus 39·2 pg/ml, P = 0·02) samples.
Anti-TPO antibodies, as anti-PLTs, were notably absent in patients with RA and pSS (Fig. 1), possibly indicating that anti-TPO antibodies represent a disease-specific immunological disturbance, constrained to SLE patients.
Fig. 1.
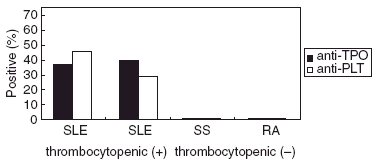
Both anti-platelet antibodies (anti-PLTs) and anti-thrombopoietin (anti-TPO) antibodies were detected in systemic lupus erythematosus (SLE) patients with or without thrombocytopenia. These antibodies were absent in patients with rheumatoid arthritis and primary Sjögren’s syndrome control sera.
The prevalence of anti-TPO antibodies did not vary between current and post-thrombocytopenic individuals compared to non-thrombocytopenic individuals (Table 2). Whether anti-TPO antibodies are truly blocking antibodies of physiological significance or are simply an epiphenomenon preventing the detection of TPO/anti-TPO complex by the ELISA used, is a matter of debate.
Table 2.
Anti-thrombopoietin (anti-TPO)-positive sera have lower TPO concentrations, although unassociated with lower platelet counts.
| Anti-TPO positive | Anti-TPO negative | P-value | |
|---|---|---|---|
| TPO-levels (mean ± s.d.) | 74 pg/ml (± 66) | 147 pg/ml (± 166) | 0·04 |
| Platelets (mean ± s.d.) | 226·2 ± 84·9 × 103/µl | 210·2 ± 116·2 × 103/µl | n.s. |
| Current thrombocytopenic | Non-thrombocytopenic and post-thrombocytopenic | ||
| Anti-TPO-positive | 4/11 | 11/27 | n.s. |
| Current thrombocytopenic and post-thrombocytopenic | Non-thrombocytopenic | ||
| Anti-TPO-positive | 9/24 | 6/14 | n.s. |
| RA | pSS | ||
| TPO levels (mean ± s.d.) | 45·7 ± 35·3 | 49·2 ± 33·5 | 0·02* |
Compared to systemic lupus erythematosus (SLE) mean TPO levels (117·3 ± 137·9 pg/ml). pSS: primary Sjögren’s syndrome; RA: rheumatoid arthritis.
A role for anti-TPO antibodies
Anti-TPO antibodies in the sera of SLE patients is seen to be associated with a lower circulating TPO level. To assess the impact of these antibodies on circulating platelets we performed a repeated-measurement data analysis of post-thrombocytopenic individuals who had monthly measurements of platelet counts from the onset of thrombocytopenia and for a total period of 10 consecutive months. Eleven post-thrombocytopenic patients were included in the analysis and individual profiles were constructed. Most of the anti-TPO positive patients had platelet kinetics fluctuating near low values (Fig. 2a).
Fig. 2.

(a) Individual profiles for anti-thrombopoietin antibodies (anti-TPOs)-positive (red lines) and anti-thrombopoietin (anti-TPO)-negative (blue lines) post-thrombocytopenic systemic lupus erythematosus (SLE) subjects for a period of 10 months. Each platelet measurement is marked with a circle or a triangle, respectively. (b) Plotting the average predicted profile of anti-TPO positive (red) versus anti-TPO negative (blue) post-thrombocytopenic individuals, according to the previous multivariate model, reveals that the former tend to have lower platelet counts throughout the 10-month observation period.
A multivariate analysis was conducted, after being adjusted for possible confounders such as corticosteroid treatment (low dose versus high dose), cyclophosphamide use, intravenous globulin administration and the presence of anti-phospholipid antibodies. The results are summarized in Table 3. The presence of anti-TPO antibodies is associated with a lower mean platelet value (− 67·16 × 103/µl, P = 0·002) during the 10-month-observation period. After plotting the average predicted platelet profile, stratified for anti-TPO negative versus anti-TPO positive individuals, the former maintain normal platelet values and the latter significantly lower values (Fig. 2b).
Table 3.
Multivariate analysis; anti-thrombopoietin (anti-TPO)-positive patients maintain lower platelet counts compared to anti-TPO negative patients (lower mean value −67·16 × 103/µl) after adjusting for other confounders (R2 0·56).
| Variable | Coefficient§ | P-value | 95% CIs* |
|---|---|---|---|
| Anti-TPO (+ versus–) | − 67·16 | 0·002 | − 110·2 to −24·1 |
| Corticosteroids (high versus low dose) | − 66·40 | 0·002 | − 108·0 to −24·7 |
| IVIG (yes versus no) | − 105·54 | 0·000 | − 155·9 to −55·2 |
| Cyclophosphamide (dose, mg) | − 0·02 | 0·377 | − 0·08 to 0·03 |
| Antiphospholipid antibodies (+ versus–) | 75·56 | 0·000 | 35·3 to 115·8 |
| Constant | 268·18 | 0·000 | 183·9 to 352·4 |
Positive, + negative, –
mean platelet difference (× 103/µl)
confidence intervals.
Bone marrow findings
BM histology in all 11 SLE patients with thrombocytopenia displayed a decrease in overall cellularity and normal or low normal MK numbers. These MKs were hypolobulated, pyknotic and often revealed denuded cytoplasm, so-called ‘naked’ MKs (Fig. 3a). Megakaryocytic atypias, including recurrent hypolobulated forms, pyknotic megakaryocytes with denuded cytoplasm and pronounced abnormalities of megakaryocyte differentiation generating bizarre cells, were detected in all patients. BM necrotic alterations were evident in all specimens (Table 4). The morphology of BM necrosis on histological sections was distinctive. A smooth homogeneous basophilic background protein staining was often present. In addition, an increase in eosinophilic granular stroma was identified, along with the ghosts of many dead haematopoietic cells (yellow arrow). Other evidence of bone marrow necrosis included the decrease in overall cellularity, prominent stromal oedema (black arrow) and variable reticulin fibrosis (Table 4, Fig. 3b). On the contrary, ITP biopsies showed a uniform pattern of increased MKs, normal cellularity and notably absence of stromal alterations, such as necrosis (Fig. 3c).
Fig. 3.

(a). Bone marrow biopsy in systemic lupus erythematosus (SLE) thrombocytopenia: hypolobulated, pyknotic megakaryocytes (arrow) [haematoxylin and eosin (H&E)]. (b). Bone marrow biopsy in systemic lupus erythematosus (SLE) thrombocytopenia: necrosis (yellow arrow) and stromal oedema (black arrow) (H&E). (c). Bone marrow biopsy in idiopathic thrombocytopenic purpura (ITP): increased number of morphologically normal megakaryocytes (arrows), normal cellularity, absence of stromal alterations are evident in an ITP patient.
Table 4.
Histological findings in thrombocytopenic systemic lupus erythematosus (SLE) patients. Dysplastic megakaryocytes (MKs) and stromal changes are dominant features.
| Necrosis | ||||||
|---|---|---|---|---|---|---|
| hr/ | ||||||
| No. Age | Erythroid | Myeloid | MKs (no. /high power field) | Global cellularity | Oedema/haemorrhage | Fibrosis |
| 1 | Increased (+) | Decreased (+) | 2/field | Normal | Present (+) | Present (+) |
| 35 | Dyserythropoiesis | Left shift | Dysplastic | |||
| 2 | Increased (+) | Normal | 3–4/field | Hypocellular | Present (3 +) | Present (2 +) |
| 22 | Dyserythropoiesis | Left shift | Dysplastic | |||
| 3 | Increased (+) | Decreased (2 +) | 3/field | Hypocellular | Present (+) | Present (+) |
| 25 | Dyserythropoiesis | Dysplastic | ||||
| 4 | Increased (2 +) | Decreased (+) | 1–2/field | Hypocellular | Present (+) | Absent |
| 24 | Dyserythropoiesis | Dysplastic | ||||
| 5 | Increased (+) | Decreased (+) | 1/field | Hypocellular | Present (+) | Present (+) |
| 32 | Dyserythropoiesis | Dysplastic | ||||
| 6 | Increased (+) | Decreased (+) | 1–2/field | Hypocellular | Present (2 +) | Present (+) |
| 45 | Dyserythropoiesis | Dysplastic | ||||
| 7 | Normal | Decreased (2 +) | 4/field | Hypocellular | Present (+) | Absent |
| 65 | Left shift | Dysplastic | ||||
| 8 | Increased (3 +) | Normal | 1–2/field | Hypercellular | Present (+) | Present (+) |
| 44 | Dyserythropoiesis | Left shift | Dysplastic | |||
| 9 | Increased (+) | Decreased (+) | 1/field | Normal | Present (3 +) | Present (2 +) |
| 34 | Dyserythropoiesis | Left shift | Dysplastic | |||
| 10 | Increased (+) | Decreased | 2–3/field | Normal | Present (+) | Present (+) |
| 50 | Dyserythropoiesis | Dysplastic | ||||
| 11 | Increased (+) | Decreased (+ 2) | 1/field | Hypocellular | Present (2 +) | Present (+) |
| 27 | Dyserythropoiesis | Left shift | Dysplastic | |||
Discussion
IIb/IIIa antigen (or more specifically αiiα3 integrin) is the most abundant platelet membrane glycoprotein, and represents the main antigenic target in autoimmune thrombocytopenias, including that of SLE. Circulating and platelet-bound anti-PLTs have been implicated in the pathogenesis of lupus thrombocytopenia, as sensitized platelets interact mainly with splenic macrophages through the Fc receptor and are eliminated from the circulation. Despite the high prevalence of these antibodies among thrombocytopenic individuals already described, a number of patients display thrombocytopenia without anti-PLT positivity, and a significant proportion of anti-PLT positive patients have never developed thrombocytopenia [23–25]. Moreover, a correlation between anti-PLT antibodies and lower platelet counts is not evident in our data, confirming previous reports [26]. However, the fact that anti-PLTs are absent in the sera of SLE patients who had recovered from thrombocytopenia after receiving immunosuppressive treatment confirms a potential pathogenetic role, although not an exclusive one. Published data have shown that circulating and platelet-bound anti-PLTs are noted in a significant proportion of patients (SLE, ITP) who have active thrombocytopenia. In contrast, when platelets normalize in responders, they become undetectable or decrease significantly and reappear in relapse [6,27]. For the detection of anti-platelet antibodies, a qualitative solid phase enzyme linked immunosorbent assay, designed for the detection of antibodies to IIb/IIIa, Ia/IIa and Ib/IX platelet glycoproteins, was used. In contrast to platelet suspension immunofluorescence test (PSIFT) assay, the method used in our study has the ability to distinguish antibodies to platelet-specific antigens. monoclonal antibody immobilization of platelet antigens (MAIPA) is an alternative method for the detection of specific anti-platelet antibodies that employs monoclonal antibodies for immobilization of platelet antigens. The method used in our study is commercially available and its ability to recognize antigenic specificities is similar to MAIPA (for details, see United States Patent 5514557).
Anti-TPO antibodies were a frequent finding in our cohort (39%), comparable with previously published data [13]. Thrombocytopenic and post-thrombocytopenic individuals did not differ from those without thrombocytopenia, indicating that anti-TPO antibodies are a feature of SLE that remains stable over time. This hypothesis is further supported as the prevalence of anti-TPO antibodies in heavily immunosuppressed, post-thrombocytopenic patients does not differ.
Remarkably, however, anti-TPO positive patients had significantly lower circulating levels of TPO compared to an anti-TPO negative group, whereas a previous study [13] displayed statistically insignificant differences. The ELISA used for the detection of anti-TPO IgG antibodies was a sandwich enzyme immunoassay technique, utilizing recombinant-TPO captured by a monoclonal anti-TPO antibody as antigen. By this specific method a monoclonal anti-TPO antibody (absorbed in the plastic surface of ELISA plates) holds the antigen (recombinant TPO) in its native conformation, avoiding major denaturation effects (that usually accompany the absorption to the plastic) and steric hindrance issues (caused by the plastic surface). Although this modified technique employs recombinant human TPO instead of human serum TPO, the ligand for monoclonal anti-TPO antibody in both cases is human TPO. Hence, it should be considered a more specific technique in detecting circulating antibodies. Therefore, a more accurate detection of anti-TPO antibodies may reveal their physiological role in reducing circulating TPO levels.
Whether anti-TPO antibodies are truly blocking antibodies with physiological importance or are simply a side epiphenomenon preventing the detection of TPO/anti-TPO complex by the ELISA used, is a matter of debate. When present in high concentrations, antibodies may bind avidly with natural TPO and their binding characteristics may favour an ability to strip and eliminate systemic TPO by capturing the protein in immune complexes. Hence, a number of SLE patients produce antibodies that are likely to neutralize their own TPO. Therefore, their contribution to thrombocytopenic states may occur in a bimodal fashion: first, by engendering immune complexes, a non-specific mechanism that enhances peripheral PLT consumption. Platelets have both immunoglobulin receptor (FcR) and complement receptors (C1q, CR2, CR4) which enable platelets to interact in immunological reactions. Normal human platelets express a single Fc gamma receptor (FcRII, designated CD32), having polymorphic variants that differ in their ability to bind IgG and complexes [28,29]. The cell membrane of platelets is also equipped with several regulatory complement proteins (DAF, MCP, MIRL, C8bp) which protect them from complement attack induced by the binding of platelet autoantibodies or non-specific immune complexes. It has been shown that blood cells and platelets act not only as effector cells in the inflammatory process but are also involved in transporting the complexes thus clearing them from circulation. This mechanism may prove clinically important when less efficient handling of immune complexes in complement deficient individuals leads to high immune complex loads or when an increased production of complexes causes a high antigen load, such as in SLE, resulting in an increased platelet turnover [30,31]. A direct association between disease activity and PLT counts was not evident in our study. Indeed, cohort studies are less efficient in detecting marginal differences, especially when sample size is relatively small. Matched case–control studies [14] are more effective in this regard. Secondly, decreasing the effective TPO concentrations for stimulating megakaryopoiesis may also contribute to thrombocytopenia. Human TPO is the crucial regulator of PLT production, acting as a growth factor in the commited progenitor cells [colony-forming units of megakaryocytes (CKU-MK)], differentiating immature megakaryoblasts, inhibiting apoptosis and leading finally to the release of normal platelets. Morphologically, it increases the number, size and ploidy of MK [32–34]. It has no impact on normal platelets. MKs and platelet mass determine TPO levels in the serum, as TPO production is constant [35,36]. Post-thrombocytopenic SLE individuals display different platelet kinetics after occurrence of thrombocytopenia in relation to their anti-TPO status. Anti-TPO positive patients had a lower mean predicted profile for platelet counts after adjusting for other confounders, providing a plausible pathogenetic role for anti-TPO in SLE thrombocytopenia.
It is evident, however, that anti-TPO antibodies alone are not associated directly with thrombocytopenia, but possibly consitute a part of a more complex mechanism. These antibodies may represent a unique phenomenon in SLE, as our data did not support their presence in other common autoimmune processes, such as RA and pSS. Both types of autoantibodies (anti-PLTs and anti-TPO) seem to participate in the complex pathophysiological events related to thrombocytopenia in SLE, leading to peripheral platelet turnover and reducing the effective circulating TPO, respectively. Seven patients had both anti-PLT and anti-TPO reactivity in their sera; however, three of them were thrombocytopenic. The absence of thrombocytopenia ( < 100 000/µl) in double-positive patients suggests that these autoantibodies do not have an exclusive pathogenetic role in platelet destruction. Half the thrombocytopenic individuals (five of 11) had no evidence of either anti-PLTs or anti-TPO antibodies in their sera. Other mechanisms, such as the early clearance and destruction of platelets coated with non-specific immune complexes, may also contribute to peripheral thrombocytopenia in SLE, as previously suggested [24].
However, a normal bone marrow can overcome these defects by increasing megakaryopoiesis. Our histological findings suggested that bone marrow in SLE patients displays decreased cellularity, lower MK concentrations and stromal alterations such as necrosis. Non-specific abnormalities, such as BM hypocellularity, increased reticulin and necrosis were underscored as the most prominent features of the BM histology in two published series of 23 and 21 SLE patients, respectively [37,38]; contradictory data were obtained by another study, in which BM findings were apparently normal [39]. BM necrosis is best defined as necrosis of haemopoietic and medullary stroma in large areas of the haemopoietic BM. The best accepted mechanism of BM necrosis is the vascular obstruction in the BM, which leads to ischaemia and subsequent necrosis. However, in our study mechanical obstruction of the microcirculation by thrombus plugs or inflammatory vascular damage was not detected making the pathogenesis of BM necrosis in SLE patients unclear. It has been shown that stromal cells exhibit a defective haematopoietic microenvironment that produces altered cytokine expression resulting in faulty haematopoiesis in SLE. Evidence for the culpability of BM stroma in SLE haemopoietic failure derived from culture experiments in which stromal cells from SLE patients failed to support allogenic progenitor cells growth [40]. It has also been shown that, as a result of a diminished activity of monocytes, the production of haemopoietic growth factors by BM fibroblasts is insufficient, a fact that could explain SLE haematological abnormalities [41]. The cause and the underlying pathogenetic mechanisms of these abnormalities are presently unknown, but it seems to be related to the presence of autoreactive lymphocytes in the BM of patients with SLE, which may affect the haemopoietic cells and supporting capacity of BM stroma not only via direct immune destruction but also by an indirect effect due to release of proinflammatory cytokines such as tumour necrosis factor (TNF)-α[42]. In this regard, BM necrosis in SLE may be the result of an abnormal microenvironment characterized by deregulated production of haematopoietic colony-stimulating factors due to damaged stromal cells. Lymphoid aggregates were present in the majority of specimens, mainly located central-perivascularly or in deeper BM spaces with a mixture of B and T lymphocytes, as well as a polyclonal expression of immunoglobulins by immunohistochemistry. The lymphocytic component did not exceed 30%. These findings suggest that bone marrow is a target organ in SLE. As TPO-receptor availability, and therefore MK mass, is the major determinant of circulating TPO, numerical and morphological alterations in MKs in conjunction with decreased global cellullarity in SLE can justify increased serum TPO concentrations. Such damage may impair TPO ability to mount an adequate compensatory response to increased platelet turnover, defining a subset of patients who, in the presence of these autoantibodies, are likely to develop thrombocytopenia. However, a central pathophysiological role of BM in SLE and decreased platelet production (resulting in true thrombocytopenia) could not be excluded.
Better understanding of the underlying mechanisms may confer new therapeutic interventions: detailed structural and functional studies of the TPO receptor, coupled with novel molecular pharmacology approaches, have led to new classes of thrombopoietic mimetics. Initial clinical trials with recombinant TPOs faltered as they encountered significant neutralizing antibodies. Ongoing studies have invigorated the field, with positive results now reported in idiopathic thrombocytopenic purpura [43]. Moreover, given that interleukin-11 is a potential growth factor for MK proliferation and maturation, a therapeutic advantage may be gained by administering this cytokine to patients with SLE and other forms of immune-mediated thrombocytopenia. Optimal dosing and indications still remain a matter of debate [44,45]. Finally, it has become clear that the B lymphocyte plays a key role in SLE pathogenesis by both autoantibody-dependent and autoantibody-independent mechanisms. New agents that directly target abnormal immune cells in SLE include the B cell depleting or modulating antibodies, rituximab (anti-CD20) and epratuzumab (anti-CD22). Another promising approach has been to block co-stimulatory interactions between T and B cells, for example by inhibiting the CD40–CD40 ligand pathway with anti-CD40 ligand monoclonal antibody [46]. All these alternative approaches may prove promising in the near future.
In summary, specific anti-PLT antibodies, non-specific, immune complex-mediated PLT destruction, anti-TPO antibodies and primary bone marrow defect constitute a puzzle of interactions related to SLE thrombocytopenia. Anti-PLT antibodies seem to have a more prominent role, as they disperse in patients who recover after cytotoxic therapy, whereas anti-TPO antibodies although weak remain persistent. Their exact role, however, warrants further investigation.
Acknowledgments
The study was kindly supported by research funding from Genesis Pharma SA.
References
- 1.Cervera R, Khamashta MA, Font J, et al. Morbidity and mortality in systemic lupus erythematosus during a 5-year period. A multicenter prospective study of 1,000 patients. Medicine (Baltimore) 1999;78:167–75. doi: 10.1097/00005792-199905000-00003. European Working Party on Systemic Lupus Erythematosus. [DOI] [PubMed] [Google Scholar]
- 2.Wang F, Wang CL, Tan CT, Manivasagar M. Systemic lupus erythematosus in Malaysia: a study of 539 patients and comparison of prevalence and disease expression in different racial and gender groups. Lupus. 1997;6:248–53. doi: 10.1177/096120339700600306. [DOI] [PubMed] [Google Scholar]
- 3.Vila LM, Alarcon GS, McGwin G, Jr, et al. for the LUMINA Study Group. Early clinical manifestations, disease activity and damage of systemic lupus erythematosus among two distinct US Hispanic subpopulations. Rheumatology (Oxford) 2004;43:358–63. doi: 10.1093/rheumatology/keh048. [DOI] [PubMed] [Google Scholar]
- 4.Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000;39:399–406. doi: 10.1093/rheumatology/39.4.399. [DOI] [PubMed] [Google Scholar]
- 5.Michel M, Lee K, Piette JC, et al. Platelet autoantibodies and lupus-associated thrombocytopenia. Br J Haematol. 2002;119:354–8. doi: 10.1046/j.1365-2141.2002.03817.x. [DOI] [PubMed] [Google Scholar]
- 6.Lipp E, von Felten A, Sax H, Muller D, Berchtold P. Antibodies against platelet glycoproteins and antiphospholipid antibodies in autoimmune thrombocytopenia. Eur J Haematol. 1998;60:283–8. doi: 10.1111/j.1600-0609.1998.tb01041.x. [DOI] [PubMed] [Google Scholar]
- 7.Macchi L, Rispal P, Clofent-Sanchez G, et al. Anti-platelet antibodies in patients with systemic lupus erythematosus and the primary antiphospholipid antibody syndrome: their relationship with the observed thrombocytopenia. Br J Haematol. 1997;98:336–41. doi: 10.1046/j.1365-2141.1997.2243038.x. [DOI] [PubMed] [Google Scholar]
- 8.Diz-Kucukkaya R, Hacihanefioglu A, Yenerel M, et al. Antiphospholipid antibodies and antiphospholipid syndrome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood. 2001;98:1760–4. doi: 10.1182/blood.v98.6.1760. [DOI] [PubMed] [Google Scholar]
- 9.Porta C, Bobbio-Pallavicini E, Centurioni R, Caporali R, Montecucco CM. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus. J Rheumatol. 1993;20:1625–6. Italian Cooperative Group for TTP. [PubMed] [Google Scholar]
- 10.Porta C, Caporali R, Montecucco C. Thrombotic thrombocytopenic purpura and autoimmunity: a tale of shadows and suspects. Haematologica. 1999;84:260–9. [PubMed] [Google Scholar]
- 11.Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633–9. doi: 10.1002/art.11368. [DOI] [PubMed] [Google Scholar]
- 12.Kuwana M, Okazaki Y, Kajihara M, et al. Autoantibody to c-Mpl (thrombopoietin receptor) in systemic lupus erythematosus: relationship to thrombocytopenia with megakaryocytic hypoplasia. Arthritis Rheum. 2002;46:2148–59. doi: 10.1002/art.10420. [DOI] [PubMed] [Google Scholar]
- 13.Fureder W, Firbas U, Nichol JL, et al. Serum thrombopoietin levels and anti-thrombopoietin antibodies in systemic lupus erythematosus. Lupus. 2002;11:221–6. doi: 10.1191/0961203302lu177oa. [DOI] [PubMed] [Google Scholar]
- 14.Ziakas PD, Giannouli S, Zintzaras E, Tzioufas AG, Voulgarelis M. Lupus thrombocytopenia: clinical implications and prognostic significance. Ann Rheum Dis. 2005;64:1366–9. doi: 10.1136/ard.2004.033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan EM, Cohen AS, Fries JF, et al. The revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 16.Pileri SA, Roncador G, Ceccarelli C, et al. Antigen retrieval techniques in immunohistochemistry: comparison of different methods. J Pathol. 1997;183:116–23. doi: 10.1002/(SICI)1096-9896(199709)183:1<116::AID-PATH1087>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Pileri S. Malignant non-Hodgkin lymphomas: from method to diagnosis. Bologna: Societa Editrice Esculapio; 1985. [Google Scholar]
- 18.Hartsock RJ, Smith EB, Petty CS. Normal variations with aging of the amount of hematopoietic tissue in bone marrow from the anterior iliac crest: a study made from 177 cases of sudden death examined by necropsy. Am J Clin Pathol. 1965;43:326–31. doi: 10.1093/ajcp/43.4.326. [DOI] [PubMed] [Google Scholar]
- 19.Tuzuner N, Cox C, Rowe JM, Watrous D, Bennett JM. Hypocellular myelodysplastic syndromes (MDS): new proposals. Br J Haematol. 1995;91:612–17. doi: 10.1111/j.1365-2141.1995.tb05356.x. [DOI] [PubMed] [Google Scholar]
- 20.Foucar K. Bone marrow pathology. Chicago: American Society of Clinical Pathologists (ASCP) Press; 1995. [Google Scholar]
- 21.Norgard MJ, Carpenter JT, Jr, Conrad ME. Bone marrow necrosis and degeneration. Arch Intern Med. 1979;139:905–11. [PubMed] [Google Scholar]
- 22.Bauermeister DE. Quantitation of bone marrow reticulin − a normal range. Am J Clin Pathol. 1971;56:24–31. doi: 10.1093/ajcp/56.1.24. [DOI] [PubMed] [Google Scholar]
- 23.Mcmillan R. Immune thrombocytopenia. Clin Haematol. 1983;12:69–88. [PubMed] [Google Scholar]
- 24.Mcmillan R. Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. Semin Hematol. 2000;37:239–48. doi: 10.1016/s0037-1963(00)90102-1. [DOI] [PubMed] [Google Scholar]
- 25.Kurata Y, Hayashi S, Kosugi S, et al. Elevated platelet associated IgG in SLE patients due to antiplatelet autoantibody: differentiation between autoantibodies and immune complexes by ether elution. Br J Haematol. 1993;85:723–8. doi: 10.1111/j.1365-2141.1993.tb03215.x. [DOI] [PubMed] [Google Scholar]
- 26.Brighton TA, Evans S, Castaldi PA, Chesterman CN, Chong BH. Prospective evaluation of the clinical usefulness of an antigen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood. 1996;88:194–201. [PubMed] [Google Scholar]
- 27.Berchtold P, Wenger M. Autoantibodies against platelet glycoproteins in autoimmune thrombocytopenic purpura: their clinical significance and response to treatment. Blood. 1993;81:1246–50. [PubMed] [Google Scholar]
- 28.Fossati G, Bucknall RC, Edwards SW. Fcgamma receptors in autoimmune diseases. Eur J Clin Invest. 2001;31:821–31. doi: 10.1046/j.1365-2362.2001.00881.x. [DOI] [PubMed] [Google Scholar]
- 29.King M, Mcdermott P, Schreiber AD. Characterization of the Fc gamma receptor on human platelets. Cell Immunol. 1990;128:462–79. doi: 10.1016/0008-8749(90)90041-o. [DOI] [PubMed] [Google Scholar]
- 30.Hed J. Role of complement in immune or idiopathic thrombocytopenic purpura. Acta Paediatr Suppl. 1998;424:37–40. doi: 10.1111/j.1651-2227.1998.tb01231.x. [DOI] [PubMed] [Google Scholar]
- 31.Spycher MO, Nydegger UE. Participation of the blood platelet in immune reactions due to platelet–complement interaction. Infusionsther Transfusionsmed. 1995;22:36–43. doi: 10.1159/000223090. [DOI] [PubMed] [Google Scholar]
- 32.Kuter DJ, Beeler DL, Rosenberg RD. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Natl Acad Sci USA. 1994;91:11104. doi: 10.1073/pnas.91.23.11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaushansky K. Thrombopoietin: the primary regulator of platelet production. Blood. 1995;86:419–31. [PubMed] [Google Scholar]
- 34.Zucker-Franklin D, Kaushansky K. Effect of thrombopoietin on the development of megakaryocytes and platelets: an ultrastructural analysis. Blood. 1996;88:1632–8. [PubMed] [Google Scholar]
- 35.Emmons RV, Reid DM, Cohen RL, et al. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood. 1996;87:4068–71. [PubMed] [Google Scholar]
- 36.Chang M, Suen Y, Meng G, et al. Regulation of TPO mRNA expression and protein production: TPO gene regulation appears post transcriptional, and endogenous levels are inverseley correlated to megakaryocyte mass and circulating platelet count. Blood. 1995;86(Suppl 1):368a. [Abstract]. [Google Scholar]
- 37.Pereira RMR, Velloso ERP, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17:219–22. doi: 10.1007/BF01451051. [DOI] [PubMed] [Google Scholar]
- 38.Feng CS, Ng MH, Szeto RS, Li EK. Bone marrow findings in lupus patients with pancytopenia. Pathology. 1991;23:5–7. doi: 10.3109/00313029109061430. [DOI] [PubMed] [Google Scholar]
- 39.Xu XM, Fan ZR, Zhou SY, Wei W, Zhou BH, Cao YF. Hematological abnormality and clinical characteristics in systemic lupus erythematosus. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2004;12:170–3. [PubMed] [Google Scholar]
- 40.Papadaki HA, Boumpas DT, Gibson FM, et al. Increased apoptosis of bone marrow CD34+ cells and impaired function of bone marrow stromal cells in patients with systemic lupus erythematosus. Br J Haematol. 2001;115:167–74. doi: 10.1046/j.1365-2141.2001.03076.x. [DOI] [PubMed] [Google Scholar]
- 41.Otsuka T, Okamura S, Harada M, et al. Multipotent hemopoietic progenitor cells in patients with systemic lupus erythematosus. J Rheumatol. 1988;15:1085–90. [PubMed] [Google Scholar]
- 42.Alvarado-de la Barrera C, Alcocer-Varela J, Richaud-Patin Y, Alarcon-Segovia D, Llorente L. Differential oncogene and TNF-alpha mRNA expression in bone marrow cells from systemic lupus erythematosus patients. Scand J Immunol. 1998;48:551–6. doi: 10.1046/j.1365-3083.1998.00427.x. [DOI] [PubMed] [Google Scholar]
- 43.Solberg LA., Jr Biologic aspects of thrombopoietins and the development of therapeutic agents. Curr Hematol Rep. 2005;4:423–8. [PubMed] [Google Scholar]
- 44.Bussel JB, Mukherjee R, Stone AJ. A pilot study of rhuIL-11 treatment of refractory ITP. Am J Hematol. 2001;66:172–7. doi: 10.1002/1096-8652(200103)66:3<172::aid-ajh1041>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 45.Feinglass S, Deodhar A. Treatment of lupus-induced thrombocytopenia with recombinant human interleukin-11. Arthritis Rheum. 2001;44:170–5. doi: 10.1002/1529-0131(200101)44:1<170::AID-ANR22>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 46.Anolik JH, Aringer M. New treatments for SLE. cell-depleting and anti-cytokine therapies. Best Pract Res Clin Rheumatol. 2005;19:859–78. doi: 10.1016/j.berh.2005.05.006. [DOI] [PubMed] [Google Scholar]