

Abstract
The B7-1/B7-2-CD28/CTLA-4 pathway is crucial in regulating T cell activation and tolerance. Autoantibodies to surface molecules on lymphocytes have already been described in various immune conditions, such as autoimmune diseases, infections and blood transfusions. The objective of this study was to test sera from healthy individuals and from patients for association of CD28 autoantibodies with inflammatory and non-inflammatory diseases. First, CD28 was obtained by digestion of CD28-Ig fusion protein with trypsin. The cleavage products were separated by sodium dodecyl sulphate–page gel electrophoresis. Additionally, a CD28/GST fusion protein was expressed in Escherichia coli and was used to establish an enzyme-linked immunosorbent assay for detection of autoantibodies against CD28. Sera from healthy individuals (n = 72) and patients with different inflammatory and non-inflammatory skin diseases (n = 196) were tested for the presence of autoantibodies against CD28. Using mixed lymphocyte reaction (MLR), purified autoantibodies against CD28 were tested for their effects on CTLA-4-Ig-induced T cell anergy. In this study, for the first time, we describe the existence of autoantibodies against CD28 in humans which are associated with atopic diseases, e.g. allergic rhinitis and asthma. These antibodies stimulate T cells and overcome the CTLA-4-Ig-induced anergy of T cells in an MLR. The existence of autoantibodies against CD28, which may have a T cell-stimulating function, has been shown. The data indicate that autoantibodies against CD28 could be a new immunological mechanism in allergic inflammation. Additionally, autoantibodies against CD28 could be an important new marker to discriminate between atopic diseases and other inflammatory skin diseases.
Keywords: allergic asthma, allergic rhinitis, atopic dermatitis, autoantibodies, CD28
Introduction
Initiation of T cell-dependent immune responses requires at least two signals by the antigen-presenting cells (APC). One signal is antigen-specific and is mediated by the ligation of the T cell receptor (TCR) by antigen–major histocompatibility complex (MHC). However, this antigen-specific signal is not sufficient to generate a full response and, in the absence of a second signal, leads to clonal anergy. Successful immune responses require a second signal mediated either by binding of CD28 or CTLA-4 on T cells to their ligands CD80 (B7-1) and CD86 (B7-2) expressed on APC [1,2].
CD28, a 44-kDa membrane protein, is expressed on resting and activated T cells whereas CTLA-4 is found on activated T cells and is expressed constitutively on regulatory T cells (CD4+, CD25+) [3]. Initial activation of T cells after antigen exposure is mediated by CD28-CD80 (B7-1) interactions and induces proliferation and differentiation of effector T cells. In contrast, CTLA-4 binding to B7 ligands inhibits the response of activated T cells. Thus, CTLA-4 serves as a negative regulator of T cell activation. The abnormal expression of these co-stimulatory receptors can lead to an activation of self-reactive T cells, resulting in autoimmunity [1]. Moreover, linkage for the CTLA-4 and CD28 gene has been demonstrated in some autoimmune diseases and in atopic dermatitis (AD) [4]. Because of these important functions, alteration of the co-stimulatory pathway could result in susceptibility to immunological diseases.
Autoantibodies to surface molecules on lymphocytes (ALA) have been described in human autoimmune diseases, infections and blood transfusions [5,6]. The presence of ALAs was shown to be correlated with disease activity [7], lymphopenia [8], lymphocyte subset distortions [9,10] and functional abnormalities of T cells, B cells and monocytes [11–13]. So far, only very few targets of ALAs have been identified in humans [CD45, β2-microglobulin and human leucocyte antigen (HLA)-class I molecules][14–17] and in animals (CTLA-4) [18].
Therefore, it was the aim of this study to develop a method for detecting autoantibodies against CD28. Then, sera from healthy individuals and from patients with different types of inflammatory or non-inflammatory skin diseases were tested for the presence of autoantibodies against CD28.
Materials and methods
Patients and samples
Serum samples were obtained after informed consent from a group of randomly selected patients (n = 196) with various skin diseases treated at the Department of Dermatology, University Hospital Eppendorf, Hamburg. All patients with AD included (n = 16) belonged to the extrinsic subtype [19]. In the group of patients with autoimmune diseases two patients had scleroderma, three had autoimmune bullous skin disease and three were diagnosed with lupus erythematosus. Additionally, sera from a group of 72 healthy individuals were tested for presence or absence of CD28 autoantibodies (for numbers of subgroups see Table 1). The study was approved by the regional ethical committee. Informed written consent was obtained from each subject.
Table 1.
Association of diagnosis with anti-CD28 autoantibodies based on results from immunoblot.
| Total | Anti-CD28+ | Anti-CD28– | OR | 95% CI | P* | |
|---|---|---|---|---|---|---|
| Healthy controls | 72 | 8 | 64 | – | – | – |
| Atopic dermatitis | 16 | 14 | 2 | 56·00 | 28·4–110·4 | < 0·0001 |
| Allergic rhinitis/asthma | 54 | 31 | 23 | 10·78 | 5·39–21·55 | < 0·0001 |
| Autoimmune disease | 8 | 5 | 3 | 13·33 | 5·72–31·1 | < 0·01 |
| Cutaneous lymphoma | 3 | 1 | 2 | 4·00 | 0·71–22·5 | n.s. |
| Non-melanoma skin cancer | 27 | 9 | 18 | 4·00 | 1·72–9·30 | n.s. |
| Leg ulcer | 53 | 13 | 40 | 2·60 | 1·16–5·82 | n.s. |
| Cutaneous infections | 24 | 5 | 19 | 2·11 | 0·76–5·82 | n.s. |
| Other inflammatory skin diseases | 49 | 10 | 39 | 2·05 | 0·87–4·83 | n.s. |
| Psoriasis | 9 | 1 | 8 | 1·00 | 0·14–7·10 | n.s. |
| Malignant melanoma | 11 | 1 | 10 | 0·80 | 0·11–5·59 | n.s. |
CI: confidence interval; n.s.: not significant; OR: odds ratio.
P: Bonferroni-corrected P-value calculated by Fisher's exact test.
Digestion of human CD28-Ig fusion protein with trypsin
CD28-Ig fusion protein (R&D Systems, Minneapolis, MN, USA) was dissolved (1 mg/ml) in phosphate-buffered saline buffer [(PBS) containing 137 mM NaCl, 2·7 mM KCl, 7·4 mM Na2HPO4, 1·5 mM KH2PO4] and 100 µl of the solution was digested with 50 µl trypsin (10 mg/ml) at 37°C for 15 min. Subsequently, 1·5 µl aprotinin (10 mg/ml) and 2·5 µl N-tosyl-L-lysinechloromethyl ketone (TLCK) (20 mg/ml) were added to inhibit trypsin. The solution was stored at −20°C.
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot analysis
The cleavage products were separated via SDS-PAGE (10·0% gel) with a 4% stacking gel (110 V, 150 min) under non-reducing conditions. The cleavage products were then transferred (50 mA, 3 h) to polyvinyl difluoride (PVDF) membranes (Segin-Blot; Biorad, Munich, Germany). After blocking with 5% skimmed milk for 60 min at room temperature (RT) and washing in PBS (3×) the membranes were cut into strips. For control experiments the strips were incubated with the following antibodies: monoclonal mouse anti-human CD28 (R&D Systems), diluted 1:5000 in phosphate-buffered saline (PBS), biotinylated polyclonal mouse anti-human CD28 (R&D Systems), diluted 1:5000 in PBS, monoclonal mouse anti-human IgG Fc fragment (Dianova, Hamburg, Germany), diluted 1:10 000 in PBS and polyclonal rabbit anti-human IgG (Sigma-Aldrich, Steinheim, Germany), diluted 1:3500 in PBS. For detection of CD28 autoantibodies, the sera were diluted 1:10 in PBS. The strips were incubated for 1 h at room temperature (RT) in either antibody solution or diluted sera. After washing three times, the second antibody was added and the membranes were incubated for 1 h at RT. For detection of monoclonal mouse anti-human CD28 and mouse anti-human IgG Fc primary antibodies an alkaline phosphatase (AP)-conjugated anti-mouse IgG (Sigma-Aldrich) diluted 1:10 000 in PBS was used. For detection of polyclonal rabbit anti-human IgG an AP-conjugated goat anti-rabbit IgG (Sigma-Aldrich) diluted 1:10 000 in PBS was used. The membranes were again washed three times, and detection was performed with 5-bromo-4-chloro-3-inodolyl phosphate/nitroblue tetrazolium (BCIP/NBT) reagent (Sigma). Binding of the biotinylated polyclonal anti-human CD28 was detected using a streptavidin–AP conjugate (Sigma-Aldrich). After proper colour development, the membrane was washed in distilled water and dried in open air.
Expression of a CD28/GST-fusion protein in Escherichia coli
The reference sequence for human CD28 is deposited at NM-006139. A fragment encoding the immunoglobulin-like domain of CD28 was amplified from a human blood cDNA library with the following primers, which included suitable restriction sites (underlined) for cloning: sense AAAGAATTCCCTTCAATTCAAGTAACAGGAAAC; anti-sense AAACCCGGGAAATAGGGGACTTGGACAAAG. The amplified fragment was then cloned via EcoRI and SmaI into the pGEX-4T-1 vector (Amersham Biosciences, Munich, Germany) and transformed into the protease-deficient E. coli strain BL21-RIL (Stratagene, La Jolla, CA, USA) for large-scale expression. Cells were grown at 37°C with shaking in 500 ml LB (Luria-Bertani) medium (1% bacto-tryptone, 1% yeast extract, 100 mM NaCl) supplemented with 150 µl/ml ampicillin. When the turbidity (A600) reached 0·6 (after approximately 2 h), isopropylthio-beta-d-galactoside (IPTG) (Biomol) was added to 0·5 mM and growth continued for 4 h. The final A600 was approximately 1·2. Cells were then harvested by centrifugation at 4000 g at 4°C. The resulting pellet was resuspended in PBS and sonicated six times for 10 s each (Branson Sonifier 250) and centrifuged at 20 000 g for 20 min. For affinity purification glutathione-sepharose (Amersham Biosciences) was loaded onto a polypropylene column (Pierce) and equilibrated with 5 volumes of PBS. The bacteria lysate was loaded onto the column and the flow-through loaded once again. The column was subsequently washed with 10 volumes of PBS. Elution was performed with 10 mM reduced glutathione, 50 mM Tris pH 7·5, 100 mM NaCl, 10% glycerol. The eluate was stored at −80°C.
Enzyme-linked immunosorbent assay (ELISA) for detection of autoantibodies against CD28
Microtitre plates (Maxisorp, Nunc, Weisbaden, Germany) were coated with 100 µl of a monoclonal mouse anti-gluthatione-S-transferase (GST) antibody (specific for GST from Schistosoma japonicum, diluted 1:2000 in PBS for 1 h at RT. Afterwards, the plates were washed three times with PBS + 0·1% Tween 20 and blocked with 250 µl PBS containing 1% skimmed milk powder and 0·1% Tween 20 for 1 h at RT. After a second washing step the microtitre plates were incubated with 100 µl of CD28/GST fusion protein diluted 1:2 in PBS + 0·1% Tween 20 for 1 h at RT. Subsequently, the plates were washed, and sera as well as the positive control (polyclonal rabbit anti-human CD28 antibody from Santa Cruz, Heidelberg, 1 µg/ml, diluted 1:200 in PBS + 0·1% Tween 20) were added. The background binding of a negative serum was taken as control. Additionally, each serum was tested without CD28/GST fusion protein as an additional negative control. After washing the plates, 100 µl of the secondary antibodies for positive control (AP-conjugated monoclonal anti-rabbit IgG Fc antibody, diluted 1:5000, Sigma) and for sera (AP-conjugated monoclonal anti-human IgG Fc antibody, diluted 1:5000, Sigma) were incubated for 1 h at RT. Subsequently, the plates were washed five times and substrate [P-nitrophenyl phosphatase (pNPP) tablets; Sigma] was added and the reaction was measured after 60 min at 405 nm. The results for each sample were calculated using the following formula:
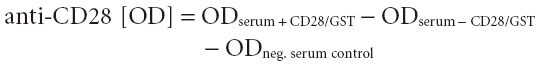 |
The cut-off point was calculated on the basis of 72 sera from healthy individuals. In this control group, in 99% of the donors negative for CD28 autoantibodies the OD was = 0·500, so that an OD > 0·500 was stated as positive for autoantibodies against CD28.
Purification of CD28 autoantibodies from patient's sera
Autoantibodies against CD28 were purified from patient's sera using Protein G beads (Dynal, Oslo, Norway). First, 100 µl of the digested CD28-Ig fusion protein was added to 200 µl of Dynabeads solution and incubated for 40 min at RT. The beads were washed twice in PBS and separated magnetically. After separation the beads were resuspended in 500 µl of 0·1 M PBS (pH 7·0). Subsequently, the labelled CD28-Ig fusion protein was cross-linked using triethanolamine (0·2 M, pH 8·2) and dimethyl pimilidate (20 mM in triethanolamine) for 30 min at RT. The reaction was stopped by resuspending the beads in 50 mM Tris-buffer (pH 7·5) for 15 min. After washing the beads three times with PBS, pooled serum (300 µl) was added to the beads and incubated for 45 min at RT. Two separate pools of serum from AD patients with CD28 autoantibodies and from AD patients without CD28 autoantibodies were used in these experiments. After separation of the beads, bound antibodies were eluted with 100 µl of 0·1 M citrate buffer (pH 2·7). After separation the supernatants were stored at −20°C.
Mixed lymphocyte reaction
Raji cells and Jurkat cells were generously provided by Dr Guse (University of Hamburg). Cell lines were maintained in RPMI-1640 supplemented with 10% FCS. MLR was conducted by culturing Jurkat cells (104 cells/ml) together with fresh irradiated (30 Gy) Raji cells (104/ml) as described previously [20]. After 3 days cellular proliferation of quadruplicate cultures was measured by uptake of bromodeoxyuridine (BrdU) during the last 5 h of culture, as described previously [21]. Purified sera (50 µl) with or without autoantibodies against CD28 were added alone or with 4 µg CTLA-4-Ig (R&D Systems) on day 0 of culture.
Statistical analysis
Univariate analyses were performed to evaluate associations between diseases and CD28 autoantibodies. Continuously measured parameters such as age and serum IgE were compared between patients with and without CD28 autoantibodies using the Wilcoxon rank sum test; dichotomous parameters (such as sex or the presence or absence of autoantibodies) were compared using Fisher's exact test. Logistic regression analysis was performed to identify factors independently predictive for the presence of CD28 autoantibodies. Bonferroni correction was used to compensate for multiple testing biases. The results of anti-CD28 autoantibody ELISA in different patient groups were compared using the Wilcoxon test. In order to assess the relationship between immunoblot and ELISA, we calculated Kendall's tau-b and the corresponding P-values to test the hypothesis that both variables are not associated.
Results
Detection of autoantibodies against CD28 by immunoblot
CD28-Ig fusion protein was digested with trypsin and the cleavage products were tested for binding of either monoclonal or polyclonal antibodies against CD28 using Western blots. SDS-PAGE and immunoblot analysis of digested CD28-Ig fusion protein showed that digestion with trypsin for 15 min at 37°C leads to a distinct cleavage product, which was detected by monoclonal and polyclonal anti-human CD28 antibodies but not by antibodies against human IgG and human Fc (Fig. 1a). The polyclonal anti-human CD28 antibody also detected smaller cleavage products. In contrast, the anti-human IgG and Fc-antibodies reacted only with fragments of the Ig-part of the CD28 fusion protein and not with CD28. These results indicated that trypsin digestion of the fusion protein leads to a distinct CD28 fragment, which did not contain residues of the Ig-molecule. Thus, possible cross-reactions between circulating antibodies directed to Fc were excluded. The secondary antibodies used did not show any unspecific binding (data not shown).
Fig. 1.

(a) Cleavage products after digestion of CD28Ig fusion protein with trypsin. Lane 1, CD28 stained with mouse anti-human CD28 mAb. Lane 2, CD28 stained with biotinylated polyclonal mouse anti-human CD28 antibody. Some smaller fragments of CD28-Ig are detected by this polyclonal antibody but not by the mAb. Lanes 3–5, detection of Ig cleavage products by rabbit anti-human IgG (lane 3), by goat anti-human IgG (lane 4) and by mouse anti-human Fc (lane 5) polyclonal antibodies. (b) Immunoblots of four patients with atopic dermatitis (AD) and autoantibodies to CD28 and one AD patient without CD28 autoantibodies. As an example of CD28 autoantibodies in autoimmune disease (AI) the immunoblot of a patient with scleroderma is presented. The patient with epidermolysis bullosa acquisita (AI) and the patient with psoriasis (PS) were negative for CD28 autoantibodies. Additionally, one immunoblot of a negative serum from a blood donor fromthe control group (N) and the molecular weight standard (MW) is shown.
For detection of autoantibodies against CD28 with this method, sera from healthy individuals and from randomly selected patients with different dermatological diseases were tested (Fig. 1b). CD28 autoantibodies were present in eight of 72 (11·1%) healthy individuals and in 53 of 196 (27·04%) of the patient group. Table 2 shows that the presence of CD28 autoantibodies was related to a slight trend for younger age and female preponderance in both groups. Univariate analysis showed that presence of CD28 autoantibodies was related highly significantly to AD [odds ratio (OR), 25·31 (95%, CI 5·52–116·11); P < 0·0001], allergic asthma/rhinitis [OR 10·78 (95%, CI 5·39–21·55); P < 0·0001] and less significantly to autoimmune diseases, e.g. scleroderma (Table 1). No significant relationship to any other inflammatory (e.g. contact dermatitis, seborrheic dermatitis or psoriasis), bacterial, viral or fungal skin infections, melanoma or non-melanoma skin cancer or leg ulcers was detected. Because several factors (e.g. serum IgE level, AD, allergic asthma/rhinitis) were related to one another, multivariate logistic regression analysis was performed. As shown in Table 3, CD28 autoantibodies were related most closely to AD and allergic asthma/rhinitis as well as to autoimmune disease, whereas the influence of other potential factors (age, sex and serum IgE) could be excluded.
Table 2.
Age and sex related to anti-CD28 autoantibodies.
| CD28+ | CD28– | P | |
|---|---|---|---|
| Age (± s.d.) (years) | 52·3 ± 18·8 | 58·7 ± 19·9 | 0·073 |
| Sex | |||
| Male | 19 | 69 | |
| Female | 34 | 74 | 0·146 |
s.d.: Standard deviation.
Table 3.
Logistic regression analysis of factors influencing anti-CD28 autoantibodies.
| Factor | OR | 95% CI | P |
|---|---|---|---|
| Age | 0·993 | 0·974–1·012 | 0·443 |
| Sex | 1·073 | 0·503–2·288 | 0·855 |
| IgE (kU/l) | 0·000 | 0·999–1·000 | 0·077 |
| Allergic rhinitis/asthma | 2·484 | 1·138–5·417 | 0·022 |
| Atopic dermatitis | 62·68 | 2–624·02 | 0·000 |
| Autoimmune disease | 8·909 | 1·572–50·48 | 0·014 |
CI: confidence interval; OR: odds ratio.
ELISA for detection of anti-CD28 autoantibodies
In order to confirm the results obtained with the immunoblot method, the sera were tested for presence of autoantibodies against the recombinant CD28/GST fusion protein.
The mean OD of sera tested without CD28-GST (negative control) was 0·21 ± 0·038. Again, testing of sera with the ELISA showed significantly (P < 0·02) enhanced titres of autoantibodies against CD28 in patients with allergic rhinitis and asthma compared with the control group (Fig. 2). A significant difference (P = 0·014) was also observed between allergic rhinitis and asthma patients compared with the group of patients with other inflammatory skin diseases. Increased titres compared with the control group were observed in patients with autoimmune diseases. Compared with the results obtained with the immunoblot method (14 of 16), fewer patients with AD (eight of 16) showed increased titres of autoantibodies against the CD28/GST fusion protein. In the other disease groups the results of the immunoblot and the ELISA were comparable (Table 4). The results of both methods were correlated significantly (τb = 0·523, P < 0·0001).
Fig. 2.

The prevalence of autoantibodies to recombinant CD28 in the sera of 196 patients with different diseases and 72 healthy donors (control) by enzyme-linked immunosorbent assay. The dotted line represents the cut-off point (OD = 0·05). *P < 0·05 significant compared with control.
Table 4.
Correlation between positive results obtained with immunoblot and enzyme-linked Immunsosorbent assay (ELISA).
| Immunoblot versus ELISA | |||||
|---|---|---|---|---|---|
| Total | Immunoblot CD28+ | ELISA CD28+ | τb | P | |
| Controls | 72 | 8 | 9 | 0·448 | < 0·0001 |
| Patients | 196 | 90 | 79 | 0·546 | < 0·0001 |
| Total | 258 | 98 | 88 | 0·523 | < 0·001 |
MLR
In order to clarify if autoantibodies against CD28 are immunologically relevant, an MLR using irradiated Raji cells and vital Jurkat cells was used. Therefore, CD28 autoantibodies were eluted from positive sera of patients with AD. Sera from patients with AD, which were negative for CD28 autoantibodies, served as controls. Optimal results were obtained on day 3 of culture. The experiments demonstrated that sera containing autoantibodies against CD28 significantly stimulated T cell proliferation, whereas sera without CD28 autoantibodies inhibited proliferation (Fig. 3). CTLA-4 engagement delivers a negative signal, inhibiting TCR- and CD28-mediated signal transduction [2]. Thus, CTLA-4-Ig was added, which is a well-established model for inhibition of T cell proliferation in MLR [20]. Elutes from sera containing autoantibodies against CD28 were able to overcome the CTLA-4-Ig-mediated inhibition of T cell proliferation, but not sera without CD28 autoantibodies. These results demonstrate that autoantibodies against CD28 are able to induce T cell proliferation in this model of anergy.
Fig. 3.

Mixed lymphocyte reaction (MLR) with irradiated Raji cells and Jurkat cells. Proliferation was measured by bromodeoxyuridine incorporation on day 2 of culture. Results are shown as percentage-stimulation of spontaneous proliferation (control). CTLA4-Ig inhibited proliferation, whereas elutes containing CD28 autoantibodies significantly stimulated T cell proliferation. Co-stimulation with CTLA4-Ig and autoantibodies against CD28 resulted in a significantly reduced inhibition of Jurkat cell proliferation. **P < 0·01 significant compared with control.
Discussion
In this study we demonstrate that autoantibodies against CD28 are detectable in human serum and hypothesize that presence of these autoantibodies is related closely to atopic diseases. Moreover, the presence of CD28 autoantibodies may play a role for persistent stimulation of T cells and lack of immunological tolerance in these diseases.
The pathophysiological concept of atopic diseases is based on the finding that allergen-specific T cells (Th2-lymphocytes) secreting mainly interleukin (IL)-4, IL-5, IL-10, IL-13 and granulocyte–macrophage colony-stimulating factor (GM-CSF) are preferentially expanded, whereas interferon (IFN)-γ-producing Th1 cells are less active [22]. Cytokines such as IL-4, IL-5 and IL-13 are, to a great extent, responsible for the eosinophilia and the switching of B cells to IgE synthesis in atopy [23]. In animal models it has been demonstrated that the Th2 response requires continuing co-stimulation via CD28, and treatment with CD80 and CD86 antibodies inhibits IgE production in mice [24,25]. Baseline expression of CD80 and CD86 on peripheral B cells has been demonstrated to be low in normal donors and increased in donors with atopic dermatitis (AD) [26]. In the presence of anti-CD28, cell proliferation and IgE synthesis were significantly enhanced in anti-CD40+ IL-4-stimulated peripheral mononuclear cells from patients with AD, supporting the assumption that CD28 activation is important in Th2-mediated inflammation [26].
CD28 signalling regulates the threshold for T cell activation and significantly decreases the number of TCR engagements needed for effective T cell activation [27]. The main effects of CD28 co-stimulation are to augment and sustain T cell responses initiated by antigen-receptor signalling, by promoting T cell survival and thereby enabling cytokines to initiate T cell clonal expansion and differentiation [28,29]. Thus, autoantibodies directed to CD28 in atopic diseases could lead to a persistent stimulation and increased survival of Th2 cells [25]. This assumption has been supported by the fact that sera from patients with AD, allergic rhinitis or asthma which contain CD28 autoantibodies significantly stimulated T cell proliferation in MLR.
CTLA-4 has a crucial role in regulating peripheral T cell tolerance [30], and CTLA-4 blockade in vivo augments anti-tumour immunity [31] and exacerbates autoimmune responses [32]. It is likely that CTLA-4 also has an inhibitory function in atopic diseases. The importance of CD28/B7 pathway in the in vivo immune response to allergen was described in recent studies examining the effects of B7 blockade with CTLA-4-Ig, a soluble fusion protein inhibitor of B7-1 and B7-2. Administration of CTLA-4-Ig to mice before allergenic stimulation led to a significant reduction of bronchoalveolar lavage (BAL) peribronchial eosinophilia, mucous cell hyperplasia, Th2 cytokine contents in BAL fluid and airway hyperreactivity (AHR) in response to airway antigen challenge [33–35]. The fact that CD28 autoantibodies overcome CTLA-4-mediated T cell inhibition in MLR indicates that the lack of Th2 tolerance in AD and other atopic diseases might be the result of chronic stimulation via stimulating CD28 autoantibodies.
Recently, it has been demonstrated that a considerable percentage of patients with AD mount IgE autoantibodies against a broad variety of human proteins [36]. Additionally, IgG-reactive autoantigens have been characterized in AD patients [37]. As the autoantibodies against CD28 are of the IgG isotype, this finding supports the suggestion that autoimmune phenomena occur frequently in AD. All patients with AD in this study belong to the ‘extrinsic’ subtype. Therefore, an association between autoantibodies against CD28 and the ‘intrinsic’ or ‘extrinsic’ subtypes cannot be concluded from our data.
In the majority of positive sera autoantibodies against CD28 could be detected both against the CD28-Ig fusion protein (immunoblot) and the recombinant CD28/GST (ELISA). Interestingly, six sera from patients with AD showed positive reactions to the CD28-Ig fusion protein but not to CD28/GST. It may be suggested that this difference is due to different epitopes or to a different spatial structure of the two proteins used. This assumption is supported by the results of Matsui et al. [38], who demonstrated the presence of different epitopes on CTLA-4 in a large number of patients with systemic autoimmune diseases, which implies that different patients may recognize different three-dimensional conformations of the protein. However, autoantibodies against CD28 were not detected in their study. In our patient group we found autoantibodies against CD28 in patients also with autoimmune diseases. These differences are probably the result of differences between the used antigens.
On the other hand, the autoantibodies produced in several diseases could recognize distinct epitopes of the recombinant CD28 molecules. Future investigations focusing on this topic may provide new insights into the pathophysiology of atopic or autoimmune diseases.
Because CD28 is a central regulatory receptor in immune-mediated diseases, we are convinced that these autoantibodies are not only serum markers for immune-mediated diseases (e.g. E-selectin [39] or soluble cytokine receptors [40]), but can also be an important tool to discriminate between specific diseases. This suggestion is supported by the fact that the difference between allergic diseases and other inflammatory skin diseases was statistically significant, although only a relatively small number of patients were tested. Additionally, it will be extremely important to investigate whether autoantibodies against CD28 are correlated with course and severity of atopic or autoimmune diseases.
Acknowledgments
This work was supported in part by grants from the Leidenberger-Müller-Foundation and from the Buch-Foundation, Germany. Gerlinde Finger is thanked for excellent technical assistance. We also thank Dr Linda Ruhde for help with the collection of blood samples.
References
- 1.Appleman LJ, Boussiotis VA. T cell anergy and co-stimulation. Immunol Rev. 2003;192:161–80. doi: 10.1034/j.1600-065x.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 2.Sharpe AH, Freeman GJ. The B7–CD28 superfamily. Nature Rev. 2002;2:116–26. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 3.Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T cell biology. Immunol Rev. 2006;212:131–48. doi: 10.1111/j.0105-2896.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 4.Jones G, Wu S, Jang N, et al. Polymorphisms within the CTLA4 gene are associated with infant atopic dermatitis. Br J Dermatol. 2005;154:467–71. doi: 10.1111/j.1365-2133.2005.07080.x. [DOI] [PubMed] [Google Scholar]
- 5.Osman C, Swaak AJ. Lymphocytotoxic antibodies in SLE: a review of the literature. Clin Rheumatol. 1994;13:21–7. doi: 10.1007/BF02229861. [DOI] [PubMed] [Google Scholar]
- 6.Swaak AJG. Lymphocytotoxic antibodies. In: Peter JB, Shoenfeld Y, editors. Autoantibodies. Amsterdam: Elsevier Science; 1996. p. 478. [Google Scholar]
- 7.Winfield J, Mimura T. Pathogenetic significance of antilymphocyte antibodies with systemic lupus erythematosus. Clin Immunol. 1992;63:13–6. doi: 10.1016/0090-1229(92)90085-3. [DOI] [PubMed] [Google Scholar]
- 8.Winfield JB, Winchester RJ, Kunkel HG. Association of cold reactive antilymphocyte antibodies with lymphopenia in systemic lupus erythematosus. Arthritis Rheumatol. 1975;18:587–94. doi: 10.1002/art.1780180609. [DOI] [PubMed] [Google Scholar]
- 9.Morimoto C, Steinberg AD, Letvin NL, et al. A defect of immunoregulatory T cell subsets in systemic lupus erythematosus patients demonstrated with anti-2H4 antibody. J Clin Invest. 1987;79:762–8. doi: 10.1172/JCI112882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka S, Matsuyama T, Steinberg AD, et al. Antilymphocyte antibodies against CD4+2H4+ cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32:398–405. doi: 10.1002/anr.1780320408. [DOI] [PubMed] [Google Scholar]
- 11.Sakane T, Steinberg AD, Reeves JP, et al. Studies of immune functions of patients with systemic lupus erythematosus. Complement-dependent immunoglobulin M anti-thymus-derived cell antibodies preferentially inactivate suppressor cells. J Clin Invest. 1979;63:954–65. doi: 10.1172/JCI109396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wernet P, Kunkel HG. Antibodies to specific surface antigen of T cells in human sera inhibiting mixed leukocyte culture reactions. J Exp Med. 1973;138:1021–6. doi: 10.1084/jem.138.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeuchi T, Abe T, Kiyotaki M, et al. In vitro immune response of SLE lymphocytes. The mechanism involved in B-cell activation. Scand J Immunol. 1982;16:369–77. doi: 10.1111/j.1365-3083.1982.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 14.Mimura T, Fernsten P, Jarjour W, et al. Autoantibodies specific for different isoforms of CD45 in systemic lupus erythematosus. J Exp Med. 1990;172:653–6. doi: 10.1084/jem.172.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czyzyk J, Fernsten P, Shaw M, et al. Cell-type specificity of anti-CD45 autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 1996;39:592–9. doi: 10.1002/art.1780390408. [DOI] [PubMed] [Google Scholar]
- 16.Revillard JP, Vincent C, Rivera S. Anti-beta2-microglobulin lymphocytotoxic autoantibodies in systemic lupus erythematosus. J Immunol. 1979;122:614–8. [PubMed] [Google Scholar]
- 17.Propper DJ, Leheny WA, Urbaniak SJ, et al. Lymphocytotoxins in sera from highly sensitized multiparous dialysis patients: antibody class, relationship with the HLA and with paternal antigens. Clin Sci. 1991;80:87–93. doi: 10.1042/cs0800087. [DOI] [PubMed] [Google Scholar]
- 18.Khatlani TS, Ma Z, Okuda M, et al. Autoantibodies against T cell co-stimulatory molecules are produced in canine autoimmune diseases. J Immunother. 2003;26:12–20. doi: 10.1097/00002371-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Fölster-Holst R, Pape M, Buss YL, et al. Low prevalence of the intrinsic form of atopic dermatitis among adult patients. Allergy. 2006;61:629–32. doi: 10.1111/j.1398-9995.2006.01076.x. [DOI] [PubMed] [Google Scholar]
- 20.Linsley PS, Brady W, Urnes M, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–9. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuber K, Schmidt S, Mensch A. Telomere length measurement and determination of immunosenescence-related markers (CD28, CD45RO, CD45RA, IFNg and IL-4) in skin-homing T cells expressing the cutaneous lymphocyte antigen: indication of a non-aging T cell subset. Immunology. 2003;109:24–31. doi: 10.1046/j.1365-2567.2003.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jujo K, Renz H, Abe J, et al. Decreased interferon gamma and increased interleukin-4 production in atopic dermatitis promotes IgE synthesis. J Allergy Clin Immunol. 1992;90:323–31. doi: 10.1016/s0091-6749(05)80010-7. [DOI] [PubMed] [Google Scholar]
- 23.Punnonen J, Aversa G, Cocks BG, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA. 1993;90:3730–4. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacPhee IAM, Turner DR, Yagitad H, et al. The Th2-response in mercuric chloride-induced autoimmunity requires continuing co-stimulation via CD28. Clin Exp Immunol. 2002;129:405. doi: 10.1046/j.1365-2249.2002.01928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapoval SP, David CS. CD28 co-stimulation is critical for experimental allergic asthma in HLA-DQ8 transgenic mice. Clin Immunol. 2003;106:83–94. doi: 10.1016/s1521-6616(03)00002-0. [DOI] [PubMed] [Google Scholar]
- 26.Oberwalleney G, Henz BM, Worm M. Expression and functional role of co-stimulatory molecules in CD40+ IL-4-stimulated B cells from atopic and non-atopic donors. Acta Derm Venereol. 2000;80:287–91. doi: 10.1080/000155500750012199. [DOI] [PubMed] [Google Scholar]
- 27.Viola A, Lanzavecchia AT. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–6. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 28.Thompson CB, Lindsten T, Ledbetter JA, et al. CD28 activation pathway regulates the production of multiple T cell-derived lymphokines/cytokines. Proc Natl Acad Sci USA. 1989;86:1333–7. doi: 10.1073/pnas.86.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boise N, Minn A, Noel P, et al. CD28 co-stimulation can promote T cell survival by enhancing the expression of Bcl-xl. Immunity. 1995;3:87–98. [Google Scholar]
- 30.Greenwald RJ, Boussiotis VA, Lorsbach RB, et al. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–55. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 31.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 32.Perrin PJ, Maldonado JH, Davis TA, et al. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–6. [PubMed] [Google Scholar]
- 33.Padrid PA, Mathur M, Li X, et al. CTLA4Ig inhibits airway eosinophilia and hyperresponsiveness by regulating the development of Th1/Th2 subsets in a murine model of asthma. Am J Respir Cell Mol Biol. 1998;18:453–62. doi: 10.1165/ajrcmb.18.4.3055. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalo JA, Tian J, Delaney T, et al. ICOS is critical for T helper cell-mediated lung mucosal inflammatory responses. Nat Immunol. 2001;2:597–604. doi: 10.1038/89739. [DOI] [PubMed] [Google Scholar]
- 35.Keane-Myers A, Gause WC, Linsley PS, et al. B7-CD28/CTLA-4 co-stimulatory pathways are required for the development of T helper cell 2-mediated allergic airway responses to inhaled antigens. J Immunol. 1997;158:2042–9. [PubMed] [Google Scholar]
- 36.Mittermann I, Aichberger KJ, Bunder R, et al. Autoimmunity and atopic dermatitis. Curr Opin Allergy Clin Immunol. 2004;4:367–71. doi: 10.1097/00130832-200410000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Ohkouchi K, Mizutani H, Tanaka M, et al. Anti-elongation factor-1alpha autoantibody in adult atopic dermatitis patients. Int Immunol. 1999;11:1635–40. doi: 10.1093/intimm/11.10.1635. [DOI] [PubMed] [Google Scholar]
- 38.Matsui T, Kurokawa M, Kobata T, et al. Autoantibodies to T cell co-stimulatory molecules in systemic autoimmune diseases. J Immunol. 1999;162:4328–35. [PubMed] [Google Scholar]
- 39.Wolkerstorfer A, Savelkoul HF, de Waard van der Spek FB, et al. Soluble E-selectin and soluble ICAM-1 levels as markers of the activity of atopic dermatitis in children. Pediatr Allergy Immunol. 2003;14:302–6. doi: 10.1034/j.1399-3038.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 40.Yoshizawa Y, Nomaguchi H, Izaki S, et al. Serum cytokine levels in atopic dermatitis. Clin Exp Dermatol. 2002;27:225–9. doi: 10.1046/j.1365-2230.2002.00987.x. [DOI] [PubMed] [Google Scholar]