

Abstract
The Saccharomyces cerevisiae gene TOR2 encodes a putative phosphatidylinositol kinase that has two essential functions. One function is redundant with TOR1, a TOR2 homolog, and is required for signaling translation initiation and early G1 progression. The second essential function is unique to TOR2. Here we report that loss of the TOR2-unique function disrupts polarized distribution of the actin cytoskeleton. A screen for dosage suppressors of a dominant negative TOR2 allele identified TCP20/CCT6, encoding a subunit of the cytosolic chaperonin TCP-1 that is involved in the biogenesis of actin structures. Overexpression of TCP20 restores growth and polarized distribution of the actin cytoskeleton in a tor2 mutant. TCP20 overexpression does not restore growth in a tor1 tor2 double mutant. We suggest that the unique function of the phosphatidylinositol kinase homolog TOR2 is required for signaling organization of the actin cytoskeleton during the cell cycle. TOR2, via its two functions, may thus integrate temporal and spatial control of cell growth.
Keywords: signal transduction, rapamycin, TCP-1 chaperonin, phosphatidylinositol kinase, cell growth
Budding yeast undergoes polarized cell growth that is dependent on an asymmetric distribution of the actin cytoskeleton. As Saccharomyces cerevisiae initiates a new cell cycle, membrane-localized actin patches concentrate at a previously selected and marked bud site, the secretory apparatus becomes oriented toward this site, and bud emergence begins (1, 2). During maturation of the bud, actin cables become prominent in the mother cell and align toward the bud. The polarized actin distribution is lost during mitosis when the actin patches redistribute to the surface of the mother and the bud. Before cytokinesis, actin patches relocate to the mother–bud neck to target secretion to this region for the formation of the septum. Mutations in genes encoding the small GTPases CDC42, RHO1, RHO3, and RHO4 disrupt polarized growth and, in some cases, the highly asymmetric distribution of actin structures (3, 4, 5, 6). However, the nature of the signal(s) received by these Rho-like GTPases and the mechanisms by which they control the actin cytoskeleton during vegetative growth are not known.
In mammalian cells, Rho-like GTPases, including Rho, Rac, and Cdc42Hs, are also involved in the control of the actin cytoskeleton (7). They are thought to be key regulators in signaling pathways linking growth factor receptors to the assembly and organization of the actin cytoskeleton. Using cell-free assays and intact cell systems, it has been shown that Rho-like GTPases interact, physically or functionally, with phosphatidylinositol (PI) kinases. For example, Rho activates PI 3-kinase and PI-4-P 5-kinase (PIP 5-kinase) (8, 9), and Cdc42Hs activates PI 3-kinase (10); PI 3-kinase activates Rac (11); Cdc42Hs, Rho, and Rac physically interact with PI kinases (10, 12, 13). PI kinases or their products, phosphorylated phosphoinositides, have also been implicated in the control of the actin cytoskeleton. Actin rearrangement in neutrophils and fibroblasts correlates with the synthesis of phosphatidylinositol-3,4,5-triphosphate, and wortmannin, a PI 3-kinase inhibitor, blocks such rearrangement (14, 15, 16). In addition, phosphorylated phosphoinositides modulate the interaction of actin-capping proteins with actin in vitro (17), and, in permeabilized platelets, activated Rac uncaps actin filament barbed ends through phosphoinositide synthesis (18). This suggests that PI kinases and Rho-like GTPases are part of the mammalian signal transduction pathways that regulate the reorganization of the actin cytoskeleton in response to external stimuli.
TOR1 and TOR2 are structurally related PI kinase homologs first identified in yeast as the targets of the immunophilin–immunosuppressant complex FKBP-rapamycin (19, 20, 21, 22). Treatment with rapamycin or combined deletion of TOR1 and TOR2 causes yeast cells to arrest in early G1, to undergo a severe reduction in protein synthesis, and to induce several other physiological changes characteristic of starved cells entering stationary phase (G0) (23). TOR1 and TOR2 are presumably part of a novel signaling pathway that activates translation initiation and, thereby, G1 progression in response to nutrient availability. It remains to be determined that TOR1 and TOR2 indeed possess kinase activity.
While combined disruption of TOR1 and TOR2 causes a G1 arrest, disruption of TOR1 alone has little or no effect and loss of TOR2 alone is lethal, with cells arresting throughout the cell cycle (20, 21, 22). The lethality of a TOR2 disruption cannot be suppressed even by overexpression of TOR1. This suggests that TOR2 has at least two essential functions (24). One function is shared with TOR1, is rapamycin-sensitive, and is required for translation initiation and G1 progression, whereas the second essential function is unique to TOR2. It is not known what the unique function of TOR2 might be and how it might be related to the shared G1 function.
Here we show that loss of TOR2 alone disrupts the polarized distribution of the actin cytoskeleton during the cell cycle. Overexpression of TCP20/CCT6 suppresses the lethality of a TOR2 mutation, but not a TOR1 TOR2 double mutation, and partially restores polarized organization of the actin cytoskeleton in strains lacking TOR2. TCP20 encodes a subunit of the TCP-1 complex, a chaperonin known to be involved in the folding and assembly of actin structures (25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36). This suggests that the unique function of TOR2 is involved in the organization of the actin cytoskeleton.
MATERIALS AND METHODS
Strains, Plasmids, and Media.
S. cerevisiae strains used in this work are listed in Table 1. All strains were isogenic JK9-3da derivates. Plasmid pJK5 is pSEYC68 galp (ampr CEN URA3 GAL1 promoter), containing the entire TOR2 gene under control of the GAL1 promotor (20). pJK9 is pSEY18 (ampr 2μ URA3) containing a 3.0-kb SalI restriction fragment encompassing the 3′-region of the TOR2 gene. pJK10 is a deletion variant of plasmid pJK9 where the internal 1.3-kb BglII restriction fragment had been deleted. pJK11 is plasmid pJK9 containing the tor2kin− allele, a single nucleotide change converting TOR2 codon 2,279 from GAC (aspartate) to GCC (alanine). pTOR2kin− (pJK66) is pAS10 containing the tor2kin− mutation. pAS10 is pSEYC68 galp (ampr CEN URA3 GAL1 promoter) containing amino-terminally 6His-tagged TOR2 under control of the GAL1 promoter; amino-terminally 6His-tagged TOR2 is functionally indistinguishable from untagged TOR2 (A.S., unpublished work). YCplac111::tor2-21ts (ampr CEN LEU2) contains the entire TOR2 gene and was isolated by hydroxylamine mutagenesis as a temperature-sensitive TOR2 allele (S. B. Helliwell and N. C. Barbet, unpublished work). pAS26 is YEplac181 (ampr 2μ LEU2) carrying a 2.8-kb HindIII–SpeI genomic DNA insert containing the TCP20 gene. pAS27 is pSEY18 (ampr 2μ URA3) carrying a 2.6-kb SacI genomic DNA insert containing the TCP20 gene. The yeast genomic library in a multicopy LEU2 vector (gift of K. Nasmyth, Institute of Molecular Pathology, Vienna) was in YEp13 and had an average insert size of 8 kb. The composition of rich medium [yeast extract/peptone/dextrose (YPD)] and synthetic minimal medium (SD, SGal/Gly) complemented with the appropriate nutrients for plasmid maintenance was as described (37). Wild-type strain JK9-3da containing pAS27 was examined for resistance to rapamycin (gift of Sandoz Pharmaceutical) at concentrations of 20 and 200 ng/ml in YPD at 30°C. 5-Fluoroorotic acid (5-FOA) was used to counterselect URA3 plasmid pJK5 in strain JK350-18a as described (37).
Table 1.
Strains used in this study
| Strain | Genotype |
|---|---|
| JK9-3da | MATa leu2-3,112 ura3-52 trp1 his4 rme1 HMLa |
| JK9-3da/α | MATa/MATα leu2-3,112/leu2-3,112 ura3-52/ura3-52 trp1/trp1 his4/his4 rme1/rme1 HMLa/HMLa |
| MH346 | JK9-3da/α ade2/ade2 tor2::ADE2-3/TOR2 |
| MH349-3d | JK9-3da tor1::LEU2-4 |
| NB17-3d | JK9-3da ade2 his3 HIS4 tor1::HIS3 |
| JK350-18a | JK9-3da ade2 tor2::ADE2-3/pJK5 (pSEYC68galp::TOR2) |
| SH121 | JK9-3da ade2 tor2::ADE2-3/YCplac111::tor2-21ts |
| SH221 | JK9-3da ade2 his3 HIS4 tor1::HIS3 tor2::ADE2-3/YCplac111::tor2-21ts |
Construction of pTOR2kin−.
To construct the TOR2 kinase-dead allele, PCR-based mutagenesis was performed with the mutagenic primers 5′-TTAGGTGCCCGCCACC and 5′-GGTGGCGGGCACCTAA and flanking primers 5′-CTACAACATGTGTCGCC and 5′-CAGCACTGACCCTTTTG. A first round of PCR was performed using a mutagenic primer in combination with a flanking primer and Taq polymerase (Boehringer Mannheim), the two independent PCR reactions were gel-purified and used in a second round of PCR with only the flanking primers. A 1.3-kb BglII fragment encompassing the mutagenized TOR2 region was excised from the 3.0-kb PCR product and subcloned into the BglII site of plasmid pJK10, yielding plasmid pJK11. The mutant DNA was inserted back into TOR2 on a 3.0-kb SalI restriction fragment yielding the full-length TOR2 kinase-dead allele (plasmid pJK66). The entire 1.3-kb BglII region derived by PCR mutagenesis was sequenced to verifiy that only the desired nucleotide change was introduced.
Genetic Techniques.
Yeast plasmid DNA was isolated as described (38). Yeast transformation was performed by the lithium acetate procedure (39). Escherichia coli strain DH5α was used for propagation and isolation of plasmids as described (40).
DNA Manipulations.
Restriction enzyme digests and ligations were done by standard methods. All enzymes and buffers were obtained commercially (Boehringer Mannheim). DNA was sequenced by the dideoxy-chain termination method with the T7 sequencing system (Pharmacia).
Fluorescence Microscopy.
Cells were grown to early logarithmic phase, fixed in formaldehyde, and stained with 4′,6-diamidino-2-phenylindole (DAPI) or rhodamine phalloidin to visualize DNA and actin, respectively, as described (41).
RESULTS
Isolation of TCP20 as a Dosage Suppressor of a Dominant Negative TOR2 Allele.
Catalytically inactive kinases often confer a dominant negative effect because they titrate away an interacting factor. Overexpression of the factor itself or other, downstream components of the pathway can suppress the dominant negative phenotype. To use this strategy to identify components in a pathway with TOR2, we constructed a TOR2 kinase-dead allele (tor2kin−) under the control of the inducible GAL1 promotor. A point mutation was introduced into the sequence encoding the ATP-binding site of the lipid kinase motif of TOR2, changing aspartate 2,279 to alanine (see Materials and Methods). An equivalent mutation was shown to abolish the enzymatic activity of the VPS34 and p110 PI 3-kinases (42, 43). tor2kin− could not provide TOR2 function as determined by complementation assays; temperature-sensitive tor2 mutants containing plasmid-borne tor2kin− did not grow on galactose medium at nonpermissive temperature, and no viable tor2 progeny were obtained on galactose medium upon dissection of a heterozygous tor2/TOR2 diploid (MH346) containing the plasmid-borne tor2kin− allele (data not shown). Overexpression of tor2kin− in our wild-type strain JK9-3da conferred a slow growth phenotype that was suppressed by overproduction of wild-type TOR2 (Fig. 1; data not shown). The above results indicated that tor2kin−, as expected, is a dominant-negative allele of TOR2.
Figure 1.
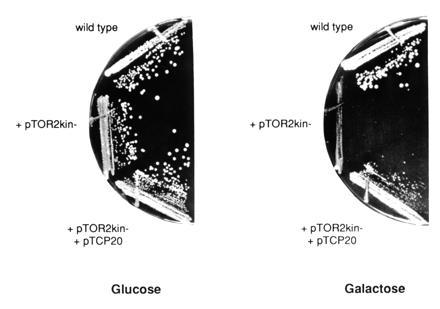
Overexpression of tor2kin− confers a growth defect that is suppressed by overexpression of TCP20. Wild-type cells (JK9-3da) transformed either with pTOR2kin− or with pTOR2kin− and pTCP20 (pAS26) were streaked on SD-URA-LEU and SGal/Gly-URA-LEU medium. The tor2kin− allele is under control of the GAL1 promoter and therefore induced on galactose and repressed on glucose.
To identify components that lie in a TOR2 signaling pathway, we screened for high copy number suppressors of tor2kin−. A yeast genomic library constructed in a high copy number LEU2 vector was introduced into JK9-3da/pTOR2kin−, and transformants were selected on SD-URA-LEU (tor2kin− repressed) and then screened for growth on SGal/Gly-URA-LEU (tor2kin− induced). Out of 7000 transformants screened, 11 transformants were isolated that showed plasmid-dependent suppression of the growth defect conferred by overexpression of tor2kin−. Plasmid DNA of these transformants was isolated, analyzed by restriction enzyme digestion, and sequenced from both ends of the insert of the genomic DNA. The obtained DNA sequence information was compared with sequences entered in the European Molecular Biology Laboratory and GenBank data bases using the blast program (44). This revealed six different genomic regions among the 11 isolates. The entire sequence of each region was already available, and the open reading frame in each region responsible for the suppression of the growth defect conferred by tor2kin− was identified by subcloning and deletion analyses. Three of the suppressors (MIG1, GAL3, and the GAL1, GAL10, and GAL7 cluster) were found to encode proteins of the galactose system, and were thus not analyzed further. Two were open reading frames (YHR127w and YBR057c) without significant homology to other genes or to each other, and one gene was TCP20/CCT6 (Fig. 1). TCP20 is an essential gene required for cytoskeletal function (see below).
TCP20 Overexpression Restores Growth in a tor2 Mutant but Not in a tor1 tor2 Double Mutant.
We examined if TCP20 overexpression could suppress recessive tor2 mutations, a temperature-sensitive allele, a promoter alteration, and a null mutation, in addition to tor2kin−. First, tor2ts strain SH121 was transformed with pAS27 (pSEY18::TCP20) and with an empty vector, and was checked for growth at restrictive temperature. Whereas transformants carrying the empty vector were inviable at 37°C, the tor2ts cells overexpressing TCP20 were able to grow at the restrictive temperature, however, not as well as TOR2 wild-type cells (Fig. 2). Second, pAS26 (YEplac181::TCP20) and an empty vector were transformed into strain JK350-18a that contained a genomic disruption of TOR2 and a plasmid-borne copy of TOR2 under the control of the GAL1 promotor (pJK5). The transformants were tested for growth on medium containing glucose as a carbon source where the GAL1 promotor is repressed. Again, tor2 cells carrying an empty vector did not grow, whereas cells overexpressing TCP20 were able to form colonies (data not shown). Finally, to determine if TCP20 overexpression could suppress a TOR2 null allele, the above strain JK350-18a containing a TOR2 disruption, TCP20 in high copy (pAS26), and plasmid-borne TOR2 (pJK5) was streaked on 5-FOA medium (see Materials and Methods) to clear the plasmid-borne copy of TOR2. JK350-18a overexpressing TCP20 was unable to grow in the presence of 5-FOA (data not shown). Thus, TCP20 overexpression could suppress the growth defect caused by a tor2ts mutation and by a severe reduction in the amount of TOR2, but could not restore growth in the complete absence of TOR2.
Figure 2.
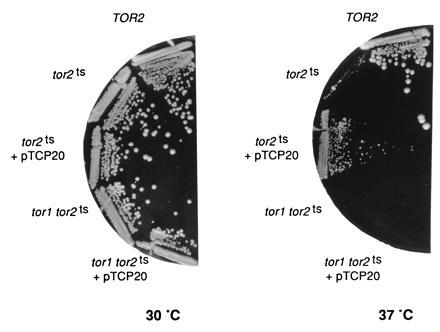
Overexpression of TCP20 suppresses the growth defect of a tor2ts mutation but not of a tor1 tor2ts double mutation. Wild-type TOR2 (JK9-3da), tor2ts (SH121), and tor1 tor2ts cells (SH221) carrying an empty vector or pTCP20 (pAS27) were streaked on YPD and incubated at 30°C or 37°C. tor2ts and tor1 tor2ts cells carrying an empty vector are not viable at 37°C.
Previous results have indicated that TOR2 has two functions: one function is redundant with TOR1 and the other function is unique to TOR2 (20, 22, 45). The observation that TCP20 overexpression suppressed the growth defect of a TOR1 tor2 strain, where TOR1 still provides the shared function, suggested that TCP20 is related to the TOR2-unique function. To investigate this further, we examined if overexpression of TCP20 could suppress either tor2kin− or a recessive tor2ts mutation in cells that lacked TOR1. Whereas TCP20 overexpression was able to rescue tor2ts cells and to suppress tor2kin− in the presence of TOR1 (TOR1 tor2ts and TOR1 TOR2/tor2kin− strains SH121 and JK9-3da/pTOR2kin−, respectively), as described above, it was unable to do so in the absence of TOR1 (tor1 tor2ts and tor1 TOR2/tor2kin− strains SH221 and NB17-3d/pTOR2kin−, respectively) (Fig. 2 and data not shown). In addition, overexpression of TCP20 could not suppress the dominant negative effect of a tor1kin− construct (A.S., J.K., S. B. Helliwell, unpublished work) (45). Overexpression of TCP20 also did not suppress the slight growth defect of a TOR1 deletion and did not confer resistance to rapamycin (data not shown). These findings indicated that TCP20 overexpression suppresses the loss of the TOR2-unique function but not the loss of the TOR1-shared function, and suggest that TCP20 is related to the TOR2-unique function.
Cells Lacking TOR2 Are Defective in the Organization of the Actin Cytoskeleton.
TCP20, recently renamed CCT6, is a member of the TCP1 gene family and encodes a subunit of the cytosolic TCP-1 chaperonin complex (25, 26). This chaperonin has been implicated specifically in the folding of actin and tubulin. Mammalian TCP-1 complexes have been shown to bind in vitro to denatured or newly synthesized actin and tubulin monomers and to release them upon ATP-hydrolysis in an altered, assembly competent conformation (27, 28, 29, 30, 31). In yeast, eight members of the TCP1/CCT gene family have been identified and the five examined by disruption analysis have all been shown to be essential (25, 26, 32, 33, 34, 35, 36). Conditional alleles of these genes all exhibit a disorganization of the actin cytoskeleton and disruption of microtubule-mediated processes such as nuclear segregation, supporting the role of the TCP-1 complex in the function of actin and tubulins.
The suggestion that TCP20 is functionally related to TOR2 (see above) led us to examine if the lack of TOR2 caused a defect in the organization of the actin cytoskeleton. Wild-type (JK9-3da) and tor2ts (SH121) cells were grown at permissive temperature (30°C) and then shifted to restrictive temperature (37°C) for 6 h. Cells were fixed and stained with phalloidin (see Materials and Methods). Whereas wild-type cells displayed the normal polarized distribution of actin at the various stages of the cell cycle, cells lacking TOR2 were increased in size and exhibited a randomized distribution of actin in all phases of the cell cycle in essentially all cells (Fig. 3). Unbudded cells just prior to entering S phase were not able to assemble (or maintain) the actin cap at the previously selected bud site. Most (>90%) of small- and middle-sized budded cells showed a random distribution of actin patches in both the mother and the daughter cell, with no concentration of patches in the daughter cell. Furthermore, no actin cables could be detected in the mother cell. Large-budded cells, in all cases, failed to display a relocation of actin patches to the mother–bud neck before cytokinesis. The same result was obtained with cells that carried the TOR2 gene under control of the GAL1 promoter (strain JK350-18a) and that were shifted to glucose for 20 h. Thus, cells lacking only TOR2 were severely defective in the organization of the actin cytoskeleton. Cells lacking only TOR1 (strain MH349-3d) displayed a completely normal, cell cycle-dependent, polarized distribution of actin (data not shown). We could not examine the cell cycle-dependent distribution of actin in cells lacking both TOR1 and TOR2 because such a strain arrests in G1.
Figure 3.
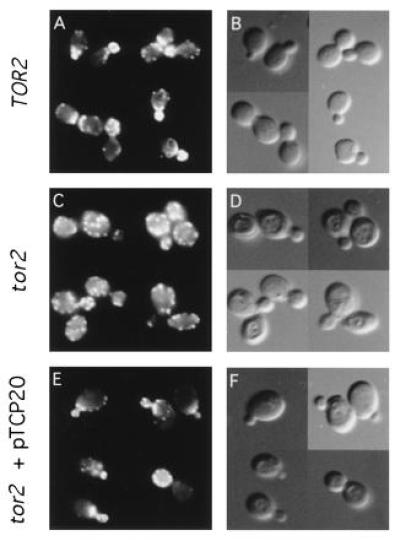
Overexpression of TCP20 suppresses the actin organization defect of tor2ts cells. Logarithmic cultures of wild-type TOR2 (JK9-3da) (A and B) and tor2ts (SH121) cells carrying an empty vector (C and D) or pTCP20 (pAS27) (E and F) were grown at 30°C, shifted to 37°C for 6 h, fixed, stained with rhodamine phalloidin, and observed by fluorescence (A, C, and E) and Nomarski (B, D, and F) microscopy. tor2ts cells carrying an empty vector have a random actin distribution.
Since the TCP-1 complex has also been implicated in the biogenesis and function of microtubules, we assessed if loss of TOR2 also caused a defect in microtubule function similar to that observed in tcp mutants (32, 33, 34, 35, 36). We examined if a tor2 mutant, like tcp mutants, had a defect in nuclear segregation by staining cells with the DNA-specific stain DAPI (see Materials and Methods). Both in wild-type cells (strain JK9-3da) and in tor2 cells (strains SH121 and JK350-18a) grown at the nonpermissive condition for the mutant cells, the DNA in large-budded cells had divided and segregated into the mother and daughter cells or was localized in the mother–bud neck. Neither in a wild-type strain nor in the tor2 strains did we find significant numbers of mononucleate large-budded cells or multi- or anucleate unbudded cells as observed with tcp mutants. From this we conclude that nuclear segregation and thus microtubules function is not affected upon loss of TOR2.
TCP20 Overexpression can Partially Restore Polarized Organization of the Actin Cytoskeleton in tor2 Cells.
We next asked if TCP20 overexpression restored organization of the actin cytoskeleton in cells lacking TOR2. The high copy number plasmid containing TCP20 (pAS27) was introduced into tor2ts strain SH121, and transformants were grown at 30°C, shifted to 37°C for 6 h, and fixed and stained with phalloidin. In comparison to tor2ts cells carrying an empty vector, which behaved as described above, tor2ts cells transformed with TCP20 exhibited less severe defects in the organization of the actin cytoskeleton (Fig. 3). Approximately 35% of the cells with small buds showed a wild-type distribution of actin; actin patches were concentrated in the bud, and actin cables were visible in the mother cells. A small percentage of large-budded cells overexpressing TCP20 exhibited the normal relocalization of actin to the mother–bud neck. Cells with middle-sized buds, however, appeared unchanged and still displayed a random distribution of actin patches in both the mother and daughter cell. Thus, TCP20 overexpression weakly suppressed both the growth defect and the actin distribution defect of a tor2 mutant. The correlation between the extent of suppression of the actin and growth defects and the fact that TCP20 affects actin function suggest that TCP20 might suppress the growth defect of a tor2 mutant by restoring actin function.
DISCUSSION
We have shown that loss of the PI-kinase homolog TOR2 results in disruption of the asymmetric distribution of the yeast actin cytoskeleton during the cell cycle. In a tor2 mutant, actin patches are not concentrated in the bud but are randomly distributed throughout the mother and the daughter cell, and no actin cables are visible in the mother cell. We isolated TCP20, which encodes a subunit of the eukaryotic cytosolic TCP-1 chaperonin, as a dosage suppressor of a dominant negative TOR2 allele (tor2kin−). Both in mammalian and yeast cells, the TCP-1 complex has been implicated specifically in the folding and biogenesis of the cytoskeletal proteins actin and tubulin (25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36). The mammalian TCP-1 complex associates with and facilitates the folding of newly synthesized actin monomers in vitro, and yeast mutants defective in genes encoding subunits of the TCP-1 complex have disorganized actin structures. Furthermore, overexpression of TCP1 or ANC2, encoding two other subunits of the TCP-1 complex, suppresses act1-1 and act1-4, mutant alleles of the actin gene that prevent actin cable formation (33, 35). Overproduction of TCP20 restores actin organization and viability in a tor2 mutant.
A tor2 mutant arrests growth throughout the cell cycle within three to five generations, whereas a tor1 tor2 double mutant arrests growth in G1 within one generation, and TOR1 overexpression cannot suppress a TOR2 deficiency (20, 22, 23). Thus, TOR2 has two essential function, one of which is TOR2-unique and the other is redundant with TOR1. Previous studies have demonstrated that a tor1 tor2 double mutant arrests growth in G1 because of a defect in translation initiation (23). Three related observation presented here suggest that a tor2 single mutant is inviable because of a defect in the actin cytoskeleton. First, a tor2 mutant displays a defect in the cell cycle-dependent distribution of the actin cytoskeleton. Second, overexpression of TCP20, previously shown to affect actin function, suppresses the lethality of a TOR2 disruption. Third, TCP20 overexpression restores actin cytoskeleton distribution in a tor2 mutant. Additional observations suggest that the essential, actin-related function of TOR2 may be the TOR2-unique function. First, the TOR2-unique function is essential, as the actin-related function appears to be. Second, cells disrupted only in TOR1 show a completely normal polarized distribution of actin throughout the cell cycle; it is not possible to determine if a tor1 tor2 double mutant has a defect in the cell cycle-dependent distribution of actin because these mutants arrest in G1. Third, overexpression of TCP20 does not suppress the lethality of a TOR1 TOR2 double mutation nor the slight growth defect conferred by a TOR1 disruption alone. Fourth, TCP20 overexpression does not confer resistance to rapamycin which is proposed to inhibit only the TOR1-shared function of TOR2 (45). Thus, it appears that the essential TOR2-unique function is to control the organization of the actin cystoskeleton. Finally, because TCP20 was isolated as dosage suppressor of the kinase-dead tor2kin− allele, we conclude that the presumed PI kinase activity of TOR2 is required for the function of TOR2 in the organization of the actin cytoskeleton. Zheng et al. (45) have suggested previously that an intact kinase domain is required for the unique function of TOR2.
How might TOR2 control organization of the actin cytoskeleton? Because PI kinases and Rho-like GTPases have previously been linked in controlling the actin cytoskeleton (see Introduction), the simplest model is that TOR2 is part of a signaling pathway involving small GTPases. This pathway might control the interactions between actin and the actin binding proteins, such as profilin, cofilin, fimbrin, capping protein, and verprolin (46, 47), which are responsible for the function and organization of the actin cytoskeleton. Increased amounts of TCP20 might suppress a defect in this signaling pathway by compensating for an altered or missing interaction between actin and one of its binding proteins. It is unlikely that TCP20 is suppressing by restoring TOR2 function directly because TCP20 suppresses several different TOR2 alleles including a promoter alteration.
Why should one protein, such as TOR2, control both translation initiation and organization of the actin cytoskeleton? This would provide a means of integrating temporal and spatial control of cell growth (Fig. 4). TOR2 might both activate synthesis of new material when nutrients are available and, via organization of the actin cytoskeleton, orient the newly synthesized material toward the appropriate site of growth, thus ensuring that growth is temporally and spatially coordinated.
Figure 4.
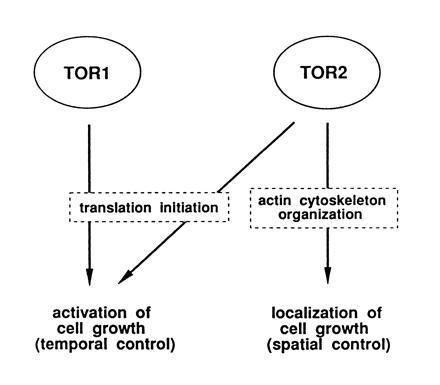
Model of TOR2 unique and redundant functions controlling cell growth.
Acknowledgments
We thank Stephen Helliwell, Marc Bickle, and Thomas Beck for comments on the manuscript. This research was supported by grants from the Canton of Basel and the Swiss National Science Foundation to M.N.H.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: PI, phosphatidylinositol.
References
- 1.Kilmartin J V, Adams A E. J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams A E, Pringle J R. J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams A E, Johnson D I, Longnecker R M, Sloat B F, Pringle J R. J Cell Biol. 1990;111:131–142. doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson D I, Pringle J R. J Cell Biol. 1990;111:143–152. doi: 10.1083/jcb.111.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamochi W, Tanaka K, Nonaka H, Maeda A, Musha T, Takai Y. J Cell Biol. 1994;125:1077–1093. doi: 10.1083/jcb.125.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui Y, Toh-e A. Mol Cell Biol. 1992;12:5690–5699. doi: 10.1128/mcb.12.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall A. Annu Rev Cell Biol. 1994;10:31–51. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, King W G, Dillon S, Hall A, Feig L, Rittenhouse S E. J Biol Chem. 1993;268:22251–22254. [PubMed] [Google Scholar]
- 9.Chong L D, Traynor-Kaplan A, Bokoch G M, Schwartz M A. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Y, Bagrodia S, Cerione R A. J Biol Chem. 1994;269:18727–18730. [PubMed] [Google Scholar]
- 11.Hawkins P T, Eguinoa A, Qiu R-G, Stokoe D, Cooke F T, Walters R, Wennström S, Claesson-Welsh L, Evans T, Symons M, Stephen L. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- 12.Ren X-D, Bokoch G M, Traynor-Kaplan A, Jenkins G H, Anderson R A, Schwartz M A. Mol Biol Cell. 1996;7:435–442. doi: 10.1091/mbc.7.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolias K F, Cantley L C, Carpenter C L. J Biol Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- 14.Eberle M, Traynor-Kaplan A E, Sklar L A, Norgauer J. J Biol Chem. 1990;265:16725–16728. [PubMed] [Google Scholar]
- 15.Arcaro A, Wymann M P. Biochem J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wymann M, Arcaro A. Biochem J. 1994;298:517–520. doi: 10.1042/bj2980517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S B, Rhee S G. Curr Opin Cell Biol. 1995;7:183–189. doi: 10.1016/0955-0674(95)80026-3. [DOI] [PubMed] [Google Scholar]
- 18.Hartwig J H, Bokoch G M, Carpenter C L, Janmey P A, Taylor L A, Toker A, Stossel T P. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 19.Heitman J, Movva N R, Hall M N. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 20.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva N R, Hall M N. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 21.Cafferkey R, Young P R, McLaughlin M M, Bergsma D J, Koltin Y, Sathe G M, Faucette L, Eng W K, Johnson R K, Livi G P. Mol Cell Biol. 1993;11:3691–3698. doi: 10.1128/mcb.13.10.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helliwell S B, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall M N. Mol Biol Cell. 1994;5:105–118. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbet N C, Schneider U, Helliwell S B, Stansfield I, Tuite M F, Hall M N. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall M N. Biochem Soc Trans. 1996;24:234–239. doi: 10.1042/bst0240234. [DOI] [PubMed] [Google Scholar]
- 25.Li W Z, Lin P, Frydman J, Boal T R, Cardillo T S, Richard L M, Toth D, Lichtman M A, Hartl F-U, Sherman F, Segel G B. J Biol Chem. 1994;269:18616–18622. [PubMed] [Google Scholar]
- 26.Stoldt V, Rademacher R, Kehren V, Ernst J F, Pearce D A, Sherman F. Yeast. 1996;12:523–529. doi: 10.1002/(SICI)1097-0061(199605)12:6%3C523::AID-YEA962%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 27.Frydman J, Nimmesgern E, Erdjument-Bromage H, Wall J S, Tempst P, Hartl F-U. EMBO J. 1992;11:4767–4778. doi: 10.1002/j.1460-2075.1992.tb05582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Y, Thomas J O, Chow R L, Lee G-H, Cowan N J. Cell. 1992;69:1043–1050. doi: 10.1016/0092-8674(92)90622-j. [DOI] [PubMed] [Google Scholar]
- 29.Gao Y, Vainberg I E, Chow R L, Cowan N J. Mol Cell Biol. 1993;13:2478–2485. doi: 10.1128/mcb.13.4.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaffe M B, Farr G W, Miklos D, Horwich A L, Sternlicht M L, Sternlicht H. Nature (London) 1992;358:245–248. doi: 10.1038/358245a0. [DOI] [PubMed] [Google Scholar]
- 31.Sternlicht H, Farr G W, Sternlicht M L, Driscoll J K, Willison K, Yaffe M B. Proc Natl Acad Sci USA. 1993;90:9422–9426. doi: 10.1073/pnas.90.20.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ursic D, Culbertson M R. Mol Cell Biol. 1991;11:2629–2640. doi: 10.1128/mcb.11.5.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinh D B-N, Drubin D G. Proc Natl Acad Sci USA. 1994;91:9116–9120. doi: 10.1073/pnas.91.19.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Sullivan D S, Huffaker T C. Proc Natl Acad Sci USA. 1994;91:9111–9115. doi: 10.1073/pnas.91.19.9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ursic D, Sedbrook J C, Himmel K L, Culbertson M R. Mol Biol Cell. 1994;5:1065–1080. doi: 10.1091/mbc.5.10.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miklos D, Caplan S, Mertens D, Hynes G, Pitluk Z, Kashi Y, Harrison-Lavoie K, Stevenson S, Brown C, Barrel B, Horwich A L, Willison K. Proc Natl Acad Sci USA. 1994;91:2743–2747. doi: 10.1073/pnas.91.7.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guthrie J, Fink G R. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic; 1991. [Google Scholar]
- 38.Hoffman C S, Winston F. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 39.Ito H, Fukuda Y, Murata K, Kiura A. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Habor Lab. Press; 1989. [Google Scholar]
- 41.Benedetti H, Raths S, Crausaz F, Riezman H. Mol Biol Cell. 1994;5:1023–1037. doi: 10.1091/mbc.5.9.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry M, Yonezawa K, Kasuga M, Waterfield M. EMBO J. 1994;13:511–521. doi: 10.1002/j.1460-2075.1994.tb06289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schu P, Takegawa K, Fry M, Stack J, Waterfield M, Emr S. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- 44.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng X-F, Fiorentino D, Chen J, Crabtree G R, Schreiber S L. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 46.Welch M D, Holtzman D A, Drubin D G. Curr Opin Cell Biol. 1994;6:110–119. doi: 10.1016/0955-0674(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 47.Donnelly S F, Pocklington M J, Pallotta D, Orr E. Mol Microbiol. 1993;10:585–596. doi: 10.1111/j.1365-2958.1993.tb00930.x. [DOI] [PubMed] [Google Scholar]