

Abstract
Apoptosis can be a potent weapon against viral infection and consequently has selected for viruses carrying antiapoptosis genes. Two baculovirus proteins, IAP and p35, can prevent insect cells from dying in response to infection. p35, which interferes with members of the Ced-3 family of cysteine proteases, can also function in mammalian cells. We investigated the ability of IAP from Orgyia pseudotsugata nuclear polyhedrosis virus to prevent death of mammalian cells. IAP was transiently expressed in mammalian cells and its ability to block cell death caused by expression of interleukin-1β converting enzyme (ICE), FADD, or the ICE homologues ICH-1 and ICE-Lap3, was investigated. IAP strongly inhibited ICE- and ICH-1-induced cell death but protected only partially against death by overexpression of FADD and not at all against death due to enforced ICE-Lap3 expression. These results demonstrate that a baculoviral IAP protein can functionally interact with conserved components of the apoptosis machinery in mammalian cells.
Keywords: interleukin 1β-converting enzyme, interleukin 1β-converting enzyme-Lap3, ICH-1, FADD
Apoptosis is used in a variety of situations. These include normal processes such as development and homeostasis, but also as a defence, both against infectious agents and dangerously altered cells of the organism itself. When the apoptosis machinery fails to act and unwanted cells survive, cancer or autoimmune disease can result. Conversely, there are examples of diseases that result from cells dying inappropriately (for review, see refs. 1 and 2). Apoptosis occurs in distantly related organisms by mechanisms that are largely conserved. There is evidence from Caenorhabditis elegans, Drosophila, and mammals for the existence of a common process in which an intracellular or extracellular trigger sends a signal that leads to activation of effector cysteine proteases in the cytoplasm. These proteases cleave substrates within the cell, ultimately causing the structural changes morphologically recognisable as apoptosis (for review, see refs. 2 and 3).
The proteases that act at the effector stage of apoptosis are members of a cysteine protease family that resemble the C. elegans protein Ced-3 (4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17). All of these cysteine proteases are produced as precursors that must be cleaved at aspartate residues to become active, and once activated each of them also cleaves to the carboxyl side of aspartic acid (15, 18, 19, 20, 21). The first described mammalian member of this family was interleukin 1β-converting enzyme (ICE). Like other family members, ICE is translated as an inactive precursor (p45). Cleavage at aspartate residues produces a shorter polypeptide that is further processed into p20 and p10 molecules. Assembly of two of each of these subunits into a heterotetramer is required to yield the active enzyme (22, 23). Further ICE homologues have been identified (4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17) that have been shown to stimulate cell death in various circumstances. The homologues described to date can be grouped, based on amino acid homology, into three subfamilies (16) with ICE and its close relatives ICE-RelII and ICE-RelIII/ICH-2/TX (8, 10, 11) as the first. ICH-1 (12) and its previously described murine homologue Nedd-2 (13) make up another of the ICE/ced-3 subfamilies. The longer transcript of ICH-1 kills when expression is enforced in cells (12), and full-length murine Nedd-2 can also trigger apoptosis upon transfection (13). ICE-Lap3/Mch-3 (16, 17), CPP32/YAMA/apopain (7, 14, 15), Mch-2 (6), and Ced-3 (4), make up the third subfamily. Overexpression of ICE-Lap3 lacking the amino-terminal 53-amino acid pro-domain in MCF7 cells causes apoptosis, whereas expression of the full-length proICE-Lap3 or a mutant in which the cysteine at the active site was changed to alanine does not cause cell death (16). The pro-form of ICE-Lap3 is cleaved into the active p12 and p20 proteins upon stimulation of Jurkat T cells and BJAB B cells with antibodies against CD95 (Fas/APO-1) or MCF7 cells with tumor necrosis factor α (TNF-α) (16).
Apoptosis is induced in many cell types by the binding of the CD95 ligand to CD95 (24), a member of the TNF receptor family of cell surface proteins. Evidence from mice in which the ICE gene has been deleted demonstrates that ICE is required for death of T-lineage cells caused by signaling through CD95 (25). The cytoplasmic domain of CD95 bears a region termed the “death domain” that allows it to interact with the death domain of another protein, FADD/MORT-1 (26, 27). It is thought that FADD may transmit the apoptotic signal from CD95, as overexpression of FADD itself can induce apoptosis (26, 27). In MCF7, BJAB, L929-Fas, and Rat-1-Fas cells, death induced by CD95 or by FADD can be blocked by the ICE inhibitor CrmA (27, 28, 29). FADD has also been implicated in signaling downstream of the TNF receptor 1 (p55) (30).
Upon viral infection, cells that die by apoptosis limit the ability of the virus to replicate. There is consequently a selective advantage for viruses that can subvert the apoptotic process. Several viruses carry genes that either resemble host antiapoptotic genes or encode proteins that interfere with the host’s apoptotic machinery (1). Genes for two distinct sets of cell death inhibitors, termed p35 and IAP proteins, have been found in insect viruses (31). p35 proteins do not resemble any other known proteins but can block apoptosis in nematode, insect, and mammalian cells (32, 33, 34, 35). p35, which acts as a protease inhibitor (36, 37), is a substrate for the Ced-3-like death effector cysteine proteases that, when cleaved, forms a stable complex with the protease. Mutation of the cleavage site of p35 prevents it from protecting against cell death.
IAP proteins contain two structural motifs: a pair of repeats designated baculovirus IAP repeats (BIRs) and a RING finger (31, 38). We were interested to determine whether IAP, like p35, could suppress apoptosis in mammalian cells. We tested the ability of IAP from Orgyia pseudotsugata nuclear polyhedrosis virus to inhibit death caused by four stimuli: enforced expression of one member of each of the ICE/ced-3 subfamilies (ICE, ICH-1, and ICE-Lap3) and of the CD95 signaling molecule FADD. The results demonstrate that baculovirus IAP can function in mammalian cells to prevent apoptosis due to ICE and ICH-1 expression but affords only partial protection against death by FADD and does not protect against death induced by enforced expression of ICE-Lap3.
MATERIALS AND METHODS
Cell Lines and Plasmids.
The p45ICE-, p32ICE-lacZ fusion plasmids, pβactM10Z and pβactM11Z (5), and ICH-LacZ plasmid, pβactH37Z (12), were provided by Junying Yuan (Massachusetts General Hospital, Boston). The FADD expression construct FADD-AU1 (27) and the ICE-Lap3 p28 cDNA (16) were obtained from Vishva Dixit (University of Michigan, Ann Arbor). The eukaryotic expression vector pEF was constructed by inserting the region from pEFBOS (39) encompassing the elongation factor EF-1α enhancer, stuffer, and poly(A) into a Bluescript plasmid (Stratagene) containing a PGKpuro cassette (D. Huang, S. Cory, and J. Adams, personal communication). The stuffer region [between the EF-1α enhancer and poly(A) sequence] was replaced by a multiple cloning site, into which fragments encoding Bcl-2, CrmA, p35 from Autographa californica nuclear polyhedrosis virus, and IAP from O. pseudotsugata nuclear polyhedrosis virus were inserted for expression from the elongation factor promoter. A fragment encoding β-galactosidase was expressed from the murine cytomegalovirus (CMV) promoter (40). The truncated OpIAP plasmid was constructed by digestion of the pEF vector containing full-length IAP with NruI and SmaI and religating. This deleted sequences 3′ of the NruI site in the OpIAP gene that encode the RING finger domain.
Transfections.
For the ICE experiments, nearly confluent HeLa or CHO cells, grown in RPMI 1640 medium with 10% fetal calf serum, were transfected with 0. 1 μg of ICE/lacZ plasmid and 1 μg of test plasmid using 3 μl of LipofectAmine (GIBCO/BRL) per well in 12-well tissue culture dishes. In the control transfections, 0. 1 μg of lacZ plasmid and 1 μg of test plasmid were used. After 16 hr of incubation, the cells were fixed (2% formaldehyde/0. 2% glutaraldehyde in PBS) for 5 min and stained for β-galactosidase expression with 0.1% 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal)/5 mM potassium ferricyanide/5 mM potassium ferrocyanide/2 mM MgCl2 in PBS. The blue cells were visually scored as alive or dead. For all experiments, transfections and scoring were carried out on randomized coded wells.
The analysis of protection against ICH-1-induced death was performed essentially as above. Cells were transfected with 0.1 μg of ICH-1/lacZ plasmid and 1 μg of test or control plasmid, allowed to express the transgenes, stained, and scored.
Analysis of protection against ICE-Lap3 induced death was performed the same way. The p28ICE-Lap3 pcDNA3 plasmid (0.1 μg), lacZ plasmid (0.1 μg), and either IAP or p35 plasmids (0.8 μg) were cotransfected into HeLa cells or the human embryonic kidney cell line 293T (a simian virus 40-transformed subline of 293 cells). For 293T transfections, 6 μl of LipofectAmine was used per well. Staining was carried out after 48 hr expression of the transfected genes.
To assess protection against FADD expression, HeLa cells or CHO cells were transfected with 0.1 μg of lacZ plasmid, 0.45 μg of a FADD expression plasmid, and 0.45 μg of test plasmid as above. In the control transfections to assess background death, 0.1 μg of lacZ plasmid and 0.9 μg of empty pEF plasmid were used.
RESULTS
DNA fragments encoding the p35 protein from A. californica nuclear polyhedrosis virus, the IAP protein from O. pseudotsugata nuclear polyhedrosis virus (OpIAP), CrmA from Cowpox virus, and human Bcl-2 were inserted into pEF, a eukaryotic expression vector. These plasmids were transfected into HeLa cells with a construct that expresses a fusion protein of ICE (p32) and β-galactosidase. Transient expression of ICE caused death of HeLa cells, as when cells were transfected with the ICE/lacZ construct, 80% of the X-Gal-staining cells had died at 18 hr, whereas when β-galactosidase was expressed alone only a quarter of the transfected (blue) cells were dead (see Fig. 1 for examples). These observations confirm the work of others who have previously characterized this system by showing that apoptosis caused by overexpression of ICE induces all of the classical morphological and biochemical changes indicative of apoptosis in both HeLa and other cell types (11, 12, 41). Furthermore, apoptosis induced in this way requires active cysteine protease to be produced from the transfected plasmid, as apoptosis does not occur if the conserved cysteine residue is mutated (5). Coexpression of either Bcl-2 or CrmA, but not an empty vector control, was able to reduce the proportion of blue cells that were dead (Fig. 2). The baculovirus apoptosis inhibitory genes p35 and OpIAP were able to reduce markedly the number of cells killed by ICE. That this protection was specific for the anti-apoptosis genes, rather than an artefact of transfection, was supported by the lack of protection afforded by an OpIAP gene from which the RING finger had been deleted (Fig. 2A) and transfections with constructs bearing other nonprotective genes (data not shown). To assess the background level of death caused by the transfection itself and to determine if any of the apoptosis inhibitory genes influenced the amount of death in the absence of transfected ICE, the vectors carrying the test genes and control plasmids were cotransfected with a vector bearing the lacZ gene alone, in a parallel set of transfections (Fig. 2A). None of the genes affected the level of cell death caused by the transfection procedure itself. OpIAP was also shown to suppress ICE-induced apoptosis in CHO cells (Fig. 2B), indicating that the protection afforded by OpIAP was not cell line-specific.
Figure 1.
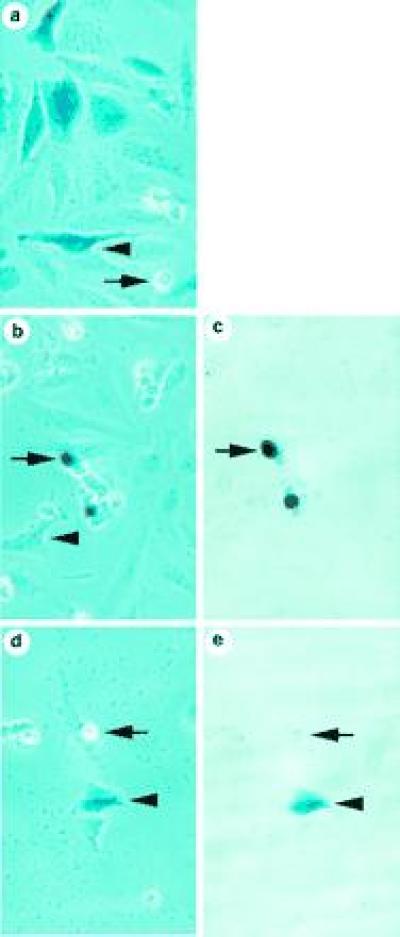
ICE-mediated apoptosis of HeLa cells. X-Gal-stained HeLa cells transiently transfected with the following plasmids: lacZ (a), ICE/lacZ (b and c), and ICE/lacZ (d and e) with pEF-OpIAP. Cells were scored as dead or alive based upon their appearance by phase contrast (Left) or conventional (Right) microscopy, as described by Miura et al. (5). Live cells (arrows without tails) were large and flat with an irregular shape and smooth edges. Dead cells (arrows with tails) were small, refractile, and round, with irregular “blebbed” membranes. Some had detached from the culture dish.
Figure 2.
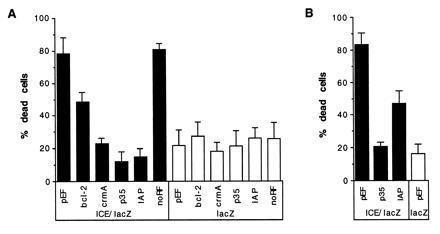
Inhibition of ICE-mediated cell death by IAP expression. (A) HeLa cells were transiently cotransfected with a p32ICE-lacZ fusion plasmid and with expression plasmids carrying either no insert, bcl-2, crmA, p35, OpIAP, or truncated IAP lacking the sequences encoding the RING finger (designated noRF). Parallel wells of HeLa cells were cotransfected with the same test plasmids and a lacZ plasmid. At least five cultures were transfected and counted blind for each test plasmid; as for all figures, values indicate the percentage of X-Gal-stained cells that were dead (mean ± 2 SEM) for each set of replicates. (B) CHO cells transfected with lacZ plasmid and empty pEF vector (lane 1) and cotransfections of ICE/lacZ and either pEF, p35, or OpIAP expression plasmids. Three transfections of CHO cells were performed for each test plasmid.
These results indicate that OpIAP is able to function in mammalian cells to block their death by ICE and, therefore, must interfere with some step in the pathway of apoptosis involving ICE. OpIAP can protect against overexpression of a fusion gene between full-length ICE (p45) and lacZ (data not shown). Because OpIAP can also confer protection against p32 ICE (Fig. 2) it does not act by inhibiting removal of this pro sequence but must act either to prevent generation of the active enzyme from the p32 subunit or prevent the active (p10p20)z enzyme from cleaving substrates.
To examine whether the protection afforded by IAP against ICE reflects a general ability to inhibit all Ced-3-related cysteine proteases, we analyzed the ability of OpIAP to prevent death due to overexpression of the cysteine proteases ICH-1 and ICE-Lap3. As in the ICE experiment, a plasmid encoding a fusion protein between ICH-1 and lacZ was introduced into HeLa or CHO cells with either OpIAP or p35 plasmids or empty vector. After 16 hr of expression time, the cells were stained and those that were blue were scored for viability. Both OpIAP and p35 were capable of substantially reducing the death caused by expression of ICH-1 in the cell lines studied (Fig. 3). A plasmid bearing the ICE-Lap3 p28 DNA was cotransfected into 293T cells or CHO cells with the lacZ plasmid and the test plasmids and controls. ICE-Lap3 requires a longer time than ICE to achieve efficient death (ref. 17 and data not shown). Two days after the cells were transfected, they were stained with X-Gal and the blue (transfected) cells were scored. In the two cells lines studied that differed in their susceptibility to killing by ICE-Lap3, p35 was able to block ICE-Lap3 induced death, but OpIAP was not (Fig. 4).
Figure 3.
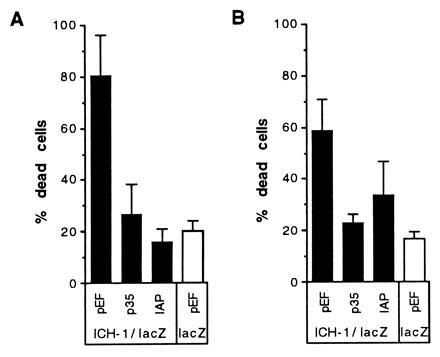
Protection from ICH-1-induced death by OpIAP. HeLa (A) or CHO (B) cells were cotransfected with the ICH-1/lacZ plasmid and empty vector, p35, or OpIAP expression constructs. A cotransfection in which the lacZ plasmid and pEF vector were introduced into cells reflects the background death due to transfection (bar on the left panel).
Figure 4.
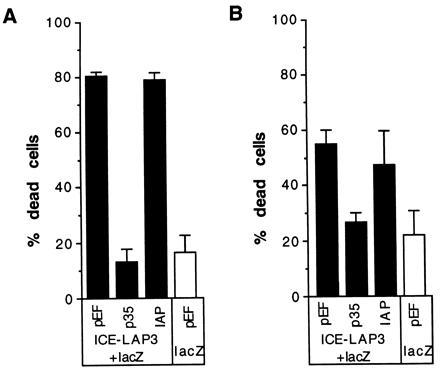
Inability of IAP to prevent death due to expression of ICE-Lap3. Cells, either 293T (A) or CHO (B) were cotransfected with ICE-Lap3 p28 plasmid, lacZ, and either empty vector or a plasmid bearing p35 or IAP. Two days after the addition of serum after transfection, the cells were stained and the blue (transfected) cells were scored for viability. At least three wells for each plasmid combination were transfected and scored blind.
FADD interacts with the cytoplasmic domain of CD95 and acts immediately downstream of it to transduce a signal leading to apoptosis after binding of CD95 to its ligand (26, 27). Transient expression of FADD has been shown to cause cell death in the absence of signaling through CD95, presumably by self-association through its death domains (26, 27). We performed cotransfections of HeLa cells with a FADD expression construct, a lacZ plasmid, and the constructs bearing the apoptosis inhibitory genes, to determine whether IAP could also protect against FADD-induced death (Fig. 5). Bcl-2, CrmA, and p35 were effective in protecting against FADD and decreased the proportion of transfected cells that were dead to background levels. OpIAP reproducibly and significantly reduced the amount of apoptosis caused by FADD, but the protection afforded was consistently only partial. In a second cell line, CHO cells, OpIAP was unable to prevent death due to expression of FADD (Fig. 5B), possibly indicating that the signal from FADD is stronger in these cells. Interestingly p35 was not able to prevent FADD-induced death in CHO cells either, presumably due to higher levels of activated cystine proteases in CHO cells after FADD expression or activation of cysteine proteases in CHO cells that are not blocked by p35.
Figure 5.
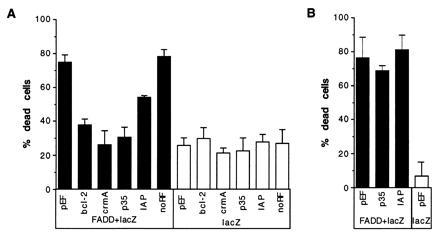
Partial suppression of death due to FADD expression by OpIAP. Plasmids expressing Bcl-2, CrmA, p35, IAP, only the amino-terminal half of OpIAP, or the empty vector were cotransfected into HeLa cells with a lacZ plasmid and a FADD expression plasmid (A). Parallel control wells were also transfected with the test plasmids and lacZ plasmid. At least five wells for each test plasmid were transfected and scored blind. The empty vector, p35, and OpIAP plasmids were also cotransfected with the lacZ and FADD plasmids into CHO cells in the same manner as the HeLa transfection (B).
DISCUSSION
These studies demonstrate that the IAP protein from O. pseudotsugata nuclear polyhedrosis virus can inhibit mammalian cell death caused by overexpression of ICE and ICH-1. Several cell types normally express ICE homologues but remain alive, so why should transfection of cells with ICE-like precursor proteins be sufficient to cause apoptosis? One possibility is that protease activation signals are constitutively present but are insufficient to activate the endogenous levels of Ced-3 homologue precursors. However, these constitutive activation signals may be able to produce enough active proteases to cause apoptosis when cells are transfected with large amounts of, e.g., p32 ICE precursor.
OpIAP could act to inhibit apoptosis caused by ICE homologues in one of two ways. The first possibility is that it could function downstream of ICE/ICH-1 proteases to block them directly or interfere with steps after protease activation. It is difficult to explain the limited ability of OpIAP to prevent FADD-induced death according to this model. As ICE is required for CD95 killing, at least in some cell types (25), and CD95 signaling appears to work via FADD (26, 27), it is reasonable to assume that FADD causes death by activating ICE. If this is also true for the cell types used in these assays and if IAP proteins act downstream of ICE, we would have expected OpIAP to provide more effective protection against FADD-mediated apoptosis.
The finding that p35 is incapable of inhibiting FADD-mediated death was somewhat surprising. p35 functions by directly interacting with Ced-3-related cysteine proteases and must be present in at least equimolar amounts to inhibit their actions (37). The ability of p35 to prevent death caused by FADD expression in HeLa but not CHO cells may reflect an increased production of active proteases in CHO cells compared with HeLa cells to the extent that the molar amount of ced-3 homologues exceeds that of p35 protein or that CHO cells use a protease that is not blocked by p35.
Rather than acting downstream of the proteases, OpIAP may act upstream of ICE and ICH-1, to inhibit steps required for their activation. According to this model, OpIAP can efficiently block the low level of constitutive endogenous activation signals that are required for activation of transfected protease precursors but are only marginally effective at blocking the greater level of signaling due to the overexpressed FADD transgene. The varying ability of IAP to protect against FADD-induced death in different cell lines may reflect differing strengths of the activation signals coming from FADD in the different cells. If OpIAP does act upstream of ICE to subdue the endogenous activation signals, the inability of OpIAP to prevent death caused by ICE-Lap3 expression may mean that the signals that lead to the activation of ICE and ICH-1 differ from those that activate ICE-Lap3.
The ability of baculovirus OpIAP protein to protect against cell death in mammalian cells is an example of conservation of apoptotic mechanisms between distantly related species. Previous demonstrations of this conservation include the ability of the mammalian protein Bcl-2 to prevent cell death in nematodes (42, 43) and the finding that the baculovirus gene p35 protects mammalian neurons from death due to growth factor withdrawal (34). That a baculovirus OpIAP protein can function to inhibit apoptosis of mammalian cells demonstrates that components of the mammalian cell death machinery exist that can interact with IAP proteins. Two groups have recently demonstrated that mammalian homologues of OpIAP can prevent cell death in mammalian cells (44, 45), confirming the functional conservation of the pathway in which OpIAP and its homologues act during evolution. Some of the mammalian IAP proteins can interact with TRAF-1 and TRAF-2 (45, 46), components of the TNF signaling complex that associate with TNFR2 (47), and although the OpIAP protein seems unable to interact with the TRAFs characterized to date (45), this may imply that OpIAP acts to block upstream signaling events leading to cell death.
Acknowledgments
We thank Miha Pakusch for excellent technical assistance, Rollie Clem and Lois Miller for providing the genes for p35 and IAP, David Pickup for the crmA gene, David Huang for pEF vector and pEF-crmA and pEF-bcl2 plasmids, Junying Yuan for the ICE/lacZ and ICH-1/lacZ expression constructs, and Vishva Dixit for the FADD-AU1 and ICE-Lap3 vectors. D.L.V. is an Investigator with the Cancer Research Institute of New York and is supported by the Anti-Cancer Council of Victoria. G.H. is supported by a fellowship from the Deutsche Forschungsgemeinschaft.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ICE, interleukin 1β-converting enzyme; TNF-α, tumor necrosis factor α; X-Gal, 5-bromo-4-chloro-3-indolyl β-d-galactoside; OpIAP, Orgyia pseudotsugata nuclear polyhedrosis virus IAP.
References
- 1.Vaux D L, Häcker G, Strasser A. Cell. 1994;76:777–779. doi: 10.1016/0092-8674(94)90350-6. [DOI] [PubMed] [Google Scholar]
- 2.Steller H. Science. 1995;267:1445–1462. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 3.Kumar S. Trends Biochem Sci. 1995;20:198–202. doi: 10.1016/s0968-0004(00)89007-6. [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Shaham S, Ledoux S, Ellis H M, Horvitz H R. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 5.Miura M, Zhu H, Rotello R, Hartweig E A, Yuan J. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 6.Fernandes-Alnemri T, Litwack G, Alnemri E S. Cancer Res. 1995;55:2737–2742. [PubMed] [Google Scholar]
- 7.Fernandes-Alnemri T, Litwack G, Alnemri E S. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 8.Faucheu C, Diu A, Chan A W E, Blanchet A M, Miossec C, Herve F, Collarddutilleul V, Gu Y, Aldape R A, Lippke J A, Rocher C, Su M S S, Livingston D J, Hercend T, Lalanne J L. EMBO J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alnemri E, Fernandes-Alnemri T, Litwack G. J Biol Chem. 1995;270:4312–4317. doi: 10.1074/jbc.270.9.4312. [DOI] [PubMed] [Google Scholar]
- 10.Kamens J, Paskind M, Hugunin M, Talanian R V, Allen H, Banach D, Bump N, Hackett M, Johnston C G, Li P, Mankovich J A, Terranova M, Ghayur T. J Biol Chem. 1995;270:15250–15256. doi: 10.1074/jbc.270.25.15250. [DOI] [PubMed] [Google Scholar]
- 11.Munday N A, Vaillancourt J P, Ambereen A, Casano F J, Miller D K, Molineaux S M, Yamin T-T, Yu V L, Nicholson D W. J Biol Chem. 1995;270:15870–15876. doi: 10.1074/jbc.270.26.15870. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 13.Kumar S, Kinoshita M, Noda M, Copeland N G, Jenkins N A. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, Munday N A, Raju S M, Smulson M E, Yamin T T, Yu V L, Miller D K. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 15.Tewari M, Quan L T, O’Rourke K, Desnoyers S, Zeng Z, Beidler D R, Poirier G G, Salvesen G S, Dixit V M. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 16.Duan H, Chinnaiyan A M, Hudson P L, Wing J P, He W W, Dixit V M. J Biol Chem. 1996;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- 17.Fernandes-Alnemri T, Litwack G, Alnemri E S. Cancer Res. 1995;55:6045–6052. [PubMed] [Google Scholar]
- 18.Sleath P R, Hendrickson R C, Kronheim S R, March C J, Black R A. J Biol Chem. 1990;265:14526–14528. [PubMed] [Google Scholar]
- 19.Howard A D, Kostura M J, Thornberry N A, Ding G J, Limjuco G, Weidner J, Salley J P, Hogquist K A, Chaplin D D, Mumford R A, Schmidt J A, Tocci M J. J Immunol. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 20.Thornberry N A, Molineaux S M. Protein Sci. 1995;4:3–12. doi: 10.1002/pro.5560040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray C A, Black R A, Kronheim S R, Greenstreet T A, Sleath P R, Salvesen G S, Pickup D J. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 22.Walker N P C, Talanian R V, Brady K D, Dang L C, Bump N J, et al. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 23.Wilson K P, Black J A F, Thomson J A, Kim E E, Griffith J P, Navia M A, Murcko M A, Chambers S P, Aldape R A, Raybuck S A, Livingston D J. Nature (London) 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 24.Nagata S, Goldstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 25.Kuida K, Lippke J A, Ku G, Harding M W, Livingston D J, Su M S, Flavell R A. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 26.Boldin M P, Varfolomeev E E, Pancer Z, Mett I L, Camonis J H, Wallach D. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 27.Chinnaiyan A M, O’Rourke K, Tewari M, Dixit V M. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 28.Enari M, Hug H, Nagata S. Nature (London) 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 29.Tewari M, Dixit V M. J Biol Chem. 1995;270:3255–3260. doi: 10.1074/jbc.270.7.3255. [DOI] [PubMed] [Google Scholar]
- 30.Hsu H, Shu H-B, Pan M-G, Goeddel D. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 31.Clem R J, Miller L K. Mol Cell Biol. 1994;14:5212–5222. doi: 10.1128/mcb.14.8.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto A, Friesen P D, Rothman J H. EMBO J. 1994;13:2023–2028. doi: 10.1002/j.1460-2075.1994.tb06475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clem R J, Fechheimer M, Miller L K. Science. 1991;254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 34.Rabizadeh S, Lacount D J, Friesen P D, Bredesen D E. J Neurochem. 1993;61:2318–2321. doi: 10.1111/j.1471-4159.1993.tb07477.x. [DOI] [PubMed] [Google Scholar]
- 35.Hay B A, Wolff T, Rubin G M. Development (Cambridge, UK) 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 36.Xue D, Horvitz H R. Nature (London) 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- 37.Bump N J, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, Licari P, Mankovitch J, Shi L, Greenberg A H, Miller L K, Wong W W. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- 38.Crook N E, Clem R J, Miller L K. J Virol. 1993;67:2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima S, Nagata S. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorsch-Haesler K, Keil G M, Weber F, Jasin M, Schaffner W, Koszinowski U H. Proc Natl Acad Sci USA. 1985;82:8325–8329. doi: 10.1073/pnas.82.24.8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miura M, Freidlender R M, Yuan J. Proc Natl Acad Sci USA. 1995;92:8318–8322. doi: 10.1073/pnas.92.18.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaux D L, Weissman I L, Kim S K. Science. 1992;258:1955–1957. doi: 10.1126/science.1470921. [DOI] [PubMed] [Google Scholar]
- 43.Hengartner M O, Horvitz H R. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 44.Liston P, Roy N, Tamai K, Lebebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda J-E, MacKenzie A, Korneluk R G. Nature (London) 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 45.Uren A G, Pakusch M, Hawkins C J, Puls K L, Vaux D L. Proc Natl Acad Sci USA. 1996;93:4976–4978. doi: 10.1073/pnas.93.10.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothe M, Pan M-G, Henzel W J, Ayres T M, Goeddel D V. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 47.Rothe M, Wong S C, Henzel W J, Goeddel D V. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]