

Abstract
Human Burkitt lymphoma cell lines give rise to progressively growing subcutaneous tumors in athymic mice. These tumors are induced to regress by inoculation of Epstein–Barr virus-immortalized normal human lymphocytes. In the present study, analysis of profiles of murine cytokine/chemokine gene expression in Burkitt tumor tissues excised from the nude mice showed that expression of the murine α-chemokine interferon-inducible protein-10 (IP-10) was higher in the regressing than in the progressive Burkitt tumors. We tested the effects of IP-10 on Burkitt tumor growth in nude mice. Inoculation of established Burkitt tumors either with crude preparations of murine IP-10 or with purified human IP-10 caused visible tumor necrosis in a proportion of the animals, although no complete tumor regressions were observed. Constitutive expression of murine IP-10 in Burkitt cells reduced their ability to grow as subcutaneous tumors, and caused visible tumor necrosis in a proportion of the animals. Histologically, IP-10-treated and IP-10-expressing Burkitt tumors had widespread evidence of tumor tissue necrosis and of capillary damage, including intimal thickening and vascular thrombosis. Thus, IP-10 is an antitumor agent that promotes damage in established tumor vasculature and causes tissue necrosis in human Burkitt lymphomas established subcutaneously in athymic mice.
The dependence of solid tumors on neovascularization for growth in vivo provides an opportunity for therapeutic intervention (1, 2, 3). Several inhibitors of angiogenesis, platelet factor 4, angiostatin, the fumagillin derivative AGM 1470, and an antibody against the vascular endothelial growth factor (also called vascular permeability factor), have shown antitumor activity in experimental systems (4, 5, 6, 7). By inhibiting tumor-induced neovascularization, the resultant tissue ischemia and starvation are thought to halt or reduce tumor growth (8, 9, 10).
Necrosis and regression of experimental Burkitt lymphoma, breast adenocarcinoma, and other human malignancies are induced reproducibly in athymic mice by intratumor inoculation of Epstein–Barr virus-immortalized lymphoblastoid cell lines (LCL) (11, 12, 13). This experimental approach to cancer treatment exploits the ability of athymic mice to reject LCL, and targets an effective antitumor response to malignant cells normally incapable of eliciting such response. The possibility of impaired angiogenesis participating in this process was suggested by the gross and microscopic morphology of regressing tumors showing central necrosis and diffuse vascular damage with endothelial thickening and capillary thrombosis, often distal to the necrotic tumor (11, 12).
Focusing on murine host factors elicited in response to inoculation of human cells, we sought to identify the causative factors of vascular injury and tumor regression in this experimental tumor model. We now report the identification of the interferon-inducible protein-10 (IP-10, also named Crg-2 in the mouse) (14, 15, 16), as one such factor that accounts for a portion of the effects typically induced by LCL treatment of Burkitt tumors. IP-10 belongs to the family of α-chemokines that includes interleukin (IL) 8 and platelet factor 4 (17).
METHODS
Cell Lines and Cell Cultures.
The human Burkitt lymphoma cell lines CA46, JD38, and BL-41 were derived by spontaneous outgrowth of single-cell suspended Burkitt lymphoma tissues (18). The Epstein–Barr virus-immortalized VDS-O cell line was obtained by spontaneous outgrowth of peripheral blood B cells from an Epstein–Barr virus-seropositive normal individual (19).
Animal Studies.
BALB/c nu/nu mice, 6 to 8 weeks of age (Taconic Farns) received 400 rad (1 rad = 0.01 Gy) total body irradiation and 24 hr later were injected subcutaneously in the right abdominal quadrant with 107 exponentially growing human Burkitt lymphoma cells in 0.2 ml RPMI medium 1640 (Biofluids, Rockville, MD) supplemented with 10% fetal bovine serum (FBS; Intergen, Purchase, NY) (11). Tumor size was estimated (in cm2) twice weekly as the product of two-dimensional caliper measurements (longest perpendicular length and width). Test mice bearing subcutaneously established Burkitt tumors (at least 0.2 cm2 in size) were injected daily (for 24 to 36 days) into the tumor with murine IP-10, human IP-10, or appropriate controls (injection volume 0.2 ml). As a source of murine IP-10, we used culture medium [serum-free MEM (GIBCO/BRL) containing 100 mM cadmium sulfate] conditioned for 24 hr with confluent cultures of Chinese hamster ovary (CHO) cells engineered to secrete murine IP-10, as detailed below. Culture supernatants from control transfected CHO cells (as detailed below) were used in control injections. Recombinant purified (>90% pure as evaluated by silver staining; endotoxin content 0.06 unit/ml) human IP-10 from PeproTech (Rocky Hill, NJ) was diluted in normal saline solution containing 1% bovine serum albumin (BSA; Boehringer Mannheim). Saline solution containing 1% BSA was used in control injections.
Expression of Murine IP-10 or Murine Mig in CHO Cells.
For transfection of murine IP-10 into CHO cells (20, 21) a 387-bp fragment containing the entire coding sequence of murine IP-10 (16) was inserted into the vector pMSXND downstream of the metallothionein 1 promoter; the vector also contained a neomycin resistance gene and the cDNA sequence for mouse dihydrofolate reductase. pMSXND/IP-10 was linearized and transfected into CHO cells by the lipofectin method (GIBCO/BRL). Transfected cells were selected in 400 mg/ml G418 (GIBCO/BRL), followed by culture without G418 but with 0.2 mM methotrexate (Sigma) in MEM supplemented with 11.5 mg/ml proline and 10% dialyzed FCS (Sigma). Methotrexate resistant colonies were analyzed for production of murine IP-10 by SDS/PAGE followed by immunoblotting. The CHO/C4 line was chosen for murine IP-10 production. Control CHO cells were transfected with pMSXND into which the cDNA for the human Mig chemokine had been inserted in the antisense orientation (20). The cell line CHO/R5 was used for production of control supernatants.
Expression of Human IP-10 in Burkitt Lymphoma Cells.
The plasmid pCEP4 IP-10 was constructed by placing a 407-bp fragment containing the entire coding region of mouse IP-10 gene (16) (along with 52 bases upstream and 54 bases downstream of the coding region) downstream of the CMV promoter in the expression vector pCEP4 (Invitrogen). CA46 Burkitt lymphoma cells were transfected with pCEP4/IP-10 or pCEP4 by electroporation. Cells were pulsed in 0.5 ml phosphate-buffered saline supplemented with 20 mM Hepes with 1.4 kV and 25 mF using a Gene Pulser (Bio-Rad). Transfected cells were selected in medium containing 0.8 mg/ml hygromycin and cloned by limiting dilution.
Reverse Transcriptase–Mediated PCR (RT-PCR) Analysis.
Total cellular RNA was isolated by guanidine thiocyanate/CsCl centrifugation, and 4 mg aliquots were reverse transcribed using an RNase H− RT (Superscript; GIBCO/BRL). The resultant cDNA (80 ng) was amplified using radiolabeled dNTP by a semiquantitative PCR (22). The number of amplification cycles was selected for each primer pair to provide the maximum signal intensity within the linear portion of a product versus template amplification curve (22). Aliquots from each amplification reaction were electrophoresed on 6% acrylamide (Long Ranger; AT Biochem, Malvern, PA) Tris·borate EDTA gels, followed by autoradiography and quantification by PhosphorImager analysis. Nucleotide sequences for murine 5′ and 3′ primers, respectively (followed in parenthesis by annealing temperature), were as follows: IP-10, ACCATGAACCCAAGTGCTGCCGTC and GCTTCACTCCAGTTAAGGAGCCCT (64°C); interferon (IFN)-γ, TGCGGCCTAGCTCTGAGACAATGA and TGAATGCTTGGCGCTGGACCTGTG (64°C); IL-12 (p35), TCCAGCATGTGTCAATCACGCTAC and GTTGATGGCCTGGAACTCTGTCTG (62°C); IL-12 (p40), CCGGTTTCCATCGTTTTGCTGG and AGAGTCAGGGGAACTGCTACTGCT (56.5°C); Mig (monokine induced by IFN-γ), ACTCAGCTCTGCCATGAAGTCCGC and AAAGGCTGCTCTGCCAGGGAAGGC (66°C); JE, ATGCAGGTCCCTGTCATG and GTTCACTGTCACACTGGT (55°C); RANTES (regulated on activation, normal T-cell expressed), GTACCATGAAGATCTCTGCA and TCTATCCTAGCTCATCTCCA (57°C); macrophage inflammatory protein (MIP)-1α, CTCAACATCATGAAGGTC and GGCATTCAGTTCCAGGTC (57°C); MIP-1β, GGCATTCAGTTCCAGGTC and CAACTCCAAGTCACTCAT (57°C); G3PDH, GCCACCCGAAAGACTGTGGATGGC and CATGTAGGCCATGAGGTCCACCAC (56.5°C). The number of amplification cycles used were 21 for G3PDH, 22 for IP-10, Mig, JE, RANTES, MIP-1α, and MIP-1β, and 31 for IFN-γ, IL-12 p35, and IL-12 p40 subunits. Nucleotide sequences (annealing temperature, amplification cycles) for 5′ and 3′ human IP-10 were as follows: GGAACCTCCAGTCTCAGCACC and CAGCCTCTGTGTGGTCCATCC (64°C, 22 cycles). The murine and human IP-10 primer pairs discriminated between murine and human IP-10.
Histology.
Tumors were removed in toto, fixed in 10% neutral buffered formalin solution (Sigma), embedded in paraffin, sectioned at 4 mm, and stained with hematoxylin and eosin or with elastin van Gieson (EVG) reagent.
Statistical Analyses.
A Student’s t test was used to evaluate the significance of group differences. The slopes of growth curves were determined from square root-transformed tumor size data.
RESULTS
Profiles of Cytokine and Chemokine Expression in Athymic Mice Bearing Burkitt Lymphoma.
Burkitt tumors were established in BALB/c nu/nu mice by subcutaneous inoculation of 107 Burkitt lymphoma cells (CA46, BL-41, and JD38 cell lines) (11, 12). Tumor regression was induced by weekly intratumor inoculation of 107 LCL (VDS cell line) as described (11, 12). Regressing tumors were harvested when LCL-treated Burkitt tumors developed typical tumor necrosis and scarring. Progressive tumors were harvested from untreated Burkitt tumors. Murine cytokine mRNA profiles of LCL-treated, regressing Burkitt lymphoma tumors were compared with those of untreated, progressive Burkitt tumors by RT-PCR analysis (Fig. 1). The PCR products for the murine cytokines/chemokines IP-10, IFN-γ, the IL-12 p35 subunit, Mig, and RANTES were found to be 8- to 15-fold more abundant in the regressing than in the progressive tumors studied. In contrast, the IL-12 p40 subunit, JE/monocyte chemoattractant protein-1 (JE/MCP-1), MIP-1α, and MIP-1β PCR products were detected at similar levels (≤ 4-fold difference) in regressing and progressive Burkitt tumors (Fig. 1). Northern blot analysis confirmed that the mRNAs for the murine chemokine IP-10 were expressed at higher levels (16.4-fold) in five LCL-treated Burkitt tumors than in five untreated control Burkitt tumors (not shown). Expression of human IP-10 was undetectable by RT-PCR in three Burkitt cell lines (CA46, BL-41, and JD38) and in the LCL VDS-O cell line (not shown).
Figure 1.
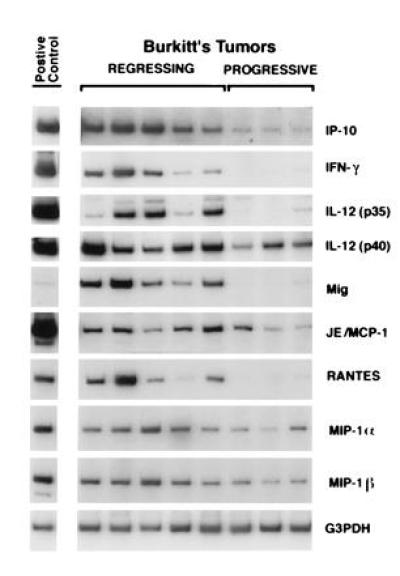
Cytokine and chemokine mRNAs in regressing and progressive Burkitt tumors revealed by RT-PCR analysis. Tissue fragments were obtained from regressing and progressive Burkitt tumors established in athymic mice. Positive controls were derived from murine macrophages incubated with 1 mg/ml lipopolysaccharide for 6 hr (IP-10, IL-12, Mig, and JE/MCP-1); murine splenocytes incubated with 4 mg/ml concanavalin A for 3 hr (IFN-γ); murine splenocytes from a 3-day mixed lymphocyte reaction (RANTES, MIP-1α, and MIP-1β). Total cellular RNA, isolated from tumor and control tissues, was subjected to RT-PCR analysis. After normalization to G3PDH, the signals derived from cytokine expression in regressing versus progressive Burkitt tumors, expressed as ratios, were as follows: IP-10, 8.2; IFN-γ, 10.6; IL-12 p35, 15.6; IL-12 p40, 4.0; Mig, 12.8; JE/MCP-1, 2.3; RANTES, 8.1; MIP-1α, 2.8; MIP-1β, 2.7.
IP-10 Treatment of Burkitt Lymphoma in Athymic Mice.
Crude supernatants of CHO cells genetically engineered to secrete high levels of murine IP-10 were each injected daily into established Burkitt lymphoma tumors. A proportion (4 of 12) of animals treated with murine IP-10, but none treated with control culture supernatants (0 of 14), developed central tumor necrosis that progressively extended superficially and deep into the tumor and was indistinguishable in appearance to that induced by weekly intratumor injections of LCL (not shown).
Purified human recombinant IP-10 was then injected daily for 30 to 35 days into subcutaneous Burkitt lymphoma tumors established in athymic mice, and the antitumor effect of IP-10 was compared with that of LCL (Table 1). IP-10 treatment was tolerated very well by all animals. Sixty-four percent of mice (9 of 14) showed a visible response to human IP-10 intratumor inoculations characterized by superficial tumor necrosis and scarring (≥4 mm2) that progressively extended during treatment to encompass larger portions of the tumor. These characteristic effects were first observed after an average of 18 IP-10 injections. In no case did the IP-10-treated tumors completely regress or stop growing. At the end of treatment, those tumors that had developed a visible response to IP-10 treatment were generally smaller than those that did not (P = 0.06). Overall, IP-10-treated tumors were not significantly smaller than control untreated and diluent-treated tumors (P = 0.46). When sectioned, the IP-10 responsive tumors were found to be 40% to 71% necrotic. Those IP-10-treated tumors that did not develop superficial necrosis and scarring (5 of 14) also showed extensive internal necrosis on tumor sections involving, on the average, 44% (range 20–56%) of the tumor tissue. In these cases, tumor tissue necrosis was deep and did not reach the epidermis. Control mice injected daily with diluent alone showed no visible responses (Table 1), and tumors showed only occasional focal necrosis that was distinguishable in size (<25% of the tumor mass) and patchy morphology from the homogeneous central necrosis of IP-10-responsive tumors. The difference in the extent of tumor necrosis in IP-10-treated and control mice was significant (P = 0.002), indicating that IP-10 caused a reduction of viable tumor tissue. As expected, 6 of 8 mice treated with intratumor injections of LCL developed visible tumor necrosis and scarring, and tumor cross-sections showed tissue necrosis involving 43–67% of the tumor mass (Table 1).
Table 1.
Burkitt tumor response to human IP-10 treatment in athymic mice
| Treatment | Tumor size at first injection,* cm2 | No. of responses/total treated† | Tumor size of responders,* cm2 | Tumor size of nonresponders,* cm2 | % tumor tissue necrosis of responders‡ | % tumor tissue necrosis of all tumors‡ |
|---|---|---|---|---|---|---|
| Experiment 1 | ||||||
| None | — | 0/2 | — | 4.7 | — | 20 |
| LCL | 0.46 | 3/3 | 3.1 | — | 56.5 | 56.5 |
| Human IP-10 50 ng per day | 0.39 | 4/5 | 5.3 | 7.2 | 55.4 | 54.5 |
| Saline | 0.22 | 0/2 | — | 7.9 | — | 22 |
| Experiment 2 | ||||||
| None | — | 0/4 | — | 9 | — | 25 |
| LCL | 0.24 | 3/4 | 4.8 | 7.6 | 54.6 | 45.2 |
| Human IP-10 200 ng per day | 0.26 | 2/4 | 5.5 | 8.5 | 43 | 50 |
| Human IP-10 400 ng per day | 0.23 | 3/5 | 6.0 | 8.6 | 50 | 40.6 |
| Saline | 0.26 | 0/4 | — | 6.31 | — | 23 |
Nude mice bearing a Burkitt tumor induced as described were injected daily into the tumor with human recombinant IP-10. In experiment 1, human IP-10 was injected at the dose of 50 ng for 2 weeks, followed by 100 ng per day for 1 week, and 150 ng per day for 1 week. In experiment 2, one group was treated with an initial dose of 200 ng per day and the other with an initial dose of 400 ng per day. IP-10 doses were doubled when the tumor reached a size ≥2 cm2. The total number of IP-10 treatment days was 30 to 35. Tumor-bearing control animals were injected into the tumor either with LCL or with normal saline containing 1% BSA.
Calculated as the product of two-dimensional caliper measurement (longest perpendicular length and width); expressed as arithmetic mean for the group.
A tumor was considered responsive when it developed a visible area of necrosis (≥4 mm2 in size) that progressively increased in size on subsequent weekly observations.
The % tumor tissue necrosis was estimated by digital analysis of tumor cross-sections. The slides were scanned using a flat-bed scanner (Scanjet II CX, Hewlett–Packard), and necrotic/total tumor area measured.
Histologically, Burkitt tumors that responded to either IP-10 or LCL (20 examined) generally displayed homogeneous central tissue necrosis extending to the epidermis and deep into the tumor mass (Fig. 2 E and I). Within the viable tumor tissue, both distant and proximal to the necrotic tumor tissue, there was vessel injury including intimal thickening with partial to complete occlusion by thrombi (Fig. 2 H and L). Lymphocyte infiltration was unremarkable in both groups, and macrophage infiltrate was limited to LCL-treated tumors (Fig. 2 G and K). Control Burkitt tumors displayed little or no tumor tissue necrosis (Fig. 2A) and had normal capillaries (Fig. 2D).
Figure 2.
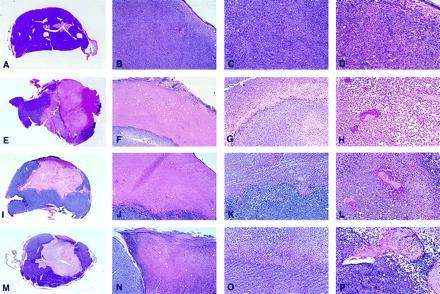
Gross and microscopic morphology of progressive and regressing Burkitt lymphoma tumors. (A–D) Tumor tissue from a mouse injected subcutaneously with CA46 Burkitt cells. (E–H) Tissue from a tumor induced by subcutaneous injection with CA46 Burkitt cells and subsequently injected with LCL (VDS line) weekly for 3 weeks. (I–L) Tissue from a tumor induced by subcutaneous injection with CA46 Burkitt cells and subsequently injected daily with human IP-10. (M–P) Tumor tissue from a mouse injected subcutaneously with murine IP-10-expressing CA46. (A, E, I, and M) Gross morphology of Burkitt tumors showing viable looking tumor tissue (A) and regressing tumors with extensive central necrosis surrounded by viable tumor (E, I, and M) (no magnification). (B, F, J, and N) microscopic morphology of Burkitt tumors extending to the epidermidis; in B viable-looking tumor tissue, and in F, J, and N the interface between necrotic (top) and viable (bottom) tumor tissue are shown. (C, G, K, and O) Higher power magnification of viable tumor tissue (C) and of the interface of necrotic (top) and viable (bottom) tumor tissue (G, K, and O). A prominent macrophage infiltration is noticeable in G. (D, H, L, and P) Higher power magnification of viable tumor tissue with patent capillaries (containing red cells) in D, and capillaries occluded with thrombi at various stages of reorganization (H, L, and P). (B, F, J, and N, ×5; C, G, K, and O, ×10; D, H, L, and P, ×20.)
Tumor sections were stained with EVG reagent specific for elastin fibers. All EVG staining vessels were evaluated for vascular damage consisting of elastin fiber fragmentation and disruption associated with tumor infiltration, fibrinoid necrosis, or fibrin thrombi. Only vessels that stained, at least in part, with EVG were evaluated, thereby excluding nonviable vessels, capillaries, and adventitial vasa vasorum. Sections from three representative diluent-treated tumors displayed evidence of vascular damage in 55% of EVG-staining vessels, as opposed to 75% of vessels in four representative IP-10-responsive tumors (P = 0.023).
We compared expression of murine cytokines/chemokines in representative IP-10-responsive Burkitt tumors and controls by RT-PCR analysis. The PCR products for murine IL-6, tumor necrosis factor α, Mig, and IP-10 were detected at similar levels (<3-fold difference) in two IP-10 responsive and in four control diluent-treated Burkitt tumors (not shown).
Tumorigenicity of IP-10 Expressing Burkitt Lymphoma Cells.
The plasmids pCEP4-murine-IP-10 and pCEP4 were transfected into CA46 Burkitt cells, and clones expressing either murine IP-10 (CA46/IP-10) or vector alone (CA46/pCEP4) were injected. Murine IP-10 expression was achieved in CA46 cells, but at lower levels than in CHO cells (Fig. 3A). Burkitt tumors derived from CA46/IP-10 cells appeared later and grew significantly slower (P < 0.001) than those from CA46/pCEP4 cells (Fig. 3B). Histological examination of some of these tumors revealed that tumor tissues derived from CA46/IP-10 cells, but not from CA46/pCEP4 cells (not shown), had microscopic evidence of central tumor tissue necrosis, capillary thrombosis, intimal thickening and degeneration (Fig. 2 M–P). In a confirmatory experiment, tumors derived from 12 different subcutaneous injections of CA46-IP-10 clones were, on the average, 38% (range 26–56%) necrotic, but four tumors derived from CA46-pCEP4 clone 1 were only 15% (range 12–18%) necrotic. Histologically, these regressing Burkitt tumors were indistinguishable from those locally treated with human IP-10, as evidenced by extensive central necrosis and capillary vessel injury (Fig. 2).
Figure 3.
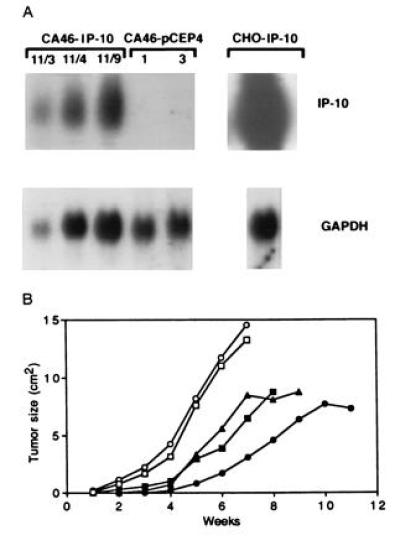
Effect of murine IP-10 expression on Burkitt lymphoma tumorigenesis. (A) Northern blot analysis of murine IP-10 mRNA expression in CA46-IP-10 clones 11/3, 11/4, and 11/9, vector control transfected and CA46 pCEP4 clones 1 and 3, and murine IP-10 expressing CHO cells. The RNA (15 mg) was run on a 1.2% agarose gel, transferred to nitrocellulose membrane, and hybridized for murine IP-10 and murine GAPDH. (B) Tumor growth in BALB/c nu/nu mice inoculated subcutaneously with murine IP-10-expressing and control CA46 Burkitt cells. Murine IP-10 expressing CA46 clones 11/3 (▪), 11/4 (▴), and 11/9 (•) and control CA46 clones 1 (□) and 3 (○) (107 cells in 0.2 ml culture medium), were each inoculated subcutaneously in irradiated (400 rad) BALB/c nu/nu mice. Tumor size was measured weekly. Each data point represents the mean tumor size of four or five mice. Because of large tumor size and mouse mobility impairment, mice injected with CA46-pCEP4 clones 1 and 3 were killed at 7 weeks.
DISCUSSION
In this study, we found levels of murine IP-10 expression to be higher in LCL-treated regressing Burkitt tumors than in untreated controls, and we provide evidence for IP-10 exerting a characteristic antitumor effect against Burkitt tumors established subcutaneously in athymic mice. Either injected into the tumor or constitutively expressed by Burkitt cells, IP-10 caused central tumor tissue necrosis in a proportion of the animals. A widespread vascular damage that included intimal thickening, fragmentation and disruption of elastin fibers, and intravascular thrombosis characterized the histology of IP-10 responsive tumors. Both the central tumor tissue necrosis and the characteristic vascular damage observed in IP-10-treated and IP-10-producing tumors were similar in appearance to those exhibited by LCL-treated Burkitt tumors. Previously, we demonstrated that IP-10 is a potent inhibitor of basic fibroblast growth factor-induced angiogenesis in vivo (23, 24) and has no direct effect on growth and survival of Burkitt cells in vitro (unpublished results). We therefore conclude that IP-10 may contribute to LCL-induced Burkitt lymphoma regression in athymic mice.
IP-10, initially described as an immediate early gene induced by IFN-γ, is expressed in activated human mononuclear cells, endothelial cells, fibroblasts, and other cells (14, 15). Biological functions attributed to IP-10 include inhibition of colony formation by human hematopoietic cells (25), chemoattraction of human monocytes, activated T cells and natural killer cells (26, 27), stimulation of T cell adhesion to endothelial cells and natural killer-mediated cytolysis (26, 27), and inhibition of angiogenesis (23, 24, 28, 29). IP-10 was reported to elicit a host-mediated and T cell-dependent antitumor effect against a murine plasmacytoma and mammary adenocarcinoma (30). This antitumor effect was accompanied by a prominent local lymphocytic and neutrophil accumulation (30). However, IP-10 has not been previously found to elicit an antitumor response associated with marked vascular damage, which is characteristically devoid of lymphocyte and macrophage infiltrate. Although we cannot exclude that low numbers of T cells known to be present in adult athymic mice might play some role in tumor necrosis in the present system, we failed to detect increased numbers of peripheral T cells in tumor bearing mice responsive to either LCL treatment (11) or IP-10 (data not shown).
The antitumor effects of IP-10 were clear and morphologically distinctive. IP-10 treatment was not toxic to the animals, but did not cause complete tumor regressions and cures as did treatment with LCL. This might be due to insufficient IP-10 levels being sustained locally. Alternatively, IP-10 may be one of several compounds participating in Burkitt tumor regression elicited by LCL. During LCL-induced tumor regression, we found that Mig, IFN-γ, IL-12p35, and RANTES are also induced.
The antiangiogenic effect of IP-10 is likely not the sole mechanism for its antitumor properties. Consistent with the effects of other inhibitors of angiogenesis (4, 5, 6), IP-10 would be expected to inhibit tumor neovascularization resulting in fewer new vessels being formed and, secondarily, diminished tumor growth. However, injury to established tumor-associated vasculature with intravascular thrombosis and extensive tumor necrosis have not generally characterized the effects of other inhibitors of angiogenesis (4, 5, 6), and, thus, may not be attributable simply to the antiangiogenic effect of IP-10. Hemorrhagic tumor necrosis by tumor necrosis factor α may be contributed by its procoagulant activity leading to intravascular thrombosis (31, 32). Microvascular injury was reported to precede tumor necrosis induced by partially purified preparations of IFN-α/β (33). But IP-10 is not known to induce tumor necrosis factor α, IFNs, or other cytokines/chemokines. Rather, IP-10 has direct effects on endothelial cells, including inhibition of proliferation and differentiation (23, 28, 29). This study extends earlier work in which the importance of tumor-induced angiogenesis in tumor progression was demonstrated (1, 2, 3, 8, 9). It may also lead to the development of more efficacious anticancer therapies, particularly in the context of T-cell immunodeficiency states.
Acknowledgments
We thank Drs. A. S. Rosenberg, L. A. Love, and K. Bhatia for their suggestions; Dr. E. Jaffe for evaluation of histology samples; D. D. Taub for generously providing human IP-10; and H. K. Kleinman and R. Yarchoan for critically reading the manuscript.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: LCL, lymphoblastoid cell lines; IP-10, interferon-inducible protein-10; IL, interleukin; CHO, Chinese hamster ovary; RT-PCR, reverse transcriptase–PCR; IFN, interferon; EVG, elastin van Gieson.
References
- 1.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Polverini P, Cotran R, Gimbrone M, Unanue E. Nature (London) 1977;269:804–806. doi: 10.1038/269804a0. [DOI] [PubMed] [Google Scholar]
- 3.Rak J, St Croix B, Kerbel R. Anticancer Drugs. 1995;6:3–18. doi: 10.1097/00001813-199502000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Maione T, Gray G, Petro J, Hunt A, Donner A, Bauer S, Carson H, Sharpe R. Science. 1990;247:77–79. doi: 10.1126/science.1688470. [DOI] [PubMed] [Google Scholar]
- 5.O’Reilly M, Holmgren L, Sing Y, Chen C, Rosenthal R, Moses M, Lane W, Cao Y, Sige H, Folkman J. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 6.Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folman J. Nature (London) 1990;348:555–557. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- 7.Kim K, Li B, Winer J, Armanini M, Gillett N, Phillips H, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 8.Folkman J. Ann Surg. 1972;175:409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folkman J, Shing Y. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 10.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 11.Tosato G, Sgadari C, Taga K, Jones K, Pike S, Rosenberg A, Sechler J, Magrath I, Love L, Bhatia K. Blood. 1994;83:776–784. [PubMed] [Google Scholar]
- 12.Angiolillo A, Sgadari C, Sheikh N, Reaman G, Tosato G. Leukemia and Lymphoma. 1995;19:267–276. doi: 10.3109/10428199509107897. [DOI] [PubMed] [Google Scholar]
- 13.Wolf J, Draube A, Bohlen H, Jox A, Mucke S, Pawlita M, Moller P, Diehl V. Int J Cancer. 1995;60:527–533. doi: 10.1002/ijc.2910600417. [DOI] [PubMed] [Google Scholar]
- 14.Luster A, Unkeless J, Ravetch J. Nature (London) 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 15.Luster A, Ravetch J. J Exp Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanguri P, Farber J. J Biol Chem. 1990;265:15049–15057. [PubMed] [Google Scholar]
- 17.Oppenheim J, Zachariae C, Mukaida N, Matsushima K. Annu Rev Immunol. 1991;9:617–648. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]
- 18.Shiramizu B, Barriga F, Neequaye J, Jafri A, Dalla-Favera R, Nieri A, Guttierez M, Levine P, Magrath I. Blood. 1991;77:1516–1526. [PubMed] [Google Scholar]
- 19.Tosato G, Blaese R M, Yarchoan R. J Immunol. 1985;135:959–964. [PubMed] [Google Scholar]
- 20.Liao F, Rabin R, Yannelli J, Koniaris L, Vanguri P, Farber J. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farber J. Proc Natl Acad Sci USA. 1990;87:5238–5242. doi: 10.1073/pnas.87.14.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burd P, Thompson W, Max E, Mills F. J Exp Med. 1995;181:1373–1380. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angiolillo A, Sgadari C, Taub D, Liao F, Farber J, Maheshwari S, Kleinman H, Reaman G, Tosato G. J Exp Med. 1995;182:155–162. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sgadari C, Angiolillo A, Tosato G. Blood. 1996;87:3877–3882. [PubMed] [Google Scholar]
- 25.Sarris A, Broxmeyer H, Wirthmueller U, Karasavvas N, Cooper S, Lu L, Krueger J, Ravetch J. J Exp Med. 1993;178:1127–1132. doi: 10.1084/jem.178.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taub D, Lloyd A, Conlon K, Wang J, Ortaldo J, Harada A, Matsushima K, Kelvin D, Oppenheim J. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taub D, Sayers T, Carter C, Ortaldo J. J Immunol. 1995;155:3877–3888. [PubMed] [Google Scholar]
- 28.Luster A, Greenberg S, Leder P. J Exp Med. 1995;182:219–231. doi: 10.1084/jem.182.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strieter R, Kunkel S, Arenberg D, Burdick M, Polverini P. Biochem Biophys Res Commun. 1995;210:51–57. doi: 10.1006/bbrc.1995.1626. [DOI] [PubMed] [Google Scholar]
- 30.Luster A, Leder P. J Exp Med. 1993;178:1057–1065. doi: 10.1084/jem.178.3.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nawroth P, Stern D. J Exp Med. 1986;163:740–745. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nawroth P, Bank I, Handley D, Cassimeris J, Chess L, Stern D. J Exp Med. 1986;163:1363–1375. doi: 10.1084/jem.163.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dvorak H, Gresser I. J Natl Cancer Inst. 1989;81:497–502. doi: 10.1093/jnci/81.7.497. [DOI] [PubMed] [Google Scholar]