

Abstract
Until recently, the degradation of aberrant and unassembled proteins retained in the endoplasmic reticulum (ER) was thought to involve unidentified ER-localized proteases. We now show that the ER-associated degradation (ERAD) of two mutant proteins that accumulate in the ER lumen is inhibited in a proteasome-defective yeast strain and when cytosol from this mutant is used in an in vitro assay. In addition, ERAD is limited in vitro in the presence of the proteasome inhibitors, 3,4-dichloroisocoumarin and lactacystin. Furthermore, we find that an ERAD substrate is exported from ER-derived microsomes, and the accumulation of exported substrate is 2-fold greater when proteasome mutant cytosol is used in place of wild-type cytosol. We conclude that lumenal ERAD substrates are exported from the yeast ER to the cytoplasm for degradation by the proteasome complex.
Quality control of newly synthesized proteins in the endoplasmic reticulum (ER) ensures that only correctly folded, processed, and completely assembled proteins exit this compartment for further transport through the secretory pathway. Most proteins that fail to reach this transport competent state are degraded (1, 2, 3, 4, 5, 6, 7). The ER-associated protein degradation pathway (ERAD) is highly selective for specific unassembled and/or aberrant proteins, while the majority of ER resident and secreted proteins are quite stable. Evidence that the ER chaperone calnexin has a role in ERAD indicates that molecular chaperones might be required for this remarkable substrate selectivity (8).
Although previous studies suggested that ERAD involves unidentified proteases localized in the ER, more recent evidence indicates that ERAD may require cytosolic proteases. Studies of ERAD in vitro suggest that degradation of aberrant ER-lumenal proteins occurs in the cytoplasm (8), and there is also evidence that the degradation of ER-retained forms of the cystic fibrosis transmembrane conductance regulator is ubiquitin- and proteasome-dependent (9, 10). In addition, cytomegalovirus-induced down-regulation of major histocompatibility complex class I molecules involves the rapid transport of the unassembled major histocompatibility complex I heavy chains from the ER to the cytoplasm for degradation by the proteasome complex (11). In this report we show that the cytosolic proteasome complex is the proteolytic component for the ER-associated degradation of two ER-lumenal proteins in yeast. Furthermore, we provide evidence that indicates ERAD substrates are exported to the cytosol for degradation by the proteasome.
MATERIALS AND METHODS
Materials and Strains.
Strains used were: RSY607 (Matα, ura3-52, leu2-3-112, pep4::URA3), provided by R. Schekman, University of California, Berkeley; AB122 (Mata, prc1-407, prb1-1122, pep4-3, leu2, ura3-52), provided by A. Brake, University of California, San Francisco; pre 1-1 pre 2-2 proteasome mutant and isogenic wild-type strain, provided by D. H. Wolf (12, 13); Δubc6 ubiquitin-conjugating enzyme mutant and isogenic wild-type strains, provided by S. Jentsch (14); and ubc4-Δ1 ubc5-Δ1 and ubc6-Δ1 ubc7-Δ1 and isogenic wild-type strains, provided by M. Hochstrasser (14). The PRE1 and PRE2 wild-type gene expression plasmids, provided by D. H. Wolf (12, 13), were transformed into the pre 1-1 pre 2-2 strain using standard procedures. The pGem2α36-3Q expression plasmid, which contains a yeast pre-pro-α factor (ppαF) gene with all three glycosylation sites altered to specify glutamine instead of asparagine (ΔGppαF), was provided by D. Meyer, University of California, Los Angeles. Ubiquitin-aldehyde was provided by R. Cohen, University of Iowa, Iowa City.
Pulse-Chase Radiolabeling of A1PiZ.
Strains expressing A1PiZ from a pYES2.0 (Invitrogen) plasmid were incubated overnight in selective medium without sulfate and methionine and were then pulse-labeled with 15–20 μCi (1 Ci = 37 GBq) of [35S]methionine per ml for 20 min and chased for 0, 60, 90, or 120 min as previously described (15). Cell lysates were immunoprecipitated with A1Pi-specific antibody and immunoreactive proteins were treated with or without endoglycosidase H (Endo H; Boehringer Mannheim) and resolved by 10% SDS/PAGE. Results were quantified by Bio-Rad phosphor analyst image analysis software as described (8).
In Vitro ERAD Assay.
The conditions of the in vitro assay were as described (8). Briefly, radiolabeled ppαF with all three glycosylation sites removed (ΔGppαF) was translocated into yeast microsomes, and the posttranslocation incubation was performed by adding 6 μg of cytosolic protein per μl of reaction using either cytosol prepared from a wild-type strain, cytosol prepared from a mutant strain, or buffer (8). Products of the posttranslocation incubations were trichloroacetic acid-precipitated and resolved by 18% SDS/PAGE containing 4 M urea. 3,4-Dichloroisocoumarin (DCI; Sigma) and lactacystin (E. J. Corey, Harvard University) were dissolved in dimethyl sulfoxide and added to the chase incubations to a final concentration of 10 nM and 10 μM, respectively. Ubiquitin-aldehyde, where indicated, was added to the chase reaction to a final concentration of 1 μM. Results were quantified by Bio-Rad phosphor analyst image analysis software.
RESULTS AND DISCUSSION
ER-Associated Degradation Is Inhibited in a Proteasome Mutant Strain.
A role for the multicatalytic proteasome complex in ER-associated degradation was first suggested by the observation that a greater number of proteasomes were associated with the ER membrane in secretory cells (16). Results from recent studies using protease inhibitors imply that certain ER membrane proteins are degraded by the proteasome (9, 10, 11). Therefore, to examine the role of the proteasome in the ER-associated degradation of lumenal substrates, a mutant form of human α-1-proteinase inhibitor, A1PiZ, a known substrate of ERAD in mammalian cells and in yeast (15, 17), was monitored in the yeast proteasome mutant, pre 1-1 pre 2-2. This strain is deficient in the “chymotrypsin-like” activity of the proteasome complex and accumulates aberrant proteins (18). Results from pulse-chase protein radiolabeling experiments showed that 94 ± 0.2% of A1PiZ was degraded over a 120-min chase in an isogenic wild-type strain, while only 39 ± 6.7% was degraded in the pre1-1 pre 2-2 mutant strain (Fig. 1). The half-life of A1PiZ calculated from these data was 163 min in the mutant, a 3-fold increase over the 56-min half-life in the wild-type strain. These results suggest that the cytosolic proteasome complex plays a role in the ER-associated degradation of A1PiZ.
Figure 1.

The proteasome complex is required for ER-associated degradation of A1PiZ. (a) Phosphorimage of pulse-chase radiolabeling of A1PiZ in the proteasome mutant (pre 1-1 pre 2-2), the isogenic wild-type strain (wild type), the proteasome mutant expressing a wild-type copy of PRE1 (pre 1-1 pre 2-2 + PRE1), and the proteasome mutant expressing a wild-type copy of PRE2 (pre 1-1 pre 2-2 + PRE2). Immunoprecipated products of chase incubations (0, 60, 90, or 120 min) were treated with (+) or without (−) endoglycosidase H (Endo H) to demonstrate removal of the three carbohydrate chains added to A1PiZ in the ER and to convert glycosylated forms of A1PiZ to a single species. Endo H digestion was incomplete in the 0 chase samples. (b) First-order decay curves generated with averaged values from at least three experiments to determine the half-life of A1PiZ. Relative amounts of A1PiZ were determined from the phosphorimages using a Bio-Rad phosphor analyst program.
Reintroduction of PRE1 and PRE2 Genes Restores ERAD.
Due to earlier findings that ER-associated degradation was proteasome-independent (5), it was important to establish that the inhibition of ER-associated degradation of A1PiZ was due to mutations in subunits of the proteasome complex and not to uncharacterized extragenic mutations. Therefore, pulse-chase experiments were performed with transformed pre 1-1 pre 2-2 strains containing centromeric expression plasmids carrying the PRE1 or PRE2 wild-type gene (Fig. 1). The half lives of A1PiZ in the pre 1-1 pre 2-2 + PRE1 and pre 1-1 pre 2-2 + PRE2 strains were 55 and 79 min, respectively. Thus, expression of the corresponding wild-type proteasome genes restored ERAD activity in the pre 1-1 pre 2-2 mutant, indicating that the inhibition of ERAD observed in this strain was due to defective proteasomes. The differences observed between the restoration of proteolysis by the PRE 1 and PRE 2 genes might be due to altered levels of gene expression or more likely, because PRE1 and PRE2 encode subunits that may differ in their specific activity. For example, a redundant and latent chymotrypsin-like activity has been identified in isolated mammalian proteasomes (19).
In Vitro ERAD Is Inhibited by Proteasome Mutant Cytosol.
To verify that the inhibition of ERAD observed in the pre 1-1 pre 2-2 mutant was a direct effect and not the result of an undefined cellular response to defective proteasome activity, ERAD was examined in vitro. We recently assembled ERAD in vitro and showed that cytosolic protein factors were required for degradation of unglycosylated pro-α factor (8). Therefore, the ability of the proteasome mutant cytosol to support ERAD could be measured using this assay. A radiolabeled glycosylation mutant of yeast ppαF (ΔGppαF) was posttranslationally translocated into wild-type microsomes and the stability of pro-α factor (pαF) was monitored in a posttranslocation chase incubation in the presence of wild-type or pre 1-1 pre 2-2 cytosol. After a 40-min incubation with wild-type cytosol, 70 ± 2.9% of pαF was degraded, while only 36 ± 3.9% was degraded in mutant cytosol. In both cases, the degradation was cytosol and ATP-dependent (Fig. 2a). Furthermore, the half-life of pαF in pre 1-1 pre 2-2 cytosol was 50 min, a 3-fold increase over the 19-min half-life observed with wild-type cytosol (Fig. 2b). This increase is identical to the difference in A1PiZ half-life seen in the in vivo experiments (Fig. 1) and supports our findings that the proteasome plays a direct role in ERAD.
Figure 2.
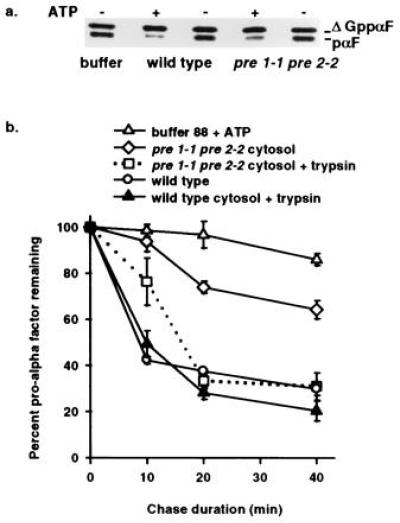
In vitro assay demonstrates ATP and proteasome-dependent ERAD and reveals export of pαF from ER-derived microsomes. (a) Phosphorimage of a 30-min posttranslocation chase incubation performed in the presence (+) or absence (−) of ATP with either proteasome mutant (pre 1-1 pre 2-2) or isogenic wild-type (wild type) cytosol. Radiolabeled ΔGppαF was translocated into wild-type microsomes and the signal sequence cleaved to generate unglycosylated pαF, a substrate for ERAD in vivo (20) and in vitro (8). (b) First-order decay curves generated with averaged values from at least three independent experiments indicate that pαF is stabilized in the presence of pre 1-1 pre 2-2 proteasome mutant cytosol. Posttranslocation chase incubations (0, 10, 20, and 40 min) in the presence of either wild-type cytosol (wild type), mutant cytosol (pre 1-1 pre 2-2 cytosol), or buffer (buffer 88 + ATP) were treated with or without trypsin before trichloroacetic acid precipitation, and samples were resolved on 18% SDS/urea-PAGE and analyzed as in Fig. 1. Degradation of pαF by trypsin treatment (0.25 g/ml) indicates that the protein substrate has been exported from microsomes, while protease protection indicates membrane-occluded pαF. All reactions were performed in the presence of ATP.
DCI and Lactacystin Inhibit in Vitro ERAD.
Because intact yeast cells are relatively impermeable, it had been infeasible to study the effect of proteasome inhibitors on ERAD until the advent of an in vitro system (8). Thus, we used this assay to examine the effects of the proteasome inhibitors DCI and lactacystin on ERAD. DCI, a general serine protease inhibitor, interacts directly with the proteasome (21) to inhibit ATP-dependent proteolysis (22), and lactacystin has been demonstrated to be a proteasome-specific inhibitor (23). When in vitro ERAD was examined in the presence of these inhibitors, pαF was stabilized (Fig. 3). The half-life of pαF in the presence of DCI and lactacystin was 55 and 56 min, respectively, a >2-fold increase over the 24-min half-life observed in the absence of inhibitor.
Figure 3.
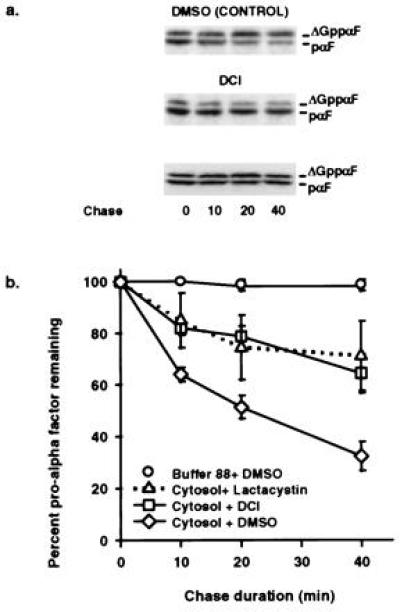
DCI and lactacystin inhibit ER-associated degradation of pαF. (a) Phosphorimage of in vitro ERAD chase reactions (0, 10, 20, and 40 min) in the presence of 10 μM lactacystin, 10nM DCI, or dimethyl sulfoxide. All reactions were performed in the presence of ATP. (b) First-order decay curves generated with averaged values from at least three experiments indicate that pαF is stabilized in the presence of proteasome inhibitors. Relative amounts of pαF were determined from the phosphorimages as in Fig. 1.
A1PiZ and pαF (these studies), Matα2 repressor (14), and cystic fibrosis transmembrane conductance regulator protein (CFTR; refs. 9 and 10) are incompletely degraded in the presence of either proteasome mutants or inhibitors. Because the proteolytic activities of the proteasome complex are redundant (16, 19, 21), this residual activity may be due to other catalytic subunits of the proteasome compensating for those mutated or inhibited.
Together, the in vivo findings that a proteasome mutant yeast strain is deficient in the degradation of a mammalian lumenal ERAD substrate, A1PiZ (Fig. 1), along with the in vitro results that proteasome mutant cytosol and inhibitors of the proteasome limit the degradation of a yeast lumenal ERAD substrate, pαF (Figs. 2 and 3), directly demonstrate that ERAD in yeast is mediated by the proteolytic activity of the proteasome. Although, it is possible that the mammalian proteasome also has a role in ERAD, the present evidence is circumstantial (9, 10, 11, 24). Thus, one cannot rule out the possibility that some ERAD substrates are removed by proteases that reside in the ER.
Export of an ERAD substrate from ER-Derived Microsomes.
How could ER proteins be degraded by the cytosolic proteasome complex? While integral ER membrane proteins could present external soluble domains to a cytosolic protease, the degradation of soluble proteins within the ER lumen requires that they first translocate back out of the ER. In retrospect, placing proteolytic machinery in a compartment distinct from that committed to protein biogenesis makes sense; nascent proteins remain isolated from the proteasome while aberrant polypeptides are banished to the cytosol and destroyed. Thus, although degradation of soluble ER proteins by the cytosolic proteasome presents a topological problem, several studies indicate ER to cytoplasm transport. First, glycopeptide export from the yeast ER can occur by a cytosol- and ATP-dependent mechanism (25). Second, free polymannose oligosaccharides synthesized in the ER are transported directly into the cytoplasm (26). Third, the ER appears to be the intracellular site for translocation of soluble 37-kDa cytotoxic proteins into the cytoplasm (27, 28, 29, 30). It was hypothesized that either the translocation machinery or components of the peptide antigen transport process are involved in the translocation of these cytotoxic proteins from the ER to the cytoplasm (31, 32). Lastly, it has been shown that the US11 gene product of human cytomegalovirus facilitates the retrograde transport of major histocompatibility complex class I heavy chains to the cytoplasm for degradation by the proteasome (11). This finding not only reveals a mechanism by which viruses can evade the immune system but has implications for the biosynthesis and degradation of ER membrane proteins, and thus underlines the significance of an ER to cytoplasm transport pathway.
In accordance with these observations, we previously demonstrated ATP-dependent export of pαF from ER-derived microsomes. The appearance of pαF in the supernatant was specific for the unglycosylated form, suggesting a selective transport of ERAD substrates (8). The in vitro experiments presented in Fig. 2b provide additional evidence for ER to cytosol protein transport. Export of pαF from microsomes can be determined either by analyzing the cytosol by SDS/PAGE or by protease protection analysis, since pαF occluded in the microsomes is shielded from exogenous protease (8). Therefore, to study pαF export in the presence of pre 1-1 pre 2-2 and wild-type cytosol, the products of the posttranslocation chase incubations were treated with or without trypsin before trichloroacetic acid precipitation (Fig. 2b and Table 1). After 10 min of chase incubation, pαF was found in the pre 1-1 pre 2-2 cytosol, while pαF was undetected in wild-type cytosol until 20 min (Table 1). Furthermore, at 20 min the amount of exported pαF detected in the pre 1-1 pre 2-2 cytosol was approximately twice that found in wild-type cytosol. These results are consistent with the evidence that defective proteasome activity causes an inhibition of ERAD. Interestingly, the inhibition of proteasome activity by DCI and lactacystin also revealed an accumulation of exported pαF (data not shown). Therefore, as expected, when ER export was examined under conditions that reduce proteasome activity, either by mutation or by a protease inhibitor, an increased cytosolic pool of ERAD substrate was evident.
Table 1.
Trypsin sensitivity reveals pαF export in vitro
| Cytosol | Chase, min | %
pαF
|
|||
|---|---|---|---|---|---|
| Untreated | Trypsin-treated | U−T | Sensitive* | ||
| pre1-1 pre2-1 | 0 | 100 | 100 | 0 | 0 |
| 10 | 93.7 ± 4.4 | 80.7 ± 6.5 | 13 | 14 | |
| 20 | 74.0 ± 2.6 | 33.3 ± 3.2 | 41 | 55 | |
| 40 | 64.2 ± 3.9 | 31.0 ± 6.0 | 33 | 52 | |
| Wild-type | 0 | 100 | 100 | 0 | 0 |
| 10 | 42.3 ± 1.5 | 49.4 ± 5.7 | 0† | 0† | |
| 20 | 37.5 ± 1.0 | 28.1 ± 2.7 | 9 | 25 | |
| 40 | 29.9 ± 2.9 | 20.3 ± 4.3 | 10 | 33 | |
Percentages were determined from results shown in Fig. 2 for untreated and trypsin-treated samples, and calculated values for the difference between trypsin-treated (T) and untreated (U) samples demonstrate the amount of pαF in the supernatant of the in vitro ERAD reactions. U − T, difference between U and T.
Amount of trypsin-sensitive pαF; value calculated as [(U−T)/U] × 100.
Within the margin of error, these values are zero.
ERAD Is Not Inhibited by Mutations in Specific Ubiquitin Conjugating Enzymes.
Many short-lived intracellular proteins are conjugated to the polypeptide ubiquitin as an obligatory step in their selective targeting to the proteasome for degradation (reviewed in refs. 33 and 34). Indeed, most known substrates of the proteasome are ubiquitinated, raising the question of whether ubiquitination of substrate is a prerequisite for proteasome-dependent ERAD. We first examined whether the ubiquitin-conjugating activity of Ubc6, an integral ER membrane protein required for the degradation of an unstable mutant form of the yeast translocation channel protein Sec61p (35, 36), was necessary for the ER-associated degradation of A1PiZ. When pulse-chase experiments were performed in a ubc6 null mutant strain, the half-life of A1PiZ in the wild-type strain was 40 ± 4.0 min, compared with 28 ± 2.0 min in the Δubc6 strain. Although the reason for the increased rate of degradation in the Δubc6 strain is not clear, this result indicates that the ER-associated degradation of A1PiZ was independent of the ubiquitin-conjugating activity of Ubc6.
Because ubiquitin conjugation may occur through the activity of other ubiquitin-conjugating enzymes, we tested the ability of cytosol and microsomes prepared from ubc6 ubc7 and ubc4 ubc5 double deleted strains to support ERAD in vitro. UBC4, 5, 6, and 7, encode E2 ubiquitin-conjugating enzymes known to be involved in the degradation of aberrant proteins (reviewed in ref. 33). In addition, Ubc6/Ubc7, acting as an enzyme complex, and Ubc4/Ubc5 define two distinct ubiquitination pathways by which Matα2 transcription factor is targeted for degradation by the proteasome (14).
However, no effect on the degradation of pαF was observed when any combinations of ubc6 ubc7 mutant membranes or cytosol were used in the in vitro ERAD assay (Fig. 4). Similar results were obtained with the ubc4 ubc5 mutant (data not shown). Furthermore, the addition of ubiquitin-aldehyde, an inhibitor of the polyubiquitin disassembling isopeptidase known to inhibit ubiquitin-mediated proteasome-dependent proteolysis (37, 38), did not have a stabilizing effect on pαF in the in vitro assay (Fig. 4).
Figure 4.

Mutations in specific ubiquitin-conjugating enzymes do not inhibit ERAD in vitro. Microsomes and cytosol from either the ubc6 ubc7 (ubc) or an isogenic wild-type strain (WT) were used in an in vitro ERAD assay as previously described (8). Addition of ubiquitin-aldehyde (UA) to wild-type cytosol to a final concentration of 1 μM, did not inhibit degradation of pαF in vitro. Results represent the means of at least two independent determinations.
Together, these results suggest that proteolysis of pαF by the proteasome complex may take place by a ubiquitin-independent pathway, as observed for ornithine decarboxylase (39, 40) and casein (41). It is also known that full-length proteins can be fully degraded to peptides in the absence of ubiquitin by the 20S proteasome if the proteins are first denatured (ref. 42; reviewed in ref. 33). However, it is possible that ubiquitination may occur through the activity of other known (43) or an as yet undiscovered ubiquitin-conjugating enzyme(s).
In conclusion, our results from both in vivo and in vitro studies of lumenal ERAD substrates from two different organisms directly demonstrate that the degradation of ERAD substrates is dependent on the proteasome complex in yeast. These results indicate that ER-associated protein degradation is not a novel intracellular degradation process, but uses the proteolytic component of a cytosolic protein degradation pathway. In addition, a unique aspect of ERAD uncovered by our studies is that ER-lumenal protein substrates are exported to the cytoplasm for degradation by the proteasome complex. This discovery has further significance in that it reveals a cellular function for a retrograde protein transporter in the ER, a mechanism implicated in the delivery of cytotoxic proteins from the ER to the cytoplasm (27, 28, 29, 30), and a transport pathway exploited by the human cytomegalovirus to mask its presence in infected cells (11). The nature of this novel protein transport mechanism will be the topic of further investigation.
Acknowledgments
We thank professor D. H. Wolf for the proteasome mutant strain and the PRE1 and PRE2 gene expression plasmids, S. Jentsch for the Δubc6 mutant strain, M. Hochstrasser for the Δubc4 ubc5 and Δubc6 ubc7 strains, R. Cohen for the ubiquitin aldehyde, and J. Sambrook, M.-J. Gething, L. Weber, K. Osteryoung, and J. Werner for critical review of the manuscript. These studies were supported by Cystic Fibrosis Foundation (G789) and National Science Foundation (MCB-951042) awards to A.A.M., National Science Foundation (MCB-9506002) and American Cancer Society (JFRA-602) awards to J.L.B., and a University of Nevada Fellowship to E.D.W.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; DCI, 3,4-dichloroisocoumarin.
References
- 1.Chen C, Bonifacino J S, Yaun L C, Klausner R D. J Cell Biol. 1988;107:2149–2161. doi: 10.1083/jcb.107.6.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amara J F, Lederkremer G, Lodish H F. J Cell Biol. 1989;109:3315–3324. doi: 10.1083/jcb.109.6.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hurtley S M, Helenius A. Annu Rev Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino J S, Lippincott-Schwartz J. Curr Opin Cell Biol. 1991;3:592–600. doi: 10.1016/0955-0674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- 5.Finger A, Knop M, Wolf D H. Eur J Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- 6.Yuk M H, Lodish H F. J Cell Biol. 1993;126:1735–1749. doi: 10.1083/jcb.123.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonifacino J S, Klausner R D. Cellular Proteolytic Systems. New York: Wiley; 1994. pp. 137–160. [Google Scholar]
- 8.McCracken A A, Brodsky J L. J Cell Biol. 1996;132:291–297. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen T J, Loo M A, Pind S, Williams D B, Goldberg A L, Riordan J R. Cell. 1995;38:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 10.Ward C L, Omura S, Kopito R R. Cell. 1995;38:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 11.Wiertz E J H J, Jones T R, Sun L, Bogyo M, Geuze H J, Ploegh H L. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 12.Heinemyer W, Kleinschmidt J A, Saidowsky J, Escher C, Wolf D H. EMBO J. 1991;10:555–562. doi: 10.1002/j.1460-2075.1991.tb07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinemyer W, Gruhler A, Mohrle V, Mahe Y, Wolf D H. J Biol Chem. 1993;268:5115–5120. [PubMed] [Google Scholar]
- 14.Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Cell. 1993;74:357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- 15.McCracken A A, Kruse K B. Mol Biol Cell. 1993;4:729–736. doi: 10.1091/mbc.4.7.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivett J A. J Biochem (Tokyo) 1993;291:1–10. [Google Scholar]
- 17.Le A, Ferrell G A, Dishon D S, Le Q Q, Siefers R N. J Biol Chem. 1992;267:1072–1080. [PubMed] [Google Scholar]
- 18.Seufert W, Jentsch S. EMBO J. 1992;11:3077–3080. doi: 10.1002/j.1460-2075.1992.tb05379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orlowski M, Cardozo C, Hidalgo M, Michaud C. Biochemistry. 1991;30:5999–6005. doi: 10.1021/bi00238a025. [DOI] [PubMed] [Google Scholar]
- 20.Caplan S, Green R, Rocco J, Kurjan J. J Bacteriol. 1991;173:627–635. doi: 10.1128/jb.173.2.627-635.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orlowski M, Cardozo C, Michaud C. Biochemistry. 1993;23:1563–1572. doi: 10.1021/bi00057a022. [DOI] [PubMed] [Google Scholar]
- 22.Waxman L, Fagan J M, Goldberg A L. J Biol Chem. 1987;262:2451–2457. [PubMed] [Google Scholar]
- 23.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;286:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 24.McGee T P, Cheng H H, Humagai H, Omura S, Simoni R D. J Biol Chem. 1996;271:25630–25638. doi: 10.1074/jbc.271.41.25630. [DOI] [PubMed] [Google Scholar]
- 25.Römisch K, Schekman R. Proc Natl Acad Sci USA. 1992;89:7227–7231. doi: 10.1073/pnas.89.15.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore S H E, Bauvy C, Codogno P. EMBO J. 1995;14:6034–6042. doi: 10.1002/j.1460-2075.1995.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogata M, Chaudhary V K, Pastan I, Fitzgerald D J. J Biol Chem. 1990;265:20678–20685. [PubMed] [Google Scholar]
- 28.Chaudhary V K, Jinno Y, Fitzgerald D J, Pastan I. Proc Natl Acad Sci USA. 1990;87:308–312. doi: 10.1073/pnas.87.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandvig K, Garred O, Prydz K, Kozlov J V, Hansen S H, van Deurs B. Nature (London) 1992;358:510–512. doi: 10.1038/358510a0. [DOI] [PubMed] [Google Scholar]
- 30.Simpson S C, Dascher C, Roberts L M, Lord J M, Balch W E. J Biol Chem. 1995;270:20078–20083. doi: 10.1074/jbc.270.34.20078. [DOI] [PubMed] [Google Scholar]
- 31.Pelham H R B, Roberts M L, Lord J M. Trends Cell Biol. 1992;2:183–185. doi: 10.1016/0962-8924(92)90230-k. [DOI] [PubMed] [Google Scholar]
- 32.Wales R, Roberts L M, Lord J M. J Biol Chem. 1993;268:23986–23990. [PubMed] [Google Scholar]
- 33.Hochstrasser M. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 34.Hilt W, Wolf D H. Trends Biochem Sci. 1996;21:96–102. [PubMed] [Google Scholar]
- 35.Sommer T, Jentsch S. Nature (London) 1993;365:176–179. doi: 10.1038/365176a0. [DOI] [PubMed] [Google Scholar]
- 36.Brodsky J L, Schekman R. The Biology of Heat Shock Proteins and Molecular Chaperones. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 85–105. [Google Scholar]
- 37.Hershko A, Rose I A. Proc Natl Acad Sci USA. 1987;84:1829–1833. doi: 10.1073/pnas.84.7.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunten R L, Cohen R E. J Biol Chem. 1989;264:16739–16747. [PubMed] [Google Scholar]
- 39.Elias S, Bercovich B, Kahana C, Coffino P, Fischer M, Hilt W, Wolf D H, Ciechanover A. Eur J Biochem. 1995;229:276–285. [PubMed] [Google Scholar]
- 40.Murakami Y, Matsufuji S, Kameji T, Iggrashi K, Tamura T, Tanaka K, Ichihara A. Nature (London) 1992;360:597–599. doi: 10.1038/360597a0. [DOI] [PubMed] [Google Scholar]
- 41.Driscoll J, Goldberg A L. J Biol Chem. 1990;265:4789–4792. [PubMed] [Google Scholar]
- 42.Dick L R, Aldrich C, Jameson S D, Moomaw C R, Pramanik B C, Doyle C K, DeMartino G N, Bevan J J, Formen J M, Slaughter C A. J Immunol. 1994;152:3884–3394. [PMC free article] [PubMed] [Google Scholar]
- 43.Jentsch S. Annu Rev Genet. 1992;26:179–207. doi: 10.1146/annurev.ge.26.120192.001143. [DOI] [PubMed] [Google Scholar]