

Abstract
A previously uncharacterized 22-kDa Ca2+-binding protein that also binds guanosine nucleotides was characterized, cloned, and analyzed by electrophysiological techniques. The cloned protein, calexcitin, contains two EF-hands and also has homology with GTP-binding proteins in the ADP ribosylation factor family. In addition to binding two molecules of Ca2+, calexcitin bound GTP and possessed GTPase activity. Calexcitin is also a high affinity substrate for protein kinase C. Application of calexcitin to the inner surface of inside-out patches of human fibroblast membranes, in the presence of Ca2+ and the absence of endogenous Ca2+/calmodulin kinase type II or protein kinase C activity, reduced the mean open time and mean open probability of 115 ± 6 pS K+ channels. Calexcitin thus appears to directly regulate K+ channels. When microinjected into molluscan neurons or rabbit cerebellar Purkinje cell dendrites, calexcitin was highly effective in enhancing membrane excitability. Because calexcitin translocates to the cell membrane after phosphorylation, calexcitin could serve as a Ca2+-activated signaling molecule that increases cellular excitability, which would in turn increase Ca2+ influx through the membrane. This is also the first known instance of a GTP-binding protein that binds Ca2+.
In neuronal cells, ion channel conductance is regulated by ligand binding, direct interaction with G proteins, or phosphorylation (1). K+ and Ca2+ channels, for example, can be phosphorylated by Ca2+/calmodulin-dependent kinase (2, 3) and/or protein kinase C (PKC) (4, 5, 6, 7, 8); however, other elements of Ca2+ signaling cascades might also regulate ion channels directly. Such a protein was suggested by a previous study in which a low molecular weight protein, designated cp20, reduced two voltage-dependent K+ currents, iA and iCa-K+, in identified molluscan neurons (9, 10, 11). These same currents were reduced in the same neurons in molluscs (Hermissenda crassicornis) exposed to a Pavlovian conditioning paradigm (9, 12). To investigate this protein, we purified calexcitin (CE) from squid optic lobe and used microsequencing and PCR techniques to obtain a DNA probe which was used to screen a cDNA library. The cloned protein had separate regions that are homologous to Ca2+-binding proteins and to GTP-binding proteins. Functionally, this protein binds both Ca2+ and GTP, is phosphorylated by PKC, and regulates K+ channels in human fibroblasts and membrane excitability in mammalian and molluscan neurons. CE, therefore, offers a molecular link between PKC, intraneuronal Ca2+, and persistent changes of membrane excitability that correlate with associative memory storage.
METHODS
Purification of CE.
Squid optic lobes were homogenized in a high speed homogenizer with 1 ml of buffer (10 mm Tris·HCl, pH 7.4/20 μg/ml leupeptin/50 μg/ml pepstatin/50 mM NaF/1 mM EDTA/1 mM EGTA) containing 1 M DTT and 0.1 mM phenylmethylsulfonyl fluoride (PMSF), followed by sonication with a 20-W probe sonicator. The homogenate was centrifuged at 100,000 × g for 20 min, and the supernatant was applied to a 10 × 250 mm HPLC column of AX-300 (Synchrom, Lafayette, IN) equilibrated with 10 mM NaF. The column was eluted with a gradient of 0–1 M KAc over 0–20 min, followed by isocratic elution of CE with 1 M KAc. The elution fraction containing CE was determined for each injection by computer-assisted pattern-matching of the A280 elution profiles profiles and correcting for the change in tR caused by the rapid column deterioration that occurred under these conditions. The partially purified CE was applied to an SDS 4–20% acrylamide gradient acrylamide gel, blotted onto a poly(vinylidene difluoride) membrane, and subjected to tryptic digestion and peptide microsequencing. The amount of CE protein was estimated to be 567 pmol by amino acid analysis. Fourteen peptide fragments determined to be pure by mass spectrometry were sequenced, and a total of 118 amino acids were obtained.
Screening.
Degenerate oligonucleotides based on the peptide sequences obtained in this and previous experiments (12) were used to amplify cDNA prepared from squid optic lobe poly(A)+ mRNA using reverse transcription–PCR. The PCR products were cloned into pCR-Script (Stratagene) and used to transform XL1 Blue MRF′ cells. Plasmids containing inserts detectable after EcoRI/BamHI digestion were sequenced. From 247 such clones, one that contained 13 of the 14 peptides was found. A 52-bp oligonucleotide based on the DNA sequence was labeled with T4 kinase and used to screen a cDNA library. A 1.2-kb phage clone found after screening of 400,000 plaques was purified on CsCl, digested with EcoRI, subcloned into pBluescript SK(+), and sequenced. NdeI and XhoI sites were introduced in the 5′ and 3′ untranslated regions (at positions 120 and 846) using PCR site-directed mutagenesis. The 722-bp product was cloned into TA3 vector (Invitrogen). The plasmid was purified, and the insert was excised with NdeI and XhoI and subcloned into Novagen pET-16b expression vector to add a 4-kDa N-terminal oligohistidine domain. This product was used to transform NovaBlue cells and BL21(DE3) cells. CE protein was expressed at 37°C with isopropyl β-d-thiogalactoside in BL21(DE3) cells growing in Luria–Bertani broth plus 100 μg/ml ampicilin, and the resulting 24-kDa fusion protein was purified on a NiSO4-charged His·Bind (Novagen) affinity column and eluted with imidazole according to the manufacturer’s directions. The homogeneous His9-CE was stored at −80° in the presence of 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml pepstatin to prevent rapid (25% in 24 h at 25°) degradation by trace proteases.
Glutathione S-Transferase (GST) Fusion Protein.
CE, with HindIII and SalI restriction sites added by PCR, was inserted in-frame into the GST fusion protein vector PGEX-KG (13). The resulting recombinants were transformed into CAG456, a htpR165 mutant that is defective in proteolysis (14), or into BL21 (DE3) cells. Expression of the GST–CE fusion protein was induced with 0.1 mM isopropyl β-d-thiogalactoside at 32°C for 2–5 h. Cultures (200 ml) were pelleted and resuspended in homogenization buffer (100 mM NaCl/1 mM EDTA/10 mM Tris, pH 7.6/10 μg/ml leupeptin/1 mM PMSF/10 μg/ml aprotinin/1 mM DTT/0.5 mg/ml lysozyme) and held at 4°C for 5 min, after which Nonidet P-40 was added to a final concentration of 10%. The suspension was homogenized and centrifuged, and the supernatant containing the soluble GST fusion protein was retained. The fusion protein was bound to 400 μl of 50% glutathione agarose beads that had been washed three times with 10 ml of wash buffer (100 mM NaCl/1 mM CaCl2/10 mM Tris, pH 7.5/5 mM DTT) by rocking of the mixture at 4°C overnight.
The beads were poured into a 26 × 26 mm column and washed with 15 ml each of water, 4 M NaCl, 0.5 M sodium citrate (pH 3.0), and 1 M DTT. The GST–CE fusion protein was then eluted with 0.1 M glutathione (GSH) (15) and precipitated by addition of 0.1 volume of buffer containing 500 mM Tris·HCl (pH 8.0), 1.5 M NaCl, 25 mM CaCl2, and 1% 2-mercaptoethanol. The GST–CE fusion protein was recovered by washing with 1 ml of H2O. The DNA sequence of the coding region of this clone was identical with that of the pET-16b clone.
Thrombin Cleavage.
Purified GST–CE fusion protein was desalted and concentrated to 100 μl using Centricon-3 ultrafiltration devices which had been pretreated with bovine serum albumin before use. The GST–CE fusion protein was reacted with 1 unit of biotinylated thrombin (Novagen) for 1 h at 30°C in thrombin cleavage buffer (50 mM Tris·HCl, pH 8.0/150 mM NaCl/2.5 mM CaCl2/0.1% 2-mercaptoethanol) in a total volume of 23 μl. The reaction mixture was passed through a small (0.1 ml) column of streptavidin-Sepharose and desalted in Centricon-3 ultrafiltration units that had been pretreated with bovine serum albumin.
Northern Blots.
Total RNA from squid optic lobe was extracted using the guanidium isothiocyanate/phenol/chloroform method of Chomczynski and Sacchi (16). Poly(A)+ RNA was then purified using oligo(dT)-cellulose columns, and its concentration was determined by absorbance at 260 nm. Poly(A)+ RNA (2 μg) was electrophoresed through a 1% agarose-formaldehyde (0.22 M) gel and transferred to a nylon membrane (Hybond-N+, Amersham). The filters were UV-crosslinked and then prehybridized in buffer containing 50% formamide, 6× standard saline citrate (SSC), 5× Denhardt’s solution, 10% dextran sulfate, and sonicated salmon sperm DNA (200 μg/ml) at 42°C for 6 h. Hybridization was performed at 42°C for 12–16 h in fresh buffer containing 1 × 106 cpm/ml 32P-labeled CE probe [full-length cDNA labeled by random priming DNA labeling (Boehringer Mannheim)]. The blot was washed twice in 2× SSC/0.1% SDS at 42°C for 30 min and once in 0.1× SSC/0.1% SDS at 51°C for 30 min, and was then subjected to autoradiography at −70°.
Iodination.
CE was iodinated by incubation with 10 μg 1,3,4,6-tetrachloro-3α,6α-diphenylglycoluril (Pierce) and 0.5 mCi (1 Ci = 37 GBq) of carrier-free Na125I at 25°C for 10 min (17). The labeled CE was isolated by repeated ultrafiltration in a Centricon-3 filter.
Heat Inactivation.
CE was desalted by passing through a G-25 spin-column (STE Select-D, 5 Prime → 3 Prime) and reduced in volume by lyophilization to 10 μl. Potassium acetate (pH adjusted to 7.4 by addition of acetic acid) was added to a 1 M final concentration. Heat-inactivated samples were then boiled for 5 min.
GTPase.
GTP hydrolysis from [γ-32P]GTP was measured in 500 μl of polypropylene centrifuge tubes containing 20 mM Tris·HCl, 1 mM EDTA, 0.8 mM NaCl, and 1 μM GTP, incubated with 0.1 μg of cloned CE in a total volume 10 μl for 2 h at 30°C. The 32Pi released was reacted with silicotungstic and molybdic acid (18), extracted into benzene, and measured by scintillation counting.
GTP Binding.
GTP binding to calexcitin was measured by incubating various concentrations of CE with 1 μM [γ-32P]GTP in 500 μl polypropylene centrifuge tubes containing 20 mM Tris·HCl, 1 mM EDTA, and 0.8 mM NaCl in a total volume 10 μl for 2 h at 30°C. The reaction mixture was filtered through a nitrocellulose filter and washed with stop solution (20 mM Tris·HCl, pH 8.0/100 mM NaCl/25 mM MgCl2), and the 32P was measured by scintillation counting.
Protein Measurement.
Protein was measured colorimetrically by reaction with colloidal gold as described (9) and with bicinchoninic acid reagent (19) (Pierce). To calculate the degree of purity, the purified protein was analyzed by SDS/PAGE followed by blotting onto nitrocellulose. The blots were stained with colloidal gold (20), scanned, and analyzed densitometrically.
PKC Measurement.
α-PKC was obtained from Upstate Biotechnology (Lake Placid, NY). PKC activity was measured as described (21) except that incubation was carried out at 30°C for 30 min and 200 mM NaF was added to inhibit endogenous phosphatases found to be present in the PKC solution.
Antibody.
Peptide EWLTKYMNFMFDVNDTSGDNIIDKHEY, corresponding to positions 72–83, was synthesized (Genosys, The Woodlands, TX), conjugated to keyhole limpet hemocyanin, and used to raise polyclonal antibodies (26a-ptd3) in three rabbits.
Western Blot Analysis.
Samples were analyzed by electrophoresis on a 4–20% acrylamide gradient SDS gel, followed by blotting onto nitrocellulose, stained with antibody, and visualized with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate.
Immunohistochemistry.
Tissues were fixed for 6 h in 10% formaldehyde in PBS and embedded in paraffin. Sections (10 μm thick) were mounted on glass slides coated with poly-l-lysine (Sigma), and the sections were deparaffinized in xylene. Endogenous peroxidase activity was blocked by a 20 min incubation in 3% H2O2/methanol at 23°C. Enzymatic predigestion was performed with 0.05% trypsin (Sigma) in 0.01 M PBS (pH 7.4) for 10 min at 37°C to enhance the staining intensity. Incubation with 1:500 diluted rabbit anti-CE was performed at 4°C overnight followed by incubation with bridging antibody (biotinylated goat anti-rabbit at 1:100) and then with avidin-biotin-peroxidase complex (Vector Laboratories) for 30 min at 23°C for each step. Slides were rinsed twice for 2 min with 0.01 M PBS, pH 7.4. Bound peroxidase was visualized using 0.05% 3,3-diaminobenzidine tetrahydrochloride (Sigma) together with a 3% aqueous solution of H2O2 as chromogenic substrate. Color development was stopped after 10 min. Sections were rinsed in PBS, counterstained with 1% Harris hematoxylin. The negative controls included replacement of the primary antibody with pre-immune serum. No staining was detected in these controls, indicating not only the specificity of the primary antibody but also the absence of endogenous avidin binding activity.
Purkinje Cell Injections.
Six rabbits were anesthetized deeply with sodium pentobarbital (30 mg/kg) and decapitated. A rapid craniotomy that removed the occipital bone and mastoid processes allowed the cerebellum and brain stem to be detached, removed, and chilled in 95% O2/5% CO2-saturated artificial cerebrospinal fluid (ACSF) within approximately 70–90 s. Next, the vermis was isolated and attached with cyanoacrylate to an agar block in the cutting chamber. The tissue was then immersed in chilled ACSF, and 400-μm parasagittal slices were cut with a vibrating slicer (Vibratome 1000). Slices were then incubated in saturated ACSF at room temperature for at least 1 h before being placed in a modified recording chamber in which the ACSF was maintained at 32°C (22). The ACSF contained 124 mM NaCl, 3 mM KCl, 1.2 mM MgSO4, 2.1 mM CaCl2, 1.2 mM Na2PO4, 26 mM NaHCO3, and 10 mM dextrose and was saturated with a mixture of 95% O2 and 5% CO2, which maintained the pH at 7.4.
Intradendritic recordings from Purkinje cell dendrites were obtained by advancing a glass microelectrode (Leitz micromanipulator) through the molecular layer of slices. Recordings were made up until 8 h after animals were killed. Microelectrodes of thick-walled glass (2 mm o.d., 1 mm i.d., FHC Inc.) were fabricated on a Narishige electrode puller (NE-2). The electrode tip was filled with 1–1.5 μl (280 ng/μl) of cloned CE and backfilled with 1 M potassium acetate (pH adjusted to 7.4 with HAc). Control injections were carried out with electrode tips filled with 1–1.5 μl of heat-inactivated CE (boiled for 5 min; n = 4) or 3 M potassium acetate (n = 2). All electrodes had a dc resistance of 60–100 MΩ. A bridge amplifier (Axoprobe-1A, Axon Instruments, Foster City, CA) was used for all intradendritic recording. The recording electrodes were positioned in the molecular layer with the aid of a binocular dissecting microscope (Wild, magnification up to ×50), which permitted visualization of the different cortical layers. Penetration of a Purkinje cell dendrite was followed by a current injection of −1.0 nA for 1–2 min. Only cells that stabilized during this current injection period were used for the present experiment. Membrane potential was determined as the potential during somatic spiking (22, 23). Input resistance measures were based on a 0.5-nA, 700-ms hyperpolarizing current step during somatic spiking. The current necessary to hyperpolarize the dendrite 20 mV below the somatic spike activity level was determined and applied to the membrane to measure the dendritic spike threshold. Measurements for dendritic spike threshold before and after injection were based on the specific 700-ms current step required to reach dendritic spikes. Injections of cloned CE or control injections were carried out for 2 min using 700-ms pulses of −1.0 nA delivered at a frequency of 1 Hz.
Calcium Binding.
45Ca (1 μCi, 1–10 μM) was incubated with 0.8 μM CE for 1 h at 30°C in 10 μl of buffer (50 mM Tris·HCl, pH 7.4/50 mM KCl/5 mM MgCl2) applied to a nitrocellulose filter, and the filter was washed three times with the same buffer. The 45Ca remaining bound to the filter was measured in a scintillation counter.
Scatchard Analysis.
45Ca (1 μCi) at various 45Ca specific activities in Ca-EGTA buffer (10 nM to 10 mM Cafree) was incubated with 0.825 μg of CE for 1 h at 30°C in 1 ml of buffer (50 mM Tris·HCl, pH 7.4/50 mM KCl/5 mM MgCl2/0.01 mM EGTA) and subjected to ultrafiltration for 18 h at 4°C. Sample treatment and calculation of bound and free 45Ca was performed using the method of Rose et al. (24).
45Ca Overlay.
His9-CE (1–5 μg) was analyzed on a 4–20% acrylamide gradient SDS polyacrylamide gel, transferred to nitrocellulose, and probed with 45CaCl2 by incubation with 1.6 μM 45Ca for 1 h at 23°C followed by four 1-min washes with PBS. The bound 45Ca was visualized by autoradiography. Numerous other proteins, including various bacteria lysate fractions, did not bind 45Ca.
Fibroblasts.
Cultured skin fibroblasts from the Coriell Cell Repositories (Camden, NJ) were grown under standardized conditions (25). Line 3652 from a young control was seeded (about five cells per square millimeter) in 35 mm Nunc Petri dishes in DMEM (GIBCO), supplemented with 10% fetal calf serum, and used for patch-clamp experiments 5–7 days after seeding.
Patch-clamp experiments were performed at room temperature (21–23°C), following standard procedures (26). Before recording, culture medium was replaced with the following solution: 150 mM NaCl/5 mM KCl/2 mM CaCl2/1 mM MgCl2/10 mM Hepes (NaOH), pH 7.4. Pipets were made from Blue Tip capillary tubes (1.1–1.2 mm i.d.) using a BB-CH Mecanex (Geneva) puller and then filled with a high-K+ solution: 140 mM KCl/2 mM CaCl2/1 mM MgCl2/10 mM Hepes (NaOH), pH 7.4. Pipette resistances were ≈8 MΩ. Records were obtained using an Axopatch-1D amplifier (filtered −3 dB at 10 kHz), digitized at 5 kHz, stored on tape (Toshiba pulse-code modulation video recorder), and later transferred to a PC using a TL-1 direct memory access interface. Amplifier, interface, and software were obtained from Axon Instruments.
Computer Analysis.
Data acquisition and single channel analysis were done using pclamp (version 6.0). Curve fitting by nonlinear regression (using the Marquardt algorithm) was done using the program nplot. Image quantitation and filtering, and molecular weight estimation were done using the image analysis program tnimage (http://las1.ninds.nih.gov).
RESULTS
CE was originally observed as a conditioning-specific increase in the phosphorylation state of a low molecular weight HPLC peak in extracts from Hermissenda neuronal proteins, and was referred to generically as cp20 (9, 27). Numerous other experiments demonstrated the existence of an identical protein in the optic lobe of squid (Loligo peaeli) (12). To characterize this protein, we purified the protein from squid optic lobe, obtained a partial amino acid sequence, and used reverse transcription–PCR to amplify a portion of the CE cDNA. Extensive screening of a squid cDNA library with an oligonucleotide based on the PCR product produced a clone that matched 13 of the 14 tryptic peptides (i.e., 100 of 118 amino acids) from the original native squid protein.
The CE cDNA sequence (Fig. 1A) showed close homology to several Ca2+-binding proteins, particularly scp1 (28, 29). There were also regions with some homology to GTP-binding proteins of the ADP-ribosylation factor family (31, 32), particularly sar1p (30, 33) (Fig. 1B). The sequence (Fig. 1 A and B) contained two EF-hand consensus domains (34) (positions 23–34 and 119–130), a possible GTP-binding consensus motif (35) (Fig. 1C), a PKC phosphorylation site (36) (positions 61–63), two possible N-myristoylation sites (37) (positions 139–144 and 179–184), and a polyisoprenylation consensus (CAAX) domain at the C-terminal (positions 185–188). CAAX is a domain shared among GTP-binding proteins used for attachment of an isoprenoid tail which facilitates membrane binding (38, 39). The theoretical pI (4.97) agreed with the pI measured by isoelectric focusing (5.3) and with that observed for purified native squid and Hermissenda CE (pI = 5.2) on two-dimensional gel electrophoresis (12). No transmembrane sequences, nuclear translocation sequences, or signal sequences were found.
Figure 1.

(A) Sequence of squid CE cDNA (GenBank accession no. U49390U49390). The coding region is in boldface type. (B) Amino acid sequence of CE compared with Amphioxus SCP I (29), yeast sar1p (30), and arf1 (31). Amino acids found by peptide sequencing of the tryptic digest are underlined. (C) Possible GTP-binding sites in CE, compared with homologous regions in sar1p.
Northern blot analysis of squid optic lobe mRNA (Fig. 2A) showed two bands (about 9.99 and 8.17 kb). Reverse transcription–PCR experiments excluded the presence of alternative splicing on the coding region. The 3′ untranslated region contained long stretches of poly(AT), a common finding in squid mRNAs (40).
Figure 2.
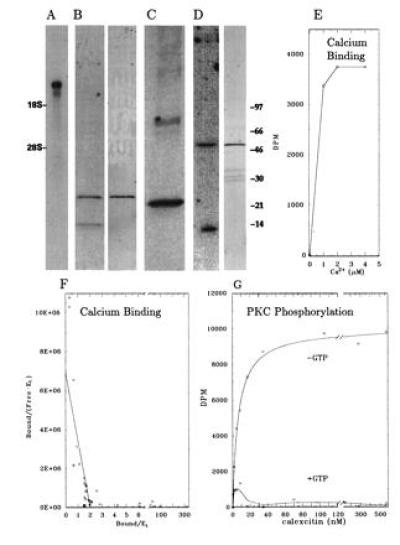
(A) Northern blot of squid CE mRNA. The positions of 18S and 28S rat rRNA are shown. CE mRNAs were approx. 9.99 and 8.17 kb long. (B Left) 45Ca overlay blot of oligohistidine–CE fusion protein (Mr = 24,000). Binding was performed in the presence of Mg2+ and the absence of GTP. Numerous other control proteins did not bind Ca2+ under these conditions. The lower band is a proteolytic degradation product. (B Right) Polyclonal antibody stained Western blot of oligohistidine–CE fusion protein. (C) Polyclonal antibody stained Western blot of squid optic lobe homogenate. (D Left) [32P]GTP overlay blot of GST–CE fusion protein (Mr ≈ 50,000). Binding was performed in the absence of Ca2+. In a separate experiment, no binding to [32P]ATP could be detected. The lower band is a proteolytic degradation product. (D Right) Polyclonal antibody stained Western blot of GST–CE fusion protein. After thrombin treatment, the band was reduced to 22 kDa, comigrating with native CE (C). (E) Ca2+ binding as a function of CE concentration. CE (0.8 μM) was incubated with various concentrations of 45Ca and filtered through a nitrocellulose membrane. 45Ca bound to the membrane was measured by scintillation counting. (F) Scatchard Ca2+-binding plot. Ca2+ binding was measured in the presence of 50 mM Tris·HCl, pH 7.4/50 mM KCl/5 mM MgCl2/0.01 mM EGTA. The dissociation constant was unchanged when measured in the absence of Mg2+. (G) Phosphorylation of cloned squid CE by α-PKC in the absence (upper curve) and presence (lower curve) of GTP. The Km of PKC for CE was estimated at 5.0 nM. Under these conditions, GTP inhibited PKC phosphorylation of histones by less than 30%.
Cloned CE bound 2 Ca2+ ions/molecule (Fig. 2 E and F), with an affinity (Ks ≈ 400 nM) similar to that of other sarcoplasmic calcium-binding protein-like proteins (28, 29, 41, 42). A 45Ca overlay blot (Fig. 2B) also demonstrated that CE was a Ca2+-binding protein. In the absence of Mg2+, CE also bound GTP (Fig. 2D) and hydrolyzed [32P]GTP at a rate of 0.14 pmol·min−1·mg−1. Other nucleotides, such as ATP, did not bind to CE (not shown). CE was an unusually high-affinity substrate for α-PKC with a Km of 5.4 nM (Fig. 2G), similar to the Km of 7.1 nM reported for native CE (12). Addition of GTP at concentrations (1 mM) that only slightly inhibited phosphorylation of other substrates completely inhibited phosphorylation of CE measured in the presence of 5 mM Ca2+ and 10 mM Mg2+ (Fig. 2G).
For measurements of unitary K+ channel conductances in inside-out patches excised from cultured human skin fibroblasts, bath addition of 0.2–2 nM CE reduced the mean channel open time (τopen) and the mean open probability for six of eight patches tested (Fig. 3 A–C) (two of the eight patches showed no changes). The currents had reversal potentials consistent with K+ conductance, and had unitary conductances of 115 ± 6 pS. CE inhibition of K+ channels (which were not dependent on extracellular Ca2+) appeared to depend on the presence of Ca2+. In preliminary experiments, CE had little or no effect on K+ channels when 10−5 M EGTA was substituted for Ca2+ (data not shown). Because endogenous kinases would be inactive in inside-out patches due to the absence of ATP, GTP, and other soluble cofactors, CE in its Ca2+-bound form is most likely acting directly on the K+ channel or its associated components. The strong inhibitory effect of squid CE on human K+ channels at these low concentrations also suggests that a mammalian homologue of CE could play a role in the normal regulation of these channels. No changes in K+ unitary currents were observed for >10 min after addition of 2 nM heat-inactivated CE.
Figure 3.
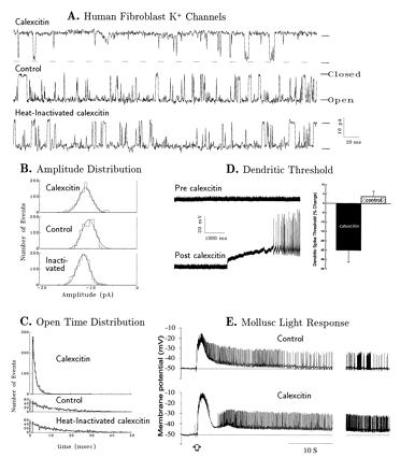
(A) Single-channel current traces of K+ channel activity in an excised inside-out membrane patch of human skin cultured fibroblast recorded in the absence of CE 7 min after excision of the membrane patch (middle trace), at 10 min after addition of heat-inactivated CE (bottom trace), and at 3 min after addition of intact CE (top trace). The channel shown was typical of those recorded at −40 mV. Pipette solution was 140 mM KCl/2 mM CaCl2/1 mM MgCl2/10 mM Hepes (NaOH)/0.001 mM tetrodotoxin, pH 7.4; bathing solution was 150 mM NaCl/5 mM KCl/2 mM CaCl2/1 mM MgCl2/10 mM Hepes (NaOH), pH 7.4. Addition of protein was made by a 20-μl plastic pipette near the tip of the patch micropipette as 10-μl aliquots of CE in 10 mM imidazole·HCl. Petri dish contained 2 ml of bath solution. CE markedly reduced the mean open time (τopen) and probability of openings. (B) Amplitude distributions of unitary K+ current records were fitted with a Gaussian function. Unitary current peak amplitudes were −10.6, −11.0, and −9.9 pA after addition of CE and heat-inactivated CE, and no addition, respectively. There were no significant differences in distribution among the three groups. (C) Open time distributions for the data collected during 30-s periods after excision of the membrane (as mentioned in A) were fitted to a single exponential. Exponential decay constants for open time distribution were 1.4, 12.0, and 12.5 ms after addition of CE and heat-inactivated CE, and no addition, respectively. CE application markedly reduced the open times of the K+ channel. (D) Membrane excitability increases due to CE. (Left) Passively propagated somatic spikes (small amplitude) occurred spontaneously in most cells, including the one illustrated here, before CE injection (pre). Spontaneous local dendritic calcium spikes (large amplitude) did not occur in some cells (upper trace, pre-calexcitin) but then did occur after injection of CE (post) through the recording electrode. In Purkinje cells hyperpolarized 20 mV below the membrane potential for somatic spikes, calcium spikes could be elicited by lower levels of injected positive current pulses after injection of CE. (Right) Mean percent change in the threshold current required to elicit local calcium spikes was lower for CE-injected cells vs. controls injected with heat-inactivated CE or 3 M potassium acetate (P < 0.001, Student’s t test). (E) Intracellular recordings of the Hermissenda type B photoreceptor response to a 1-s flash of light (103 erg/cm2·sec) before (Upper) and after (Lower) injection of purified cloned CE. The Hermissenda photoreceptors were isolated and submerged in artificial sea water (430 mM NaCl/10 mM KCl/10 mM CaCl2/50 mM MgCl2/10 mM Hepes Na, pH 7.4). The CE (intraelectrode concentration, 364 nM) was brought to 1 M in KAc (pH 7.4) and injected (3 min, 2 nA) into the photoreceptor with the recording electrode. Recordings were obtained using intracellular amplifier (Axopatch 2A), digitized at 50 Hz (Digidata 1200), and analyzed by computer. The normal light response returned to the original resting potential within a minute (Upper). Injection of CE (n = 5 cells) enhanced the light response and the cell remained depolarized for more than 5 min (Lower, only 1 min is represented). Injection of heat-inactivated CE (n = 6 cells) using the above protocol did not alter the light response. Time scale at right is compressed by 3×.
Microinjection of CE into rabbit Purkinje cell dendrites produced a substantial increase in membrane excitability, consistent with reduction of voltage-dependent K+ currents. First, there was a 25% reduction in the current required to elicit local, dendritic Ca2+ spikes (see Fig. 3D for conditions) after injection of CE (n = 8), compared with a 3.9% increase after injections of heat-inactivated CE (n = 6; Fig. 3D Right; P < 0.001). Identical changes are observed after tone-eyeblink conditioning (23). Second, in cells that showed no spontaneous dendritic activity, injection of CE produced spontaneous dendritic spike activity (n = 3; Fig. 3D Left). In contrast, injection of CE had no effect on membrane potential, input resistance, or current required to hyperpolarize the membrane 20 mV below somatic spiking. These results also suggest that a protein similar to CE may exist in mammalian neurons.
Microinjection of cloned CE (n = 5) but not heat-inactivated CE (n = 6) into Hermissenda type B photoreceptor neurons also caused an effect consistent with reduction of voltage-dependent K+ currents. After microinjection of CE, a test light stimulus produced a long-lasting depolarization (Fig. 3E) similar to the enhanced depolarizing responses observed after associative conditioning (43). Previous results have established that this long-lasting depolarization is mediated by inhibition of the outward K+ currents iA and iCa-K+ (9, 43). Because the iA and iCa-K+ K+ channels are conserved among species, it is likely that the mechanism of action of CE is similar for Hermissenda, squid, and mammalian K+ channels.
Immunohistochemical labeling of squid optic lobe sections demonstrated that CE was localized predominantly to the plexiform layer, which contains fibers originating from the retina and optic nerve, as well as amacrine cells with spreading tangential processes (Fig. 4). A small percentage of neuronal cell bodies also stained intensely for CE. Antibodies against scp1 are reported to stain the cytoplasm but not the nuclear matrix of Aplysia neurons, and staining was similarly specific to a fixed subpopulation of cells (44). This localization of CE in regions of terminating axonal processes is consistent with a function at the synaptic level and is also consistent with previously observed effects of CE on axonal transport and neuronal branching (11).
Figure 4.

Immunohistochemical localization of CE in the squid optic lobe. Sections were incubated with CE antibody and visualized by immunoperoxidase staining. Nuclei were counterstained blue with hematoxylin (g.o., outer granule cell layer; g.i., inner granule cell layer; p, plexiform layer). Arrow points to an amacrine cell. The plexiform layer containing neuronal axons and branches is darkly stained with the immunolabel.
DISCUSSION
In neuronal cells, activation of PKC (45) or sufficient elevation of intracellular Ca2+ can induce long-term inactivation of the K+ currents iA and iCa-K+ (43, 46, 47). PKC activation and Ca2+ elevation in neuronal compartments have been implicated in the induction of long-term changes of membrane excitability produced by associative conditioning (43, 48, 49, 50) and other protocols such as long-term potentiation or long-term depression (51, 52, 53). CE injection into neuronal structures was shown here to reproduce some of the same changes induced by Pavlovian conditioning: (i) lowered threshold of dendritic spiking (in rabbit Purkinje cells) and (ii) reduced iA and iCa-K+ and enhanced depolarizing response (in molluscan neurons).
Since CE is a high affinity substrate for PKC, CE phosphorylation would be also one of the earliest effects of activation or membrane translocation of PKC induced by learning. Phosphorylation of CE has been previously shown to cause its translocation to the membrane (21). Previous results showed that phosphorylation of CE by PKC had no effect on GTPase or GTP-binding activity. Thus, one function of phosphorylation may be to translocate CE to the membrane. At the membrane, CE would inactivate K+ channels and thereby enhance membrane excitability. Membrane translocation of CE could also be gated by GTP’s inhibition of PKC phosphorylation of CE. The increased membrane excitability would also result in positive feedback on both CE and PKC by increasing Ca2+ influx.
The results described here were obtained with cloned CE, which was in a dephosphorylated state. Fully phosphorylated CE, because of its higher membrane affinity, would be expected to have an even stronger inhibitory effect on K+ channels. Additional experiments are needed to address this question.
Further experiments are also required to clarify the complex relationships between phosphorylation, GTP binding, GTPase, Ca2+, Mg2+, and membrane translocation of CE, and its ability to block K+ channels.
In cells from human patients with Alzheimer disease, a neurodegenerative disorder characterized by memory loss, CE-like immunoreactivity, as well as specific K+ currents, were markedly decreased (54). The same effects were also observed in human fibroblast cells treated with low (≤10 nM) concentrations of β-amyloid protein (25). Because CE appears to directly inhibit K+ channels, the ion conductance changes induced by β-amyloid may be related to alterations in CE. It is not yet known, however, whether CE binds directly to β-amyloid in the same manner that APLP2 binds to the Go G protein (55).
While indirect interactions of GTP-binding proteins with Ca2+ have been observed (56), CE appears to be a member of a class of proteins that bind both Ca2+ and guanine nucleotides. CE is thus ideally situated to respond to a temporal association of two or more signals (as in associative conditioning) by serving as a point of convergence between these two critical signaling pathways.
Acknowledgments
We thank Drs. L. Hudson, S. Gutkind, A. L. Burns, and J. Axelrod for invaluable advice; Dr. J. Battey for supplying the squid optic lobe cDNA library; Dr. G. Magro for assitance with the immunocytochemistry; and Dr. Giuseppina Tesco for assistance with the fibroblast cell culture. S.C. acknowledges a fellowship from the Associazone Italiana Ricerca sul Cancro.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Wickman K, Clapham D E. Physiol Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 2.Siekevitz P. Proc Natl Acad Sci USA. 1991;88:5374–5378. doi: 10.1073/pnas.88.12.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taft W, Goldenring J, DeLorenzo R. Adv Exp Med Biol. 1987;221:409–425. doi: 10.1007/978-1-4684-7618-7_30. [DOI] [PubMed] [Google Scholar]
- 4.Alkon D L, Rasmussen H. Science. 1988;239:998–1005. doi: 10.1126/science.2830669. [DOI] [PubMed] [Google Scholar]
- 5.Matzel L, Lederhendler I, Alkon D. J Neurosci. 1990;10(7):2300–2307. doi: 10.1523/JNEUROSCI.10-07-02300.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasinelli P, Ramakers G, Urban I, Hens J, Oestreicher A, de Graan P, Gispen W. Behav Brain Res. 1995;66:53–59. doi: 10.1016/0166-4328(94)00124-x. [DOI] [PubMed] [Google Scholar]
- 7.Hell J, Yokoyama C, Wong S, Warner C, Snutch T, Catterall W. J Biol Chem. 1993;268:19451–19457. [PubMed] [Google Scholar]
- 8.Numann R, Catterall W, Scheuer T. Science. 1991;254:115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- 9.Nelson T J, Collin C, Alkon D L. Science. 1990;247:1479–1483. doi: 10.1126/science.247.4949.1479. [DOI] [PubMed] [Google Scholar]
- 10.Alkon D L, Nelson T J. FASEB J. 1990;4:1567–1576. doi: 10.1096/fasebj.4.6.2108074. [DOI] [PubMed] [Google Scholar]
- 11.Alkon D L, Ikeno H, Dworkin J, McPhie D, Olds J, Lederhendler I, Matzel L, Schreurs B, Kuzirian A, Collin C, Yamoah E. Proc Natl Acad Sci USA. 1990;87:1611–1614. doi: 10.1073/pnas.87.4.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson T J, Yoshioka T, Toyoshima S, Han Y-F, Alkon D L. Proc Natl Acad Sci USA. 1994;91:9287–9291. doi: 10.1073/pnas.91.20.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan K, Dixon J. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 14.Baker T, Grossman A, Gross C. Proc Natl Acad Sci USA. 1984;81:6779–6783. doi: 10.1073/pnas.81.21.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 16.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 17.Fraker P, Speck J C. Biochem Biophys Res Commun. 1978;80:849–857. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 18.Fiske C, SubbaRow Y. J Biol Chem. 1929;81:629–631. [Google Scholar]
- 19.Smith P, Krohn R I, Hermanson G T, Mallia A K, Gartner F H. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Nelson T J. Anal Biochem. 1993;214:325–328. doi: 10.1006/abio.1993.1496. [DOI] [PubMed] [Google Scholar]
- 21.Nelson T J, Alkon D L. J Neurochem. 1995;65:2350–2357. doi: 10.1046/j.1471-4159.1995.65052350.x. [DOI] [PubMed] [Google Scholar]
- 22.Schreurs B G, Sanchez-Andres J V, Alkon D L. Brain Res. 1991;548:18–22. doi: 10.1016/0006-8993(91)91100-f. [DOI] [PubMed] [Google Scholar]
- 23.Schreurs B, Oh M M, Alkon D L. J Neurophysiol. 1996;75:1051–1060. doi: 10.1152/jn.1996.75.3.1051. [DOI] [PubMed] [Google Scholar]
- 24.Rose T, Sebo P, Bellalou J, Ladant D. J Biol Chem. 1995;270:26370–26376. doi: 10.1074/jbc.270.44.26370. [DOI] [PubMed] [Google Scholar]
- 25.Etcheberrigaray R, Ito E, Kim C S, Alkon D L. Science. 1994;264:276–279. doi: 10.1126/science.8146663. [DOI] [PubMed] [Google Scholar]
- 26.Hamill D P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 27.Neary J T, Crow T, Alkon D L. Nature (London) 1981;293:658–660. doi: 10.1038/293658a0. [DOI] [PubMed] [Google Scholar]
- 28.Cox J A, Stein E A. Biochemistry. 1981;20:5430–5436. doi: 10.1021/bi00522a012. [DOI] [PubMed] [Google Scholar]
- 29.Takagi T, Konishi K, Cox J A. Biochemistry. 1986;25:3585–3592. [Google Scholar]
- 30.Shen K A, Hammond C H, Moore H-P. FEBS Lett. 1993;335:380–385. doi: 10.1016/0014-5793(93)80423-r. [DOI] [PubMed] [Google Scholar]
- 31.Serventi I M, Cavanaugh E, Moss J, Vaughan M. J Biol Chem. 1993;268:4863–4872. [PubMed] [Google Scholar]
- 32.Randazzo P A, Terui T, Sturch S, Kahn R A. J Biol Chem. 1994;269:29490–29494. [PubMed] [Google Scholar]
- 33.Nakano A, Muramatsu M. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kretsinger R H. Cold Spring Harbor Symp Quant Biol. 1987;52:499–510. doi: 10.1101/sqb.1987.052.01.057. [DOI] [PubMed] [Google Scholar]
- 35.Walker J E, Saraste M, Runswick M J, Gay N J. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner R S, Kemp B, Su H-D, Kuo J F. J Biol Chem. 1985;260:11503–11507. [PubMed] [Google Scholar]
- 37.Musil L, Carr C, Cohen J, Merlie J. J Cell Biol. 1988;107:1113–1121. doi: 10.1083/jcb.107.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glomset J A, Gelb M H, Farnsworth C C. Trends Biochem Sci. 1990;15(4):139–142. doi: 10.1016/0968-0004(90)90213-u. [DOI] [PubMed] [Google Scholar]
- 39.Lowy D, Willumsen B. Nature (London) 1989;341:384–385. doi: 10.1038/341384a0. [DOI] [PubMed] [Google Scholar]
- 40.Hara-Nishimura I, Kondo M, Nishimura H, Hara R, Hara T. FEBS Lett. 1993;317:5–11. doi: 10.1016/0014-5793(93)81480-n. [DOI] [PubMed] [Google Scholar]
- 41.Cox J A, Wnuk W, Stein E A. Biochemistry. 1976;15:2613–2620. doi: 10.1021/bi00657a021. [DOI] [PubMed] [Google Scholar]
- 42.Hermann A, Cox J A. Comp Biochem Physiol B. 1995;111:337–345. doi: 10.1016/0305-0491(94)00218-j. [DOI] [PubMed] [Google Scholar]
- 43.Alkon D L. Science. 1984;276:1037–1045. doi: 10.1126/science.6093258. [DOI] [PubMed] [Google Scholar]
- 44.Pauls T L, Cox J A, Heizmann C W, Hermann A. Eur J Neurosci. 1993;5:549–559. doi: 10.1111/j.1460-9568.1993.tb00520.x. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka C, Nishizuka Y. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- 46.Connor J, Alkon D L. J Neurophysiol. 1984;51:745–752. doi: 10.1152/jn.1984.51.4.745. [DOI] [PubMed] [Google Scholar]
- 47.Bank B, DeWeer A, Kuzirian A, Rasmussen H, Alkon D. Proc Natl Acad Sci USA. 1988;85:1988–1992. doi: 10.1073/pnas.85.6.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matzel L D, Collin C, Alkon D L. Behav Neurosci. 1992;106:954–963. doi: 10.1037//0735-7044.106.6.954. [DOI] [PubMed] [Google Scholar]
- 49.Alkon D L, Sakakibara M. Biophys J. 1985;48:983–995. doi: 10.1016/S0006-3495(85)83861-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McPhie D, Matzel L, Olds J, Lester D, Kuzirian A, Alkon D L. J Neurochem. 1993;60:646–651. doi: 10.1111/j.1471-4159.1993.tb03196.x. [DOI] [PubMed] [Google Scholar]
- 51.Collingridge G, Randall A, Davies C, Alford S. Ciba Found Symp. 1992;164:162–171. doi: 10.1002/9780470514207.ch11. [DOI] [PubMed] [Google Scholar]
- 52.Bliss T, Collingridge G. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 53.Ramakers G, de Graan P, Urban I, Kraay D, Tang T, Pasinelli P, Oestreicher A, Gispen W. J Biol Chem. 1995;270:13892–13898. doi: 10.1074/jbc.270.23.13892. [DOI] [PubMed] [Google Scholar]
- 54.Kim C S, Han Y-F, Etcheberrigaray R, Nelson T J, Olds J, Yoshioka T, Alkon D L. Proc Natl Acad Sci USA. 1995;92:3060–3064. doi: 10.1073/pnas.92.7.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wasco W, Gurubhagavatula S, Paradis M, Romano D, Sisodia S, Hyman B, Neve R, Tanzi R. Nat Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- 56.Farnsworth C L, Freshney N W, Greenberg L B, Feig L A. Nature (London) 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]