

Abstract
Despite intensive efforts, the intracellular signaling pathways that mediate apoptosis remain unclear. The human B lymphoma cell line, B104, possesses characteristics that make it an attractive model for analysis of receptor-mediated apoptosis. Although these cells express both membrane IgM (mIgM) and membrane IgD (mIgD) crosslinking mIgM results in significant apoptosis while crosslinking mIgD does not. Our results show that crosslinking mIgM but not mIgD induced a delayed and sustained activation of the mitogen-activated protein kinase (MAPK) family members stress-activated protein kinase (SAPK) and p38 MAPK. The calcium ionophore ionomycin, which also induces apoptosis in B104 cells, stimulated a similar SAPK and p38 MAPK response. Cyclosporin A, a potent inhibitor of apoptosis induced by either mIgM or ionomycin, inhibited activation of both SAPK and p38 MAPK, suggesting that stimulation of these kinases may be required for induction of apoptosis. Collectively, our results indicate that SAPK and p38 MAPK may be downstream targets during mIgM-induced, calcium-mediated, apoptosis in human B lymphocytes.
Keywords: programmed cell death, calcium, cyclosporina, B cell
Apoptosis is an active process, invoked during development, proliferation, and disease, whereby defective, destructive, or redundant cells are eliminated from the body. The process of apoptosis consists of an evolutionarily conserved cascade of characteristic changes including, the induction of critical genes, metabolic and electrochemical changes, condensation of chromatin, and the activation of endonucleases, activation of proteases, and changes in membrane morphology (1). The molecular mechanisms that regulate apoptosis have been the subject of intense investigation (2, 3). An understanding of these mechanisms will be vital to our understanding of the development and function of cell systems such as the nervous and immune systems.
Lymphoid development is tightly controlled by both positive and negative selection to ensure that individuals will be able to respond appropriately to self and foreign antigens (4). Negative selection of B lymphocytes occurs by multiple mechanisms including antigen-induced inactivation or clonal anergy and antigen-induced deletion or apoptosis. Factors thought to influence the outcome of receptor ligation include the strength of the interaction with antigen, the involvement of auxiliary signals, and the developmental state of the lymphocyte (4, 5). During development of B cells their surface Ig expression pattern changes; immature mIgM+ B cells express little or no mIgD, while mature mIgM+D+ B cells express more mIgD than IgM (4, 6). This has led to the suggestion that mIgM and mIgD may transduce qualitatively different signals during B cell development (7). However, the role of mIgD and whether mIgM and mIgD transmit distinct signals remain controversial.
The mitogen-activated protein kinase (MAPK) family of enzymes, activated by dual phosphorylation on both tyrosine and threonine residues, has been implicated in the transduction of a wide variety of extracellular signals (8). The first members of this family to be identified were p42 and p44 extracellular signal-regulated kinases (ERKs) that are activated by growth factors and many other mitogenic stimuli (9). Subsequently, p54 and p46 stress-activated protein kinases (SAPKs) were identified (10, 11). These kinases, also referred to as c-Jun N-terminal kinases, are strongly activated by stressful stimuli such as pro-inflammatory cytokines, UV light, and protein synthesis inhibitors (10, 11). However, they are also responsive to comitogenic stimuli in lymphocytes (12, 13). Recently, a third group of MAPKs, termed p38 MAPKs, has been identified (14, 15, 16). The p38 MAPK cascade is also responsive to stress-related stimuli such as osmotic shock, arsenite, and lipopolysaccharide as well interleukin 1 and thrombin (17, 18, 19, 20).
The cellular roles of the respective MAPK pathways are not understood. The ability of stressful signals to stimulate SAPK and p38 MAPK activity has led to the suggestion that these pathways may function to communicate growth inhibitory and apoptotic signals within the cell. Activation of SAPK and p38 MAPK has been shown to occur during apoptosis of PC12 cells induced by withdrawal of nerve growth factor (21). In this system, stimulation of p38 and SAPK activities appear to be critical for the induction of apoptosis. However, the signal transduction pathways involved are uncharacterized, and it is not clear whether these observations can be extended to other cell types. We present evidence that SAPK and p38 MAPK are activated during mIgM-induced apoptosis of B lymphocytes, by a calcium-mediated, cyclosporin A (CsA)-sensitive, pathway.
MATERIALS AND METHODS.
Reagents and Cells.
Goat F(ab′)2 anti-μ, anti-δ, and anti-γ sera were obtained from Jackson ImmunoResearch and Southern Biotechnology Associates. CsA was obtained from Sandoz Pharmaceutical. Phorbol 12-myristate 13-acetate and ionomycin were purchased from Calbiochem–Behring. Anisomycin was obtained from Sigma. Rabbit polyclonal antiserum raised against SAPK (C17) was obtained from Santa Cruz Biotechnology. Rabbit polyclonal against p38 has been described (20). Myelin basis protein was from GIBCO/BRL, glutathione S-transferase-Jun, and glutathione S-transferase-activating transcription factor-2 were prepared as described (18, 22). The B104 human B lymphoma line, kindly provided by Mitsufumi Mayumi (Kyoto University Hospital), was grown in culture as described (23).
Immunoprecipitation and Kinase Assays.
Following incubation with the indicated stimuli, 5–10 × 106 cells were lysed in 1 ml of lysis buffer (20 mM Hepes, pH 7.4/2 mM EGTA/50 mM β-glycerophosphate/1% Triton X-100/10% glycerol/1 mM DTT/1 mM phenylmethylsulfonyl fluoride/25 μg/ml leupeptin/25 μg/ml aprotinin/2 mM Na2VO4/10 mM NaF) for 15 min on ice. Cell debris was removed by centrifugation at 10,000 × g for 10 min at 4°C. SAPK or p38 MAPK was then immunoprecipitated by 0.5 μg of the appropriate antiserum for 3 hr with the addition of 50 μl of 1:1 protein-A Sepharose slurry for the final hour. The beads were pelleted by centrifugation and then washed three times each in lysis buffer, wash buffer (500 mM LiCl/100 mM Tris·Cl, pH 7.6/0.1% Triton X-100/1 mM DTT), and assay buffer (20 mM Mops, pH 7.2/2 mM EGTA/10 mM MgCl2/0.1% Triton X-100/1 mM DTT). The beads were left as a 1:1 suspension in assay buffer and 20 μl (0.3 mg/ml) of either glutathione S-transferase-Jun (for SAPK) or glutathione S-transferase-ATF2 (for p38 MAPK) was added. Kinase reactions were initiated by the addition of 15 μl of 32P-labeled Mg.ATP solution (50 mM MgCl2/500 μM ATP/10 μCi [γ-32P]ATP; 1 Ci = 37 GBq) and were carried out at 30°C for 20 min. Reactions were stopped by the addition of 25 μl of 6 × Laemmli sample buffer and boiling for 3 min. Samples were separated on a SDS/10% polyacrylamide gel and, after drying, were subjected to autoradiography. Quantitation by densitometry was performed with an imaging densitometer (Bio-Rad).
ERK activity was measured as described previously (24). Briefly, cells were lysed in 1 ml of buffer H+ (50 mM β-glycerophosphate, pH 7.4/1.5 mM EGTA/0.1 mM Na3VO4/1 mm DTT/25 μg/ml aprotinin/25 μg/ml leupeptin/0.5 mM phenylmethysulfonyl fluoride). ERK activity was determined by the incorporation of radioactivity from [γ-32P]ATP into myelin basic protein after a 15-min incubation at 30°C.
Cell Death and Flow Cytometric Assays.
Dead cells were quantified using flow cytometry as described (25). Briefly, the cells were washed and then resuspended in PBS containing 1% BSA. Hoechst 33342 was added to a final concentration of 1 μg/ml × 106 cells, and the cells were placed in a humidified incubator for 7 min. The cell suspension was then placed on ice and 1 μg/ml 7-amino-actinomycin D added prior to analysis using a FACStar Plus flow cytometer (Becton Dickinson). Apoptotic cells were considered to be the population that exhibited DNA damage or loss of plasma membrane integrity. Flow cytometric analyses were performed with a FACScan analyzer (Becton Dickinson) and [Ca2+]i was monitored using an Indo-1 assay as described (26).
Statistical Analysis.
All experiments shown are representative of between three and five repeats. Where applicable, data are shown ± SEM.
RESULTS
Characteristics of the Human B Lymphoma Cell Line B104.
The human B lymphoma B104 expressed about 20-fold more mIgM than mIgD (Fig. 1A). As previously reported, crosslinking mIgM with anti-IgM antisera induced B104 cells to die within hours, whereas crosslinking mIgD resulted in a small decline in cell viability (Fig. 1B). IgG was used as an irrelevent control and had no effect on cell viability (Fig. 1B) (23). Similar results were obtained with isotype-matched mAb to μ or δ chains and with heterologous F(ab′)2 anti-μ or anti-δ sera (data not shown). Treatment of B104 cells with the calcium ionophore ionomycin also induced a rapid loss of viability (Fig. 1B) (27). Graded doses of mAb to μ or δ chains induced a dose-dependent elevation of [Ca2+]i, as determined by changes in Indo-1 ratios (Fig. 1 C and D). Despite the fact that B104 cells express substantially less mIgD than mIgM (Fig. 1A), the calcium responses to ligation of either mIgD or mIgM were quantitatively and qualitatively similar.
Figure 1.
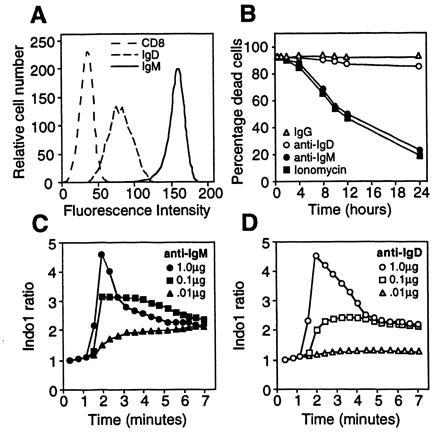
Characteristics of the mIgM+IgD+ human B cell lymphoma line, B104. (A) Flow cytometric analysis of B104 cells with mAbs specific for IgM (4B8), IgD (δTA-1), or CD8 (G10-1). (B) Effect of treatment with anti-IgM (•) (1 μg/ml), anti-IgD (▵) (1 μg/ml), IgG (○) (1 μg/ml), or ionomycin (▪) (200 mg/ml) on B104 cell viability. (C and D) Effect of graded doses of anti-IgM (solid symbols) or anti-IgD (open symbols) on B104 [Ca2+]i levels as measured by changes in Indo-1 ratio. Antibody doses were 1.0 μg/ml (circles), 0.1 μg/ml (squares), and 0.01 μg/ml (triangles).
Activation of ERKs by Either Anti-IgM or Anti-IgD.
To further characterize the signaling pathways stimulated by mIgM and mIgD, we measured p42 and p44 ERK activity induced by either anti-IgM or anti-IgD. Crosslinking either mIgM or mIgD stimulated a time-dependent activation of ERKs (Fig. 2A). The ERK response to anti-IgD was consistently observed to be weaker and somewhat shorter than that stimulated by the same dose of anti-IgM. However, doses of anti-IgM and anti-IgD that produced quantitativelty comparable ERK responses retained their differential ability to induce apoptosis (Fig. 2B). Ionomycin treatment alone did not induce measurable ERK activity (data not shown).
Figure 2.
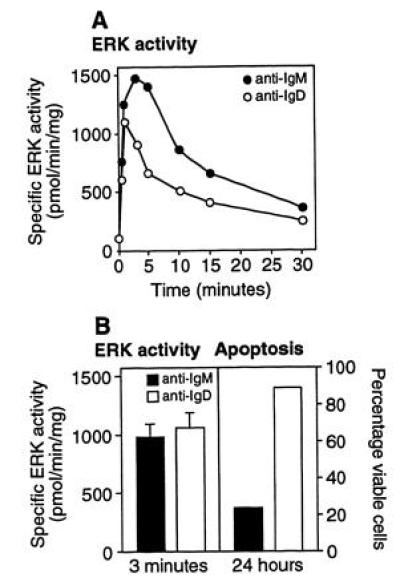
Crosslinking of mIgM or mIgD stimulates ERK activity. (A) Effect of treatment of B104 cells with anti-IgM (•) or anti-IgD (○) (1 μg/ml) for the indicated times on ERK activity. (B) B104 cells were treated with either 0.5 μg/ml anti-IgM or 1.0 μg/ml anti-IgD. (Left) ERK activity measured at 3 min. (Right) Cell viability measured at 24 hr.
Activation of SAPK and p38 MAPK by Anti-IgM or Ionomycin But Not Anti-IgD.
To determine if SAPK and p38 MAPK kinases were stimulated when B104 cells undergo apoptosis, we measured the activities of these kinases at various times after ligation of either mIgM or mIgD. No detectable SAPK activity in response to anti-IgM was detected time points from 5 min to 1 hr. However, anti-IgM-did stimulate a late and sustained increase in SAPK activity (Fig. 3A). This increase was detectable after 2–4 hr of mIgM crosslinking and was maximal by 8 hr. A similar time-dependency was observed for activation of SAPK by treatment with the calcium ionophore ionomycin. Treatment of cells with anti-IgD had a small effect on SAPK activity. SAPK activity in response to mIgD crosslinking was much weaker than was observed with either ionomycin or anti-IgM. For example, 8 hr after mIgD crosslinking, SAPK activity was 9% of that observed in response to anti-IgM at this time point. Crosslinking mIgM or treatment with ionomycin also stimulated a late and sustained increase in p38 MAPK activity (Fig. 3B). The time-dependency of anti-IgM- and ionomycin-induced p38 MAPK activity was similar to that observed with SAPK activity (Fig. 3A). A weak effect of anti-mIgD on p38 MAPK activity was observed.
Figure 3.
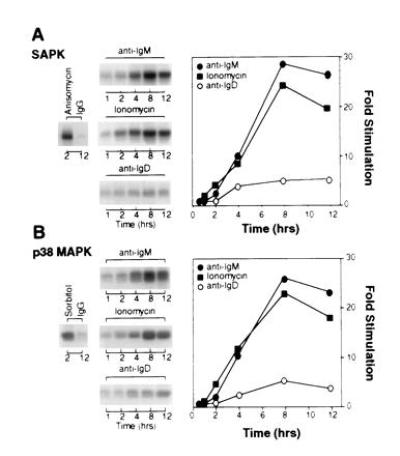
Crosslinking of mIgM or treatment with ionomycin stimulates activation of SAPK and p38 MAPK. B104 cells were treated for the indicated time with either anti-IgM (•; 1 μg/ml), anti-IgD (○; 1 μg/ml), or ionomycin (▪; 200 ng/ml). SAPK (A) or p38 MAPK (B) activities were measured by immune complex kinase assay. (Left) Autoradiograms. (Right) Activities, as determined by densitometric quantification. The effect of treatment with anisomycin (20 μg/ml) (A) or sorbitol (500 mM) (B) for 2 hr are also shown.
Fig. 4 shows a comparison of the effects of anti-IgM on the activities of ERKs, SAPK, p38 MAPK, and B104 cell apoptosis. Activation of SAPK and p38 MAPK occur significantly later than activation of ERKs in response to anti-IgM. However, the increase in SAPK and p38 MAPK activities are amongst the earliest biochemical events that we have observed to correlate with apoptosis in these cells and may temporally precede induction of DNA damage and loss of viability. Western blotting with reagents specific for either SAPK or p38 MAPK indicated that no change in the level of expression of these protein kinases occurred during the time course of these experiments (data not shown).
Figure 4.
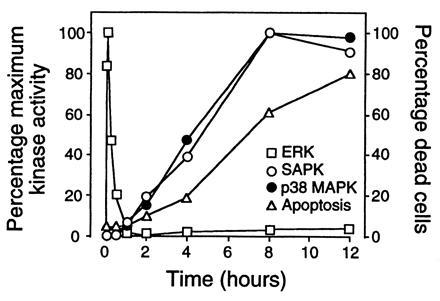
Comparison of the kinetics of induction of ERK, SAPK, p38 MAPK activities, and with apoptosis induced by crosslinking of mIgM. The effect of anti-IgM on induction of ERK (□), SAPK (•), and p38 MAPK (○) activities and apoptosis (▵), expressed as percentages of the maximum response at the indicated times.
CsA Inhibits SAPK and p38 MAPK Activation in B104 Cells.
CsA inhibited both anti-IgM- and ionomycin-induced apoptosis of B104 cells (Fig. 5A) (28, 29). Apoptosis induced by mIgM crosslinking was almost completely blocked by treatment with CsA while a partial inhibition of ionomycin-induced apoptosis was observed (Fig. 5A). Since CsA inhibits both anti-IgM- and ionomycin-induced apoptosis of B104 cells, we investigated the effect of CsA on the activation of SAPK and p38 MAPK by these stimuli. Treatment of B104 cells with CsA almost completely blocked the late SAPK response to anti-IgM (Fig. 5B). Consistent with its partial inhibition of ionomycin-induced apoptosis, CsA only partially blocked ionomycin-induced SAPK activation. Similar results were obtained when the effects of CsA on activation of p38 MAPK were investigated (Fig. 5C). CsA significantly blocked the ability of mIgM crosslinking to stimulate p38 MAPK and also partially inhibited ionomycin-induced p38 MAPK activity. CsA had no effect on ERK activity observed in response to crosslinking mIgM or mIgD (data not shown).
Figure 5.
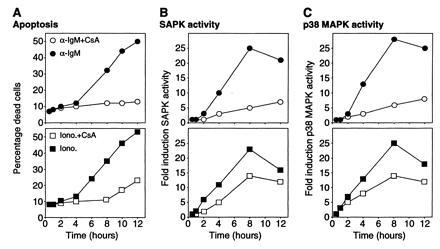
Effect of CsA on induction of apoptosis, SAPK, and p38 MAPK activities by crosslinking of mIgM or ionomycin treatment. B104 cells were treated with anti-IgM (squares) (1 μg/ml) or ionomycin (circles) (200 ng/ml) for the indicated times in the presence (solid symbols) or absence (open symbols) of CsA (150 ng/ml). At the indicated times, the following characteristics were measured: (A) Cell viability; (B) SAPK activity; and (C) p38 MAPK activity.
DISCUSSION
Why some B cells express both mIgM and mIgD and what function mIgD serves remains unclear. While some studies have suggested that crosslinking either mIgM or mIgD induced similar responses, other studies have reported clear differences (7). In B104 cells, crosslinking either mIgM or mIgD induces tyrosine phosphorylation, phosphatidylinositol breakdown, elevation of intracellular free calcium concentration ([Ca2+]i), activation of protein kinase C, and induction of immediate early genes (30, 31). However, while mIgM ligation strongly induces apoptosis in these cells, crosslinking mIgD provides a very weak apoptotic signal. The ability of either anti-IgM or anti-IgD to induce comparable elevation in [Ca2+]i suggests that this early calcium signal is not a determinant factor in the transduction of apoptotic signals. Crosslinking either mIgM or mIgD also resulted in a rapid, transient activation of p42 and p44 ERKs. The response to anti-IgD was somewhat weaker and more transient than that observed upon crosslinking of anti-IgM. However, a lower dose of anti-IgM that produced an ERK response quantitatively and qualitatively similar to that obtained with the indicated dose of anti-IgD still induced apoptosis. This result suggests that the differential ability of mIgM and mIgD to activate the ERKs does not account for the contrasting effects of mIgM and mIgD crosslinking on B104 cell apoptosis.
A clear difference was observed in the abilities of anti-IgM and anti-IgD to induce activation of SAPK and p38 MAPK. Crosslinking of mIgM induced activation of both SAPK and p38 MAPK that was detectable 2–4 hr after stimulation and was maximal by 8 hr. In contrast, anti-IgD had a weak stimulatory effect on SAPK and p38 MAPK activity. These results are consistent with the fact that mIgD crosslinking induces a small amount of apoptosis in B104 cells (Figs. 1B and 2B) (23). The relative abilities of anti-IgM and anti-IgD to stimulate SAPK and p38 MAPK activities correlated well with their effects on B104 cell viability, suggesting that these kinases may function in signaling pathways by which mIgM induces apoptosis.
Calcium signaling has been implicated in the induction of apoptosis in response to a variety of stimuli (32). Calcium ionophores induce apoptosis in many cell systems suggesting that targets of calcium signaling play an important role in apoptotic pathways. Persistent elevation of [Ca2+]i may lead to stimulation of the activities of proteases, phospholipases, and endonucleases. The calcium ionophore ionomycin induces apoptosis of B104 cells (Fig. 1A) (27). Ionomycin stimulated a delayed and sustained activation of SAPK and p38 that was quantitatively and qualitatively similar to that observed upon mIgM crosslinking. These results are consistent with SAPK and p38 MAPK representing downstream targets for Ca2+ signaling pathways in the induction of apoptosis by both ionomycin and mIgM. However, both anti-IgM and anti-IgD elicit increases in [Ca2+]i, suggesting that this early calcium signal is insufficient to induce apoptosis. Although our data suggests that calcium plays an important role in signaling apoptosis in response to anti-IgM, it appears likely that other signals are also required.
The ability of CsA to inhibit apoptosis in many cells suggests that one important target may be the calcium-dependent phosphatase calcineurin (32, 33, 34). CsA inhibited apoptosis as well as activation of SAPK and p38 MAPK by either ionomycin or anti-IgM. However, CsA was observed to be a much more effective inhibitor of apoptosis and SAPK/p38 MAPK activation induced by mIgM than by ionomycin. One possible explanation for this differential sensitivity is that ionomycin treatment results in the activation of multiple calcium-mediated pathways, only some of which are sensitive to inhibition by CsA. Collectively, these data suggest an important role for calcineurin, upstream of SAPK and p38 MAPK, in a calcium-mediated pathway of apoptosis stimulated by mIgM.
Several lines of evidence suggest that p42 and p44 ERKs do not function in the pathway whereby mIgM induces apoptosis in B104 cells. First, ERK activity is stimulated in response to ligation of either mIgM, or mIgD. Second, CsA treatment did not inhibit ERK activation upon crosslinking of mIgM. Third, ionomycin does not significantly induce ERK activity at either early or late timepoints. These results do not, however, rule out the possibility that ERK activation may regulate the induction of apoptosis. In this respect, the ERKs have been suggested to deliver a signal that promotes survival in antagonism of signals derived from SAPK and p38 MAPK (21).
One interesting feature of the SAPK and p38 MAPK responses measured in this study is their comparatively delayed and sustained kinetics. Most stimuli that have been identified to stimulate SAPK or p38 MAPK activity show rapid and transient kinetics (10, 11, 18). Although anti-IgM- and ionomycin-induced SAPK and p38 MAPK activation were delayed, they appeared to precede induction of DNA damage and loss of cell viability. One possibility is that temporal differences in the activation of these kinases may lead to different cellular outcomes. A similar paradigm has been suggested for the ERK pathway to explain its role in both proliferative and differentiative responses (35). This model would reconcile the apparently contradictory roles that have been suggested for SAPK in both mitogenic and growth inhibitory responses. Thus, delayed and persistent activation of SAPK and p38 MAPK may be an essential requirement for apoptosis.
A major question concerns the mechanism of induction of the delayed SAPK/p38 MAPK response by calcium and mIgM crosslinking. This process could involve either persistent activation of upstream kinases or inhibition/down-regulation of specific phosphatases. One possibility is that mIgM crosslinking induces an autocrine factor that is responsible for activation of SAPK/p38 MAPK and triggers apoptosis. Alternatively, SAPK and p38 MAPK may be activated as a direct consequence of the apoptotic program initiated by mIgM. Although activation of these kinases appears to precede induction of DNA damage, we cannot rule out the possibility that a very low threshold of DNA damage is required to initiate SAPK and p38 MAPK activation. Consistent with a role for DNA damage, SAPK activity is known to be activated during apoptosis induced by DNA damaging agents such as gamma irradiation (36). Since activation of a highly conserved cascade of proteases is another early event observed during apoptosis, it is also possible that SAPK/p38 MAPK activation is the result of the proteolytic cleavage of upstream regulatory components.
The differential abilities of mIgM and mIgD to induce apoptosis may provide an opportunity to elucidate the mechanism by which SAPK and p38 MAPK are activated during apoptosis. The resulting persistent activation of SAPK and p38 MAPK might serve to amplify the apoptotic response initiated by mIgM. In this regard, it will be important to determine the precise roles of SAPK and p38 MAPK in mIgM-induced and Ca2+-mediated apoptosis. Identification of critical substrates will also facilitate a greater understanding of the role of these stress-activated kinases in apoptotic responses. Such information might significantly advance our ability to develop treatments that may alter the pathogenesis of human diseases, such as autoimmune diseases and lymphomas, where changes in cell survival have been implicated.
Acknowledgments
We thank Jeffrey Ledbetter and David Hockenberry for critical reading of the manuscript, and members of the Krebs and Clark labs for helpful discussions. This work was supported by National Institutes of Health Grant GM42508.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: mIgM, membrane IgM; mIgD, membrane IgD; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; SAPK, stress activated protein kinase; CsA, cyclosporin A; [Ca2+]i, intracellular free calcium concentration.
References
- 1.Steller H. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 2.Wyllie A H. Curr Opin Gen Dev. 1995;5:97–104. doi: 10.1016/s0959-437x(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 3.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 4.Goodnow C C. Proc Natl Acad Sci USA. 1996;93:2264–2271. doi: 10.1073/pnas.93.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melchers F, Rolink A, Grawunder U, Winkler T H, Karasuyama H, Ghia P, Andersson J. Curr Opin Immunol. 1995;7:214–227. doi: 10.1016/0952-7915(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 6.Havran W L, DiGiusto D L, Cambier J C. J Immunol. 1984;132:1712–1716. [PubMed] [Google Scholar]
- 7.Monroe J G. J Immunol. 1996;156:2657–2660. [PubMed] [Google Scholar]
- 8.Graves, J. D., Campbell, J. S. & Krebs, E. G. (1995) Ann. N.Y. Acad. Sci. 766, pp. 320–343. [DOI] [PubMed]
- 9.Cobb M H, Boulton T G, Robbins D J. Cell Regul. 1991;2:965–978. doi: 10.1091/mbc.2.12.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Erijard B, Hibi M, Wu I H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 11.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. Nature (London) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 12.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben Neriah Y. Cell. 1994;77:727–36. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 13.Berberich I, Shu G, Siebelt F, Woodgett J R, Kyriakis J M, Clark E A. EMBO J. 1996;15:92–101. [PMC free article] [PubMed] [Google Scholar]
- 14.Han J, Lee J D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 15.Rouse J, Cohen P, Trigon S, Morange M, Alonso Llamazares A, Zamanillo D, Hunt T, Nebreda A R. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 16.Lee J C, Laydon J T, McDonnell P C, Gallagher T F, Kumar S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, Strickler J E, McLaughlin M M, Siemens I R, Fisher S M, Livi G P, White J R, Adams J L, Young P R. Nature (London) 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 17.Freshney N W, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Cell. 1994;78:1039–49. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 18.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 19.Kramer R M, Roberts E F, Strifler B A, Johnstone E M. J Biol Chem. 1995;270:27395–27398. doi: 10.1074/jbc.270.46.27395. [DOI] [PubMed] [Google Scholar]
- 20.Saklatvala J, Rawlinson L, Waller R J, Sarsfield S, Lee J C, Morton L F, Barnes M J, Farndale R W. J Biol Chem. 1996;271:6586–6589. doi: 10.1074/jbc.271.12.6586. [DOI] [PubMed] [Google Scholar]
- 21.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 22.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 23.Kim K M, Yoshimura T, Watanabe H, Ishigami T, Nambu M, Hata D, Higaki Y, Sasaki M, Tsutsui T, Mayumi M, Mikawa H. J Immunol. 1991;146:819–825. [PubMed] [Google Scholar]
- 24.Seger R, Seger D, Reszka A A, Munar E S, Eldar Finkelman H, Dobrowolska G, Jensen A M, Campbell J S, Fischer E H, Krebs E G. J Biol Chem. 1994;269:25699–25709. [PubMed] [Google Scholar]
- 25.Schmid I, Uittenbogaart C H, Giorgi J V. Cytometry. 1994;15:12–20. doi: 10.1002/cyto.990150104. [DOI] [PubMed] [Google Scholar]
- 26.Rabinovitch P S, June C H, Grossmann A, Ledbetter J A. J Immunol. 1986;137:952–961. [PubMed] [Google Scholar]
- 27.Kim K M, Ishigami T, Hata D, Yamaoka K, Mayumi M, Mikawa H. J Immunol. 1992;148:1797–1803. [PubMed] [Google Scholar]
- 28.Ishigami T, Kim K M, Horiguchi Y, Higaki Y, Hata D, Heike T, Katamura K, Mayumi M, Mikawa H. J Immunol. 1992;148:360–368. [PubMed] [Google Scholar]
- 29.Bonnefoy Berard N, Genestier L, Flacher M, Revillard J P. Eur J Immunol. 1994;24:325–329. doi: 10.1002/eji.1830240208. [DOI] [PubMed] [Google Scholar]
- 30.Hata D, Kawakami T, Ishigami T, Kim K M, Heike T, Katamura K, Mayumi M, Mikawa H. Int Immunol. 1992;4:797–804. doi: 10.1093/intimm/4.7.797. [DOI] [PubMed] [Google Scholar]
- 31.Kanazashi S, Hata D, Ishigami T, Jung E Y, Shintaku N, Sumimoto S, Heike T, Katamura K, Mayumi M. Mol Immunol. 1994;31:21–30. doi: 10.1016/0161-5890(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 32.McConkey D J, Orrenius S. Curr Top Microbiol Immunol. 1995;200:95–105. doi: 10.1007/978-3-642-79437-7_7. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y F, Sahai B M, Green D R. Nature (London) 1989;339:625–626. doi: 10.1038/339625a0. [DOI] [PubMed] [Google Scholar]
- 34.Shibasaki F, McKeon F. J Cell Biol. 1995;131:735–743. doi: 10.1083/jcb.131.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Biochem J. 1992;288:351–355. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y R, Meyer C F, Tan T H. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]