

Abstract
Severe combined immunodeficient (SCID) mice display an increased sensitivity to ionizing radiation compared with the parental, C.B-17, strain due to a deficiency in DNA double-strand break repair. The catalytic subunit of DNA-dependent protein kinase (DNA-PKCS) has previously been identified as a strong candidate for the SCID gene. DNA-PK phosphorylates many proteins in vitro, including p53 and replication protein A (RPA), two proteins involved in the response of cells to DNA damage. To determine whether p53 and RPA are also substrates of DNA-PK in vivo following DNA damage, we compared the response of SCID and MO59J (human DNA-PKcs-deficient glioblastoma) cells with their respective wild-type parents following ionizing radiation. Our findings indicate that (i) p53 levels are increased in SCID cells following ionizing radiation, and (ii) RPA p34 is hyperphosphorylated in both SCID cells and MO59J cells following ionizing radiation. The hyperphosphorylation of RPA p34 in vivo is concordant with a decrease in the binding of RPA to single-stranded DNA in crude extracts derived from both C.B-17 and SCID cells. These results suggest that DNA-PK is not the only kinase capable of phosphorylating RPA. We conclude that the DNA damage response involving p53 and RPA is not associated with the defect in DNA repair in SCID cells and that the physiological substrate(s) for DNA-PK essential for DNA repair has not yet been identified.
Keywords: ionizing radiation, ataxia telangiectasia, MO59J
Severe combined immunodeficient (SCID) mice are sensitive to ionizing radiation (IR) because of their reduced efficiency in performing DNA double-strand break repair (1, 2, 3). They also lack B- and T-cell immunity as a result of a deficiency in performing V(D)J recombination, the process of assembling immunoglobulin and T-cell receptor genes from gene segments by site-specific recombination (4, 5, 6).
Repair of DNA double-strand breaks following IR is essential for the survival of the cell. The mechanisms by which DNA double-strand break repair occur in mammalian cells are only beginning to be elucidated. The catalytic subunit of DNA-dependent protein kinase (DNA-PKCS) was identified as a strong candidate for the SCID gene by genetic complementation (7). Several additional lines of evidence support DNA-PKcs as the molecular defect in SCID cells. DNA-PKCS protein levels are severely reduced in SCID cells compared with cells from the wild-type parent, C.B-17 (7, 8), and the kinase activity of DNA-PK measured in cell extracts from SCID cells in vitro is absent (8, 9). DNA-PK is a serine-threonine protein kinase that is dependent on DNA double-stranded ends for its activity, with the Ku proteins being the DNA binding partner of DNA-PKCS (10, 11).
DNA-PK has been suggested as one of the central players in the DNA damage response (12), possibly linking transcription and repair. DNA-PK phosphorylates many substrates in vitro such as the transcription factors Sp1, fos, jun, Oct 1 and 2; RNA polymerase II; and proteins involved in the response of cells to DNA damage, such as p53 and replication protein A (RPA) (for a review, see ref. 13). The DNA-PKcs-deficient SCID cells are a powerful model system for investigating the role of DNA-PK in vivo.
In addition to SCID cells, another useful cell line to examine the role of DNA-PKcs in the cellular response to DNA damage is the human malignant glioma cell line MO59J. It is considerably more sensitive to IR than its sister cell line, MO59K, which was isolated from a different area of the same human malignant glioma biopsy sample (14). Similar to SCID cells, MO59J cells show no DNA-PK activity when assayed in cell extracts in vitro (15). Furthermore, the molecular defect in MO59J cells underlying the inactivation of DNA-PK activity is better characterized than in SCID cells, as there is no DNA-PKcs mRNA expression in MO59J cells (15).
One potential substrate for DNA-PK in the cellular DNA damage response is the p53 tumor suppresser gene product. Following treatment with IR, p53 protein levels are elevated via an unidentified posttranscriptional mechanism (16). This induction of p53 levels leads to a cell-cycle arrest at the G1/S phase checkpoint, presumably allowing DNA repair to occur before progression into S phase (17). One likely mechanism that may partly explain the post-IR increase in p53 protein levels is phosphorylation of p53 by an IR-activated Ser/Thr kinase (18). Studies employing cell extracts in vitro have shown that DNA-PK phosphorylates human p53 at Ser-15 and Ser-37 residues, and mouse p53 at Ser-4 and Ser-15 residues. Interestingly, Ser-4 and Ser-15 in mouse p53 have also been found to be phosphorylation sites in vivo (19, 20, 21), suggesting that DNA-PK may be a true physiological modulator of p53.
A second substrate of DNA-PK that has been implicated in DNA repair is RPA [human single-stranded DNA-binding protein (HSSB)] (for a review, see ref. 22). RPA is a trimeric protein complex that binds to single-stranded DNA (ssDNA) (22). This protein has multiple activities in DNA replication (22), recombination (23), and repair (24). Although the p70 subunit is known to bind ssDNA (22), the roles of the p34 and p14 subunits, which are essential for RPA to function in replication, are not yet known. RPA p34 is phosphorylated in a cell-cycle-dependent manner at the onset of S phase (25). Experiments have demonstrated that the p34 subunit of RPA can be phosphorylated by DNA-PK and cyclin-dependent kinase in cell extracts in vitro (26, 27). Similar “hyperphosphorylation” of RPA p34 has also been observed in extracts of cells following IR (28, 29), again implicating DNA-PK in the phosphorylation of RPA p34 following DNA damage.
We report that p53 levels are induced in both SCID and C.B-17 mouse embryo fibroblasts (MEFs), and that RPA p34 is hyperphosphorylated in the DNA-PKCS-deficient cell lines, SCID and MO59J, following IR in vivo. However, no kinase activity on an RPA substrate was found with SCID extracts in vitro. We also show that the hyperphosphorylation of RPA p34 in vivo correlates with a biological function, as it is concordant with a decrease in the ability of RPA to bind ssDNA in extracts from both SCID and C.B-17 cells. These results suggest that although DNA-PK appears capable of phosphorylating p53 and RPA in cell extracts in vitro, it might not be the only or the physiologically important kinase involved in phosphorylating these substrates in vivo in response to IR.
MATERIALS AND METHODS
Cell Lines.
SCID, C.B-17, MO59J, and MO59K cells were grown as described (7, 14). MEFs were isolated from mouse embryos at gestation days 16–18. MEFs were grown in Eagle’s medium with 10% fetal bovine serum, 2 mM glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml) for not more than three passages.
Immunoblot Analysis.
SCID, C.B-17, MO59J, and MO59K cells were grown in 100-mm2 dishes to 50–80% confluence and irradiated at either 6 Gy (for the p53 analysis) or 50 Gy (for RPA phosphorylation) in a cesium irradiator. Control cells (0 h time point) were mock-irradiated. Whole-cell extracts were prepared as described (7). Samples (50 μg total protein) were electrophoresed through a 15% (for RPA detection) or 10% (for p53 detection) denaturing SDS/polyacrylamide gel at 50 mA/gel. Proteins were then transferred to Hybond ECL nitrocellulose paper (Amersham) using a semidry apparatus (Bio-Rad) at a maximum of 250 mA/gel and 15 V for 1–1.5 h. After transfer, the gels (both RPA and p53) and the nitrocellulose membranes (p53) were stained with 0.15% Coomassie blue stain and Ponceau S solution (Bio-Rad), respectively, to ensure equal loading and transfer of the protein samples. Membranes were then blocked with TBS-T (20 mM Tris·HCl/ 137 mM NaCl/1% Tween-20/and either 2% BSA or 5% milk) and were incubated with either a p53-specific monoclonal antibody (Ab-1; Oncogene Science) or a polyclonal antibody specific for RPA p34 raised in rabbits against synthetic peptides of the conserved C terminus (SP509). Immunoreactive bands were visualized using the ECL system (Amersham). To verify the identity of the phosphorylated RPA forms protein extracts were treated with 100 units of λ phosphatase (New England Biolabs) for 0.5 h at 30°C before electrophoresis.
In Vitro Protein Kinase Assays.
Cell extracts were prepared as described (9) with the exception that 0.5 M NaCl was used to extract the isolated nuclei. Recombinant human RPA was expressed in Escherichia coli and purified by Affigel Blue (Bio-Rad) column chromatography as described (30). DNA-PKCS and Ku 70/80 was purified from HeLa cells by immunoaffinity chromatography using an anti-Ku 80 monoclonal antibody column. Briefly, HeLa cell nuclear extract was mixed for 16 h with 2 ml of anti-Ku 80 affinity matrix (2 mg IgG/ml) at 4°C. Weakly bound proteins were eluted sequentially with 10 ml of a buffer containing 25 mM Tris·HCl (pH 7.9) and 0.1 M, 0.2 M, or 0.5 M KCl. The DNA-PKCS eluted from the column at 0.2 M KCl and was further purified by gel filtration chromatography using a superdex 200 16/60 column (Pharmacia). The Ku 70/80 was eluted from the affinity matrix using 10 ml of 1.75 M MgCl2 in 50% ethylene glycol, 25 mM Tris·HCl (pH 7.9). The Ku complex was further purified by superdex 200 chromatography. DNA-agarose was prepared by coupling sheared salmon sperm DNA to CNBr activated Sepharose CL6-B (Pharmacia). To enrich for DNA-PK in the mouse cell extracts, 50 μl of the DNA-agarose beads (1:1 slurry in water) was mixed with 50 μl (100 μg) of C.B-17 or SCID cell extract and incubated for 30 min at 25°C. The DNA-beads were then washed twice with 1 ml of TM buffer (50 mM Tris·HCl, pH 7.9/12.5 mM MgCl2/1 mM EDTA/20% glycerol) containing 100 mM KCl and once with 0.5× concentrated TM buffer containing 50 mM KCl to remove weakly bound proteins. The washed DNA–protein complexes were then incubated for 60 min at 30°C with 12.5 μM ATP containing 5 μCi of [γ-32P]ATP (1 Ci = 37 GBq). As indicated in Fig. 2, 500 ng of recombinant RPA and purified DNA-PKCS was included in some of the reactions. The kinase reactions were terminated by boiling in SDS/PAGE sample buffer and the denatured proteins resolved by 10% SDS/PAGE.
Figure 2.
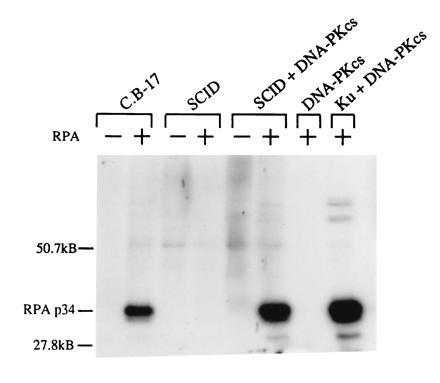
Kinase activity of DNA-PK on an RPA substrate in vitro. C.B-17, SCID, and SCID extracts (partially purified to enrich DNA-PK) were incubated with or without RPA, as indicated, and with [γ-32P]ATP. Proteins were resolved by 10% SDS/PAGE followed by autoradiography. Purified DNA-PKCS was analyzed alone and in conjunction with the Ku autoantigen as negative and positive controls, respectively.
Gel-Shift Assays.
A 17-bp DNA oligonucleotide (5′-GCC GCC GCC GCC GCC AT-3′) was end-labeled by T4 polynucleotide kinase using 5 pmol of [γ-32P]ATP and was purified by centrifugation through a size exclusion column (Bio-Rad). Cell extracts were prepared as described (C.K., S.L.G., R.F., and A.J.G., unpublished data). Binding reactions were performed for 15 min at 37°C with ≈30 fmol of radiolabeled probe and 5 μg of total cell protein as described by Seroussi and Lavi (31). Bound and free probe were separated on 10% native polyacrylamide gels in 0.25× TBE. Gels were dried and exposed to autoradiography film. The supershift was performed with a polyclonal antibody (α-RPA) to the trimeric RPA complex. As a control, recombinant human RPA was used.
RESULTS
DNA-PK extracted from SCID cells does not phosphorylate a p53 substrate in vitro (8). We investigated whether IR is capable of inducing increased p53 levels in SCID cells in vivo. p53 protein levels in MEFs were analyzed on immunoblots at different times after IR at a dose of 6 Gy. To confirm the identity of the p53 immunoreactive band, extracts from p53 −/− (knockout; no p53 protein is present) and +/+ mouse fibroblasts over expressing wild-type p53 were electrophoresed in adjacent lanes (Fig. 1, lanes 1 and 2). In C.B-17 MEFs, p53 protein levels increased after 1 h following treatment with IR and returned to control levels by 3 h. In SCID MEFs, p53 induction was also observed and appeared to be somewhat more intense, persisting at 3 h postirradiation. This apparently stronger response in SCID cells might be due to its defective ability to rejoin DNA double-strand breaks and therefore to the sustained presence of DNA damage, or to the fact that p53 levels in control untreated SCID MEFs were already higher compared with control C.B-17 MEFs (Fig. 1).
Figure 1.
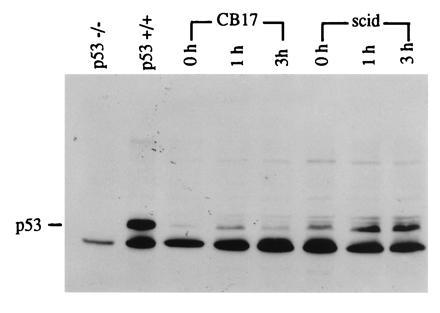
Effect of IR on p53 levels demonstrated by immunoblot analysis. C.B-17 and SCID mouse embryo fibroblasts were given a 6-Gy dose of IR. Crude protein extracts were obtained at the times indicated after irradiation. Proteins were separated by 10% SDS/PAGE. Blots were probed with a p53-specific monoclonal antibody (Ab-1) and immunoreactive bands were detected by ECL. As a negative control, protein extracts from p53 −/− (knockout) mice were used. As a positive control, extracts from p53 +/+ mouse fibroblasts were analyzed. In all samples, including the negative control, an unspecific band running just below p53 is observed.
Another DNA-PK substrate implicated in DNA repair and recombination is RPA (26, 27). Purified RPA was used as a substrate for a kinase assay to ascertain whether SCID fibroblasts have functional DNA-PK activity in vitro for this substrate (Fig. 2). Extracts from C.B-17 cells demonstrate functional kinase activity on an RPA substrate. However, the ability to phosphorylate RPA is absent in SCID extracts. Adding purified DNA-PKCS to SCID extract restores the kinase function in SCID cells on the RPA substrate, while DNA-PKcs by itself does not have kinase activity. As a positive control, purified DNA-PKcs was combined with the purified Ku subunits, resulting in kinase activity and RPA phosphorylation.
We next examined whether RPA p34 is a substrate in SCID fibroblasts in vivo. SCID cells and C.B-17 cells in log phase were irradiated with 50 Gy. Immunoblot analysis of crude extracts of the wild-type C.B-17 cells shows that there is an increase in hyperphosphorylation of RPA p34 by 1 h following IR compared with the unirradiated control (Fig. 3A Left). The hyperphosphorylation persists at 3 h postirradiation and returns to control levels by 6 h. In SCID cells, the same p34 hyperphosphorylation can also be observed at 1 h, but increases to a maximum at 3 h post-irradiation (Fig. 3A Left). RPA p34 hyperphosphorylation was reduced, but still apparent at 6 h postirradiation. The seemingly stronger hyerphosphorylation of RPA p34 in SCID cells was observed in repeat experiments. We do not know if this difference results from simple variation between cell lines or if this stronger response is inherent to the SCID cells. At 3 and 6 h postirradiation, the damage persistent in SCID cells is higher than in C.B-17 cells and, hence, might correlate with the observed further increase in RPA p34 hyperphosphorylation, again linking a stronger response of SCID cells to its DNA repair defect. The identity of the phosphorylated RPA forms in the protein extracts was verified by treatment with lambda phosphatase, which abolished the shifted RPA p34 bands (Fig. 3A Right).
Figure 3.
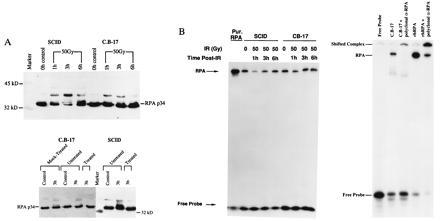
Effect of IR on RPA in SCID cells. C.B-17 and SCID fibroblasts were given a 50-Gy dose of IR. Crude protein extracts were obtained at the times indicated after irradiation. (A Upper) p34 hyperphosphorylation demonstrated by immunoblot analysis. Proteins were separated by 15% SDS/PAGE. Blots were probed with a polyclonal antibody specific for RPA p34 and immunoreactive bands were detected by ECL. (A Lower) Dephosphorylation of hyperphosphorylated RPA p34 in SCID and C.B-17 cells. Crude protein extracts were prepared from both control (unirradiated) SCID and C.B-17 fibroblasts and from cells 3 h after receiving a dose of 50-Gy IR. Control and irradiated extracts were untreated, mock-treated (incubated with 2 mM MnCl2 and λ protein phosphatase buffer), or treated [incubated with 2 mM MnCl2, λ protein phosphatase buffer, and λ protein phosphatase (New England Biolabs)]. Dephosphorylation was observed in both SCID and C.B-17 extracts with lambda protein phosphatase concentrations ranging from 100 to 600 units. (B Left) Binding of RPA p70 to ssDNA in vitro. A ssDNA binding assay was performed on the extracts using a radiolabeled 25-bp DNA oligonucleotide. Gel-shift analysis was performed on bound and free probe as described. (B Right) Gel-shift analysis of RPA binding to the radiolabeled probe. A polyclonal RPA antibody (polyclonal α-RPA) slows the mobility of the RPA–probe complex in C.B-17 cells as well as a recombinant human RPA (rhRPA)–probe complex.
As further evidence we sought to determine whether hyperphosphorylation correlates with the ability of RPA to bind to ssDNA. We assayed RPA ssDNA binding affinity before and after IR in SCID and C.B-17 cells. Fig. 3B shows the result of the gel shift analysis. Purified RPA binds to the radiolabeled single-stranded oligonucleotide and retards the mobility of this probe (lane 1). An antibody to RPA supershifts the DNA–protein complex (Fig. 3B Right). RPA binding to ssDNA decreases in extracts from irradiated cells 1 h after radiation, and mostly returns to control levels at 3 h and 6 h after radiation in both SCID and C.B-17 cells (Fig. 3B Left). These results demonstrate not only that p34 becomes hyperphosphorylated in response to IR in SCID cells, but also that RPA shows a biological response to radiation treatments that is similar, albeit quantitatively different, to that observed in C.B-17 cells.
Immunoblot analysis was also performed to study the effect of IR on RPA p34 hyperphosphorylation in another DNA-PKcs deficient cell line, MO59J, and in its wild-type parent, MO59K. In the MO59K extracts (Fig. 4 Lower), RPA p34 hyperphosphorylation is first visible at 1 h after irradiation. The hyperphosphorylation increases to a maximum at 3 h and remains present through 6 h postirradiation. In the MO59J extracts, RPA p34 hyperphosphorylation is seen by 3 h and increases by 6 h following IR (Fig. 4 Upper). These kinetics are slightly different than those seen with SCID cells; hyperphosphorylated RPA p34 is seen slightly later in MO59J cells. Because there is no DNA-PKcs mRNA and hence no protein in MO59J cells, it appears that a kinase other than DNA-PK is required for RPA phosphorylation in these cells.
Figure 4.
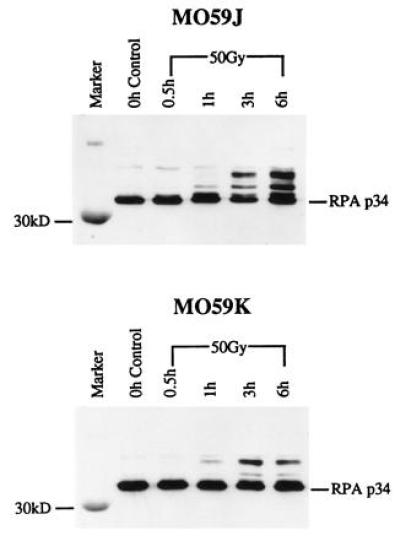
Effect of IR on RPA p34 hyperphosphorylation in MO59J cells demonstrated by immunoblot analysis. MO59K and MO59J human glioblastoma cells were given a 50-Gy dose of IR. Crude protein extracts were obtained at the times indicated after irradiation. Proteins were separated by 15% SDS/PAGE. Blots were probed with a polyclonal antibody specific for RPA p34 and immunoreactive bands were detected by ECL.
DISCUSSION
DNA-PK has been suggested as one of the central players in the cellular response to DNA damage (12). To investigate the physiological significance of mutant DNA-PKcs, we studied two potential substrates of this enzyme in intact SCID cells, p53 and RPA, both involved in the response of cells to DNA damage.
The increase in p53 protein levels observed following treatment with IR is thought to be, in part, a consequence of protein stabilization by posttranslational modification, most likely by phosphorylation (12, 18). However, no direct evidence exists supporting this mechanism for the accumulation of wild-type p53, and despite the existence of a number of kinases that are capable of phosphorylating p53 in vitro [e.g., DNA-PK (20, 21), casein kinase I and II (32, 33, 34), mitogen-activating protein kinase (35), and JNK1 (36)], no single kinase has been convincingly demonstrated to phosphorylate p53 in response to DNA damage in vivo. Since IR is an efficient producer of DNA double-strand breaks in cells, DNA-PK is a strong candidate for the phosphorylation and stabilization of p53 following DNA damage in vivo (12, 18). However, we found that wild-type p53 levels increase in both SCID and C.B-17 MEFs following irradiation, although we observed slight differences in the kinetics of induction in these two cell lines. Therefore, we would expect SCID MEFs also to display cell-cycle arrest following IR. Normal cell cycle checkpoints in SCID MEFs following DNA damage have recently been demonstrated by Huang et al. (37). Our result suggests that phosphorylation of p53 by DNA-PK is not the mechanism responsible for the induction of p53 levels following IR. It does not exclude the possibility that redundant pathways exist for p53 induction, and that in SCID cells an alternate pathway is recruited in the absence of functional DNA-PK.
Another substrate of DNA-PK which is phosphorylated in vitro is the p34 subunit of RPA. Our results indicate that RPA is hyperphosphorylated in SCID cells in vivo as well as in its wild-type parental cell line, C.B-17. Yet, SCID extracts show no kinase activity on an RPA substrate in vitro. The physiological significance of the hyperphosphorylation of RPA p34 is not clearly understood. RPA p34 has been hypothesized to be the coordinator of DNA repair and recombination with replication during the cell cycle. However, the experimental evidence varies depending on the assay system used and is not conclusive: extracts from UV-irradiated HeLa cells, which mostly contain hyperphosphorylated forms of RPA, were incapable of supporting simian virus 40 DNA replication (38), while experiments relying on phosphorylation of RPA p34 in vitro showed that replication and nucleotide excision repair were not influenced by the phosphorylation state of p34 (26, 39). We report here that RPA p34 hyperphosphorylation induced in vivo by IR is concordant with a decrease in binding of RPA to ssDNA in cell extracts of both SCID and CB-17. To our knowledge, this is the first time such a decrease in RPA binding following IR has been observed (C.K., S.L.G., R.F., and A.J.G., unpublished data). While the physiological significance of this decrease in binding for replication and repair is not yet understood, it was important for our study to extend the immunoblot analysis to a functional assay.
Since the exact nature of the SCID mutation is not known, we cannot exclude the possibility that there is some remaining DNA-PK activity in vivo which is lost in the purification and, hence, results in the observed lack of kinase activity in vitro. To eliminate this possibility, we used MO59J, which not only is devoid of DNA-PK activity in vitro, but does not express mRNA for the catalytic subunit. RPA p34 is hyperphosphorylated in MO59J cells as well as in the wild-type control cell line, MO59K, although the kinetics appear to be slower in the DNA-PKcs-deficient cells. This result suggests that RPA p34 can be phosphorylated by a kinase other than DNA-PK following IR. One candidate for such a kinase is the product of the AT gene, a gene mutated in patients with ataxia telangiectasia. AT cells show diminished and/or delayed responses of RPA hyperphosphorylation (29) and of p53 induction (40, 41) following IR. The AT-gene has recently been cloned (42) and shown to contain a kinase domain which shares homology with the phosphotidylinositol-3 (PI-3) kinase domain, a domain that is also present in DNA-PKcs (43). Although no substrates for the AT gene have been identified, it is believed that it functions as a protein kinase similar to DNA-PK, and not as a lipid kinase like PI-3 kinase. It is possible that the AT gene product and DNA-PK play roles in either parallel or overlapping pathways. Such a redundancy of mechanisms dealing with the lethal and mutagenic potential of DNA damage might ensure cell survival and genomic stability.
RPA hyperphosphorylation in SCID cells is unexpected given the lack of DNA-PK activity in extracts of SCID cells in vitro. Our results contradict a recent report claiming an apparent lack of hyperphosphorylation of RPA in SCID cells in vivo (44). We believe there are at least two reasons for this discrepancy. First, our studies employ a more direct approach of immunoblot analysis to observe hyperphosphorylation, whereas the study of Boubnov and Weaver (44) uses immunoprecipitations, which may sample only a small fraction of RPA in vivo and includes manipulations carried out in vitro. Second, our study compares the response to IR of SCID cells with the wild-type parental cells, C.B-17, whereas the experiment of Boubnov and Weaver compares SCID cells with SCID cells containing human chromosome 8, which contains human DNA-PKCS, following IR. Since human DNA-PKCS is expressed at much greater levels than mouse DNA-PKCS (7, 8, 9, 13), it is perhaps not surprising that the hyperphosphosphorylated RPA p34 can be observed in SCID/ch.8 cells earlier and stronger than in SCID cells. More importantly, this particular experiment of Boubnov and Weaver shows the presence of hyperphosphorylated p34 at a later time in SCID cells as well, suggesting a delayed phosphorylation kinetic in SCID instead of a complete absence as the authors claim.
It appears that the SCID phenotype does not stem from an inability to phosphorylate p53 or RPA, two proteins involved in the DNA damage response. Our results suggest that the physiological substrate for DNA-PK essential for DNA repair has not yet been identified and that redundant pathways may exist for p53 induction and RPA hyperphosphorylation in response to IR. As the defect in the SCID mouse appears to be restricted to the rejoining of DNA ends in both V(D)J recombination and DNA double-strand break repair, a phenotype more consistent with a defect in a structural protein than in a promiscuous regulatory protein, it is also possible that the SCID phenotype stems from a secondary function of DNA-PKcs.
Acknowledgments
We thank Jennifer Derr and Doug Menke for their technical assistance, Kerry Copeland for preparation of RPA antibodies, and Dr. Marc Wold for the gift of the RPA expression vector p11d-tRPA and recombinant human RPA. This work was supported, in part, by National Institutes of Health Grants CA15201 (J.M.B.), CA64489 (A.J.G.), CA50519 (D.J.C.), and CA56542 (R.F.); the U.S. Department of Energy (D.J.C.); American Cancer Society Junior Faculty Research award (A.J.G.); and Public Health Service Grant CA09302 (L.M.F.), awarded by the National Cancer Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: SCID, severe combined immunodeficient; RPA, replication protein A; DNA-PK, DNA-dependent protein kinase; IR, ionizing radiation; ssDNA, single-stranded DNA; MEF, mouse embryo fibroblast.
References
- 1.Fulop G M, Phillips R A. Nature (London) 1990;347:479–482. doi: 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- 2.Biedermann K A, Sun J R, Giaccia A J, Tosto L M, Brown J M. Proc Natl Acad Sci USA. 1991;88:1394–1397. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrickson E A, Qin X Q, Bump E A, Schatz D G, Oettinger M, Weaver D T. Proc Natl Acad Sci USA. 1991;88:4061–4065. doi: 10.1073/pnas.88.10.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malynn B A, Blackwell T K, Fulop G M, Rathbun G A, Furley A J, Ferrier P, Heinke L B, Phillips R A, Yancopoulos G D, Alt F W. Cell. 1988;54:453–460. doi: 10.1016/0092-8674(88)90066-9. [DOI] [PubMed] [Google Scholar]
- 5.Lieber M R, Hesse J E, Lewis S, Bosma G C, Rosenberg N, Mizuuchi K, Bosma M J, Gellert M. Cell. 1988;55:7–16. doi: 10.1016/0092-8674(88)90004-9. [DOI] [PubMed] [Google Scholar]
- 6.Hendrickson E A, Schatz D G, Weaver D T. Genes Dev. 1988;2:817–29. doi: 10.1101/gad.2.7.817. [DOI] [PubMed] [Google Scholar]
- 7.Kirchgessner C U, Patil C K, Evans J W, Cuomo C A, Fried L M, Carter T, Oettinger M A, Brown J M. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 8.Blunt T, Finnie N J, Taccioli G E, Smith G C, Demengeot J, Gottlieb T M, Mizuta R, Varghese A J, Alt F W, Jeggo P A, Jackson S P. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 9.Peterson S R, Kurimasa A, Oshimura M, Dynan W S, Bradbury E M, Chen D J. Proc Natl Acad Sci USA. 1995;92:3171–3174. doi: 10.1073/pnas.92.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb T M, Jackson S P. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 11.Dvir A, Stein L Y, Calore B L, Dynan W S. J Biol Chem. 1993;268:10440–10447. [PubMed] [Google Scholar]
- 12.Anderson C W. Trends Biochem Sci. 1993;18:433–437. doi: 10.1016/0968-0004(93)90144-c. [DOI] [PubMed] [Google Scholar]
- 13.Anderson C W, Lees-Miller S P. Crit Rev Eukaryotic Gene Expression. 1992;2:283–314. [PubMed] [Google Scholar]
- 14.Allalunis-Turner M J, Barron G M, Day R, III, Dobler K D, Mirzayans R. Radiat Res. 1993;134:349–354. [PubMed] [Google Scholar]
- 15.Lees-Miller S P, Godbout R, Chan D W, Weinfeld M, Day R, III, Barron G M, Allalunis-Turner M J. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 16.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 17.Hartwell L H, Kastan M B. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 18.Meek D W. Semin Cancer Biol. 1994;5:203–210. [PubMed] [Google Scholar]
- 19.Lees-Miller S P, Chen Y R, Anderson C W. Mol Cell Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lees-Miller S P, Sakaguchi K, Ullrich S J, Appella E, Anderson C W. Mol Cell Biol. 1992;12:5041–5049. doi: 10.1128/mcb.12.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Eckhart W. Proc Natl Acad Sci USA. 1992;89:4231–4235. doi: 10.1073/pnas.89.10.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stillman B. Annu Rev Cell Biol. 1989;5:197–245. doi: 10.1146/annurev.cb.05.110189.001213. [DOI] [PubMed] [Google Scholar]
- 23.Moore S P, Erdile L, Kelly T, Fishel R. Proc Natl Acad Sci USA. 1991;88:9067–9071. doi: 10.1073/pnas.88.20.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coverly D, Kenny M K, Lane D P, Wood R D. Nucleic Acids Res. 1992;20:3873–3880. doi: 10.1093/nar/20.15.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Din S, Brill S J, Fairman M P, Stillman B. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- 26.Brush G S, Anderson C W, Kelly T J. Proc Natl Acad Sci USA. 1994;91:12520–12524. doi: 10.1073/pnas.91.26.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan Z Q, Amin A A, Gibbs E, Niu H, Hurwitz J. Proc Natl Acad Sci USA. 1994;91:8343–8347. doi: 10.1073/pnas.91.18.8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fishel R, Kaplan D, Morrison D. J Cell Biochem. 1992;16B:102. (abstr.). [Google Scholar]
- 29.Liu V F, Weaver D T. Mol Cell Biol. 1993;13:7222–7231. doi: 10.1128/mcb.13.12.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henricksen L A, Wold M S. J Biol Chem. 1994;269:24203–24208. [PubMed] [Google Scholar]
- 31.Seroussi E, Lavi S. J Biol Chem. 1993;268:7147–7154. [PubMed] [Google Scholar]
- 32.Milne D M, Palmer R H, Meek D W. Nucleic Acids Res. 1992;20:5565–5570. doi: 10.1093/nar/20.21.5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milne D M, Palmer R H, Campbell D G, Meek D W. Oncogene. 1992;7:1361–1369. [PubMed] [Google Scholar]
- 34.Meek D W, Simon S, Kikkawa U, Eckhart W. EMBO J. 1990;9:3253–3260. doi: 10.1002/j.1460-2075.1990.tb07524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milne D M, Campbell D G, Caudwell F B, Meek D W. J Biol Chem. 1994;269:9253–9260. [PubMed] [Google Scholar]
- 36.Milne D M, Campbell L E, Campbell D G, Meek D W. J Biol Chem. 1995;270:5511–5518. doi: 10.1074/jbc.270.10.5511. [DOI] [PubMed] [Google Scholar]
- 37.Huang L, Clarkin K C, Wahl G M. Cancer Res. 1996;56:2940–2944. [PubMed] [Google Scholar]
- 38.Carty M P, Zernik K M, McGrath S, Dixon K. EMBO J. 1994;13:2114–2123. doi: 10.1002/j.1460-2075.1994.tb06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Z Q, Park C H, Amin A A, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 1995;92:4636–4640. doi: 10.1073/pnas.92.10.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kastan M B, Zhan Q, el-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 41.Khanna K K, Lavin M F. Oncogene. 1993;8:3307–3312. [PubMed] [Google Scholar]
- 42.Savitsky K, Bar S A, Gilad S, Rotman G, Ziv Y, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 43.Hartley K O, Gell D, Smith G C, Zhang H, Divecha N, Connelly M A, Admon A, Lees M S, Anderson C W, Jackson S P. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 44.Boubnov N V, Weaver D T. Mol Cell Biol. 1995;15:5700–5706. doi: 10.1128/mcb.15.10.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]