

Abstract
Numerous lines of evidence indicate that calcium signaling is essential for nerve growth factor (NGF)-directed neuronal cell differentiation. We report here that blocking production of the plasma membrane Ca2+-ATPase isoform 1 (PMCA1) in PC6 cells with antisense RNA impairs their ability to extend normal neurites in response to NGF. This result does not appear to be due to loss in NGF signaling as NGF-dependent tyrosine phosphorylation of erk1 and erk2, as well as expression of the NGF-inducible immediate early gene, NGFI-A, was observed in these cells. Resting cytosolic calcium levels did not appear to be altered in the antisense transfectants and release of calcium from internal bradykinin-sensitive calcium pools was unchanged. However, the rate of removal of free cytosolic calcium following this release was reduced in the antisense-transfected cells compared with controls. It is concluded that PMCA1 is involved in neurite extension and/or stabilization either through moderation of local calcium levels, or by some other mechanism.
The process of neurite extension, whether axonal or dendritic, is essential in forming synaptic connections. The fact that the divalent cation, Ca2+, is central to this process is now well established (1, 2, 3, 4, 5). However, the exact mechanisms underlying this phenomenon are not known. Calcium has been proposed to exert effects during neurite extension by altering the polymerization/depolymerization of microtubules (6) and actin fibers (4). Calcium also has been shown to be involved in regulating the expression of immediate early genes involved in neuronal function (7, 8). Therefore, proper regulation of cytosolic calcium in both the neuronal cell body and in the axonal or dendritic shaft is essential for proper neuritic growth.
One of the major proteins involved in regulating calcium levels in the cell is the plasma membrane Ca2+-ATPase (PMCA). In mammals, this is actually a family of enzymes encoded by four genes designated PMCA1-4. The primary RNA transcript from each gene can be alternately spliced to encode PMCA isoforms that differ primarily in their regulatory regions. To date, at least 25 different mRNAs encoding PMCAs have been reported in mammals (for review, see ref. 9) and each mRNA has a unique tissue distribution suggesting specific functions for each isoform of the pump (10, 11). PMCA1b and 4b are present in most, if not all, mammalian cell types. Other isoforms derived from these genes by alternative splicing, as well as multiple products from the PMCA2 and 3 genes, are expressed predominately in excitable tissues, suggesting that they may play essential, specific roles in these physiological settings. This is further suggested by the observation that the expression of specific isoforms is highly regulated during brain (12) and muscle (13) development.
The traditional approach for determining the specific physiological functions of different forms of ion translocases [including Ca2+-channels, the Na+/K+-ATPase, and the sarcol endoplasmic reticulum Ca2+ ATPase (SERCA) family of calcium pumps] has been to use specific pharmacological inhibitors of their activities. Unfortunately, no well-characterized, truly specific inhibitors of the plasma membrane calcium pumps have been identified to date. Therefore, we have utilized genetic manipulation of expression in the pheochromocytoma cell line PC6 to gain insight into possible specific physiological roles of PMCA1 isoforms in neuronal function. PC6 cells, a subline of PC-12, produce and release neurotransmitters and undergo extensive neurite formation in response to growth factors including nerve growth factor (NGF). We demonstrate here that blockade of PMCA1 expression with antisense methods impairs NGF-dependent neurite extension in PC6 cells without affecting downstream biochemical markers of NGF response, gross cell morphology, and growth or resting cytosolic calcium levels.
MATERIALS AND METHODS
General Methods. Cell culture.
The cell line used in these experiments was the pheochromocytoma cell line PC6. PC6 is a subline of PC-12 cells which produces neurites in response to NGF but grows as well-isolated cells rather than in clumps. The PC6 line used in these studies was the parental line described by Pittman et al. (14) (the generous gift of Kate Ivins, University of California, Irvine). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) containing 10% horse serum, 5% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (GIBCO/BRL). Cells were grown in humidified 5% CO2/95% air on standard plastic tissue culture dishes (Falcon) with no additional substrate. After stably transfected cell lines were established (vida infra), all experiments were performed without G418 (GIBCO/BRL) in the media. However, cell lines were tested periodically for continued resistance to G418.
RNA extraction.
RNA was extracted from cells at ≈80% confluence. Cultures were washed twice with ice-cold Dulbecco’s modified PBS (DPBS; Sigma) then scraped into 1 ml ice-cold DPBS and centrifuged at 12,000 × g for 30 sec at 4°C. The RNA in the cell pellets was isolated using a micro-isolation system from GIBCO/BRL.
Specific Methods. Establishment of PC6 cell lines stably transfected with sense and antisense mRNA producing constructs.
PC6 cell lines that stably expressed sense or antisense RNA derived from a 446-nt region of the 5′ end of the human PMCA1 mRNA were established using the constructs shown in Fig. 1A. The 5′ end of hPMCA1 was obtained by using reverse transcriptase-coupled PCR (RT-PCR) performed on human fibroblast (WI-38) RNA. The oligonucleotide primers used in the RT-PCR were as follows: oligonucleotide 1, 5′-ATTCTTTCCAAACACTGCTTCTCT-3′; and oligonucleotide 2, 5′-AACCGCGGCCAAAGGTCAAGATACTTCTCTG-3′. PCR conditions were as follows: 94°C, 1 min; 60°C, 2 min; 72°C, 3 min for 40 cycles. The PCR product was ligated in the SmaI site of pUC18 and sequenced to verify integrity and determine orientation in the vector. The PMCA1 5′ end was then cloned as a KpnI/XbaI fragment behind the Rouse sarcoma virus (RSV) promoter of the plasmid pRSV9 (for antisense RNA production) or pRSV14 (for sense RNA). A third plasmid which contained the G418 resistance gene behind the RSV promoter (pRSVneo) was cotransfected with either the sense or the antisense construct. Cells were transfected using Lipofectin (GIBCO/BRL) following the manufacturer’s suggested protocol. Two micrograms of pRSVneo and 16 μg of the pRSV9–PMCA1-5′E or pRSV14–PMCA1-5′E were cotransfected into PC6 cells plated at ≈70% confluence in a 10-cm plate. Drug resistant cells were selected with 800 μg/ml G418.
Figure 1.
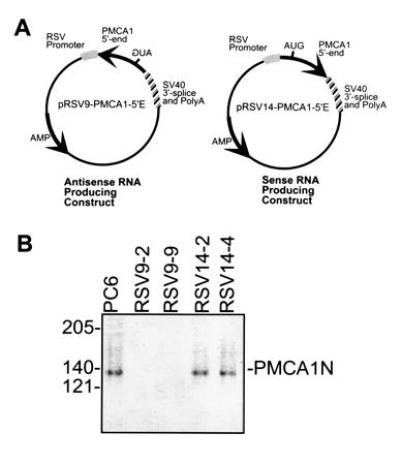
(A) Maps of plasmids and PCR primers used in this report. Arrows point in the direction of transcription of the wild-type PMCA1 gene. The approximate position of the initiation codon is indicated by “AUG,” which is at position 182 in the human PMCA1 mRNA. The fragment contains 181 bp of 5′-untranslated sequence and 265 bp of coding sequence. (B) Western blot of PMCA1 isoforms in PC6 cells and transfectants. Partially purified calmodulin-binding proteins from membrane preparations of PC6 cells and transfectants expressing PMCA1 antisense RNA (RSV9-2, RSV9-9) or sense RNA (RSV14-2, RSV14-4) was resolved on a 7.5% SDS/PAGE and transferred to an Immobilon membrane. The blot was probed with an antibody that recognizes all PMCA1 isoforms (designated PMCA1N; ref. 15).
G418-resistant cells were assayed for RSV promoter-directed synthesis of PMCA1 sense and antisense RNAs. RT-PCR using oligonucleotide 1 or 2 with a primer within transcribed RSV sequences (oligonucleotide R, 5′-ATTGGACGAACCACTGAATTC-3′) allowed detection of specific sense or antisense RNAs as described in Results. The PCR conditions for these experiments were as follows: 94°C, 1 min; 60°C, 1 min; 72°C, 1 min for 35 cycles.
Immunoassay of PMCA1 content of transfected cells.
PMCA1 protein content was determined by using a polyclonal antibody raised against the amino terminus of human PMCA1 and which reacts with rat PMCA1 isoforms (ref. 15; the generous gift of Ernesto Carafoli).
Cell extracts were prepared for PMCA1 determinations as follows. Eight 15-cm plates of each cell line (≈80% confluence) were washed twice with PBS, cells were collected in 5 ml PBS, and pelleted by centrifugation at 500 × g at 4°C for 5 min. Twenty times the cell pellet volume of 10 mM Tris (pH 7.8) containing 1 μg/ml leupeptin and pepstatin A, 1 mM phenylmethylsulfonyl fluoride and 0.1 trypsin inhibitory unit aprotinin (all from Sigma) was added to each pellet and the samples were incubated for 15 min on ice. The samples were then Dounce homogenized on ice, and membranes were collected by centrifugation at 100,000 × g for 1 hr.
The membrane pellets were each suspended in a buffer containing 20 mM Tris (pH 7.8), 130 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 0.4% Triton X-100, and protease inhibitors and the detergent solubilized fraction was recovered after centrifugation at 1000 × g at 4°C for 5 min. Protein content of this soluble fraction was determined and calmodulin-binding proteins, which include PMCAs, from 18 mg of total membrane protein from each sample were isolated by chromatography on calmodulin-agarose essentially as described (16). The EGTA eluants were loaded on a 7.5% SDS/polyacrylamide gel and immunoblotted with anti-PMCA1 antiserum diluted 1:1000 followed by alkaline phosphatase-coupled goat anti-rabbit IgG and color development with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate (BCIP) (17).
Immunoassay of total PMCA content of transfected cells.
Total PMCA protein content was determined by using a polyclonal antibody raised against a synthetic peptide based on the PMCA calmodulin binding domain identified by James et al. (18). The peptide sequence chosen was ELRRGQILWFRGLNR, which is conserved in all species and isoforms of the PMCA examined thus far. The peptide was coupled to keyhole limpet hemocyanin at a 3:1 mass ratio in 0.1% glutaraldehyde as described previously (17) and injected subcutaneously into New Zealand White rabbits as described (19). Peptide-specific antibodies were purified by immunoaffinity chromatography on peptide-Sepharose prepared by coupling the peptide to cyanogen bromide-activated Sepharose 4B (19).
Cell extracts were prepared for PMCA determinations as follows. One 10-cm plate of each cell line (≈80% confluence) was washed twice with ice-cold PBS, then cells were collected by scraping into 1 ml PBS followed by centrifugation at 12,000 × g at 4°C for 5 min. To each cell pellet was added 0.5 ml of 10 mM Tris, 10 mM EDTA (pH 7.5) containing 1% SDS, and the samples were sonicated on ice for 30 s. One-half microgram of protein from each sample was spotted onto a nitrocellulose filter. The filter was blocked in PBS containing 5% (wt/vol) milk powder and 0.1% (vol/vol) Triton X-100 for 1 hr. Fifty micrograms of affinity-purified primary antibody was added to 10 ml of blocking solution which was then incubated with the filter for 2 hr. The filter was washed four times with PBS, then 106 dpm of 125I-labeled donkey anti-rabbit IgG secondary antibody (Amersham) in 10 ml blocking solution was added, and incubation performed for 2 hr at room temperature. The filter was washed four times in PBS, air dried, and placed on x-ray film overnight. After development of the film, the corresponding spots on the nitrocellulose filter were cut out and 125I quantified in a γ counter. The amount of PMCA in the samples was determined by comparison with a standard curve prepared by using known amounts of purified human erythrocyte PMCA processed in the same manner and at the same time as the experimental samples.
Measurement of Ca2+ removal from transfected PC6 cells.
Free cytosolic calcium was measured by using cells scrape-loaded with aequorin as the calcium indicator. All protocols were as described by Newcomb et al. (20), expect that cells were not serum starved, but instead were incubated overnight in DMEM (without phenol red) containing 10% horse serum and 5% fetal bovine serum after plating in the Sykes–Moore chamber. Studies of removal of cytosolic calcium from bradykinin-induced internal release were conducted in calcium-free medium to prevent capacitative calcium entry, thereby allowing only the removal kinetics to be observed. After recovering overnight from the scrape-loading procedure, the cells were placed in a luminometer, washed with serum- and calcium-free DMEM then treated with 100 nM bradykinin in serum- and calcium-free DMEM.
NGF responsiveness of stably transfected PC6 cell lines.
The ability of sense or antisense PMCA1 RNA expressing cells to extend neurites was assessed in the presence of 100 ng/ml 2.5S murine NGF (Promega). Medium and NGF were changed every 3 days. Six days after NGF addition, random fields of cells were photographed, and neurite lengths were determined. Neurite lengths were measured in cell body diameters of the cell from which the neurite was derived. When a neurite was branched, only the longest branch was scored. Processes were only considered neurites if they were greater than two cell bodies in length and had a well-defined growth cone as described by Wu and Bradshaw (21).
Northern blot detection of NGFI-A mRNA.
Cells were treated with 100 ng/ml 2.5S NGF for 1 hr at 37°C, and RNA was harvested as described under General Methods. Ten micrograms of total cellular RNA was resolved on a 1% agarose gel in formaldehyde-Mops buffer (22). The RNA was transferred to a nitrocellulose membrane (Micron Separations Inc., Westboro, MA) by capillary action and fixed by baking at 80°C in vacuo (22). The blot was blocked and hybridized at 42°C overnight with a cDNA probe to NGFI-A [kindly provided by Ralph Bradshaw (University of California, Irvine) and by Thomas Soderling (Oregon Health Sciences University)] that had been randomly labeled with [32P]dCTP. The hybridized blot was washed two times for 5min at room temperature in 2× standard saline citrate (SSC)/0.1% SDS and once in 1× SSC/0.1% SDS at 42°C for 15 min then placed on XAR-5 x-ray film (Kodak) at −70°C with two intensifying screens for 18 hr.
Determination of tyrosine phosphorylated proteins in NGF-treated and untreated cells.
Cells were grown to 70–80% confluence in 10-cm culture dishes with DMEM containing 5% fetal bovine serum and 10% horse serum. They were then placed in DMEM containing 1% horse serum for 24 hr at which point NGF was added to a final concentration of 100 ng/ml. An equal volume of vehicle (PBS) alone was used for controls. Cells were then incubated for 5 min at 37°C and harvested. The medium was removed, the cells washed twice with room temperature PBS, and lysed in RIPA buffer (ref. 17; 0.15 mM NaCl/0.05 mM Tris·HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS) containing 1 mM sodium orthovanadate (Sigma), 1 mM phenylmethylsulfonyl fluoride, and 30 trypsin inhibitary units of aprotinin. The protein concentration of each sample was determined by the Bradford dye binding method (23) and 100 μg of protein was resolved on a 10% SDS/polyacrylamide gel and electroblotted to a nitrocellulose membrane. The immunoblot was incubated with the antiphosphotyrosine antibody, RC20, directly coupled to horseradish peroxidase (Transduction Laboratories, Lexington, KY) and phosphotyrosine containing proteins were detected by enhanced chemilumenescence (Amersham). The blot was stripped of primary antibody by incubation in 0.1 M 2-mercaptoethanol, 2% SDS, and 62.5 mM Tris (pH 6.7) at 50°C for 30 min. After stripping, the blot was reprobed with an antibody specific for erk1 and erk2 coupled to horseradish peroxidase (MK12; Transduction Laboratories).
RESULTS
Characterization of PMCA Expression in Wild-Type, Sense, and Antisense Transfected PC6 Cells.
The sense and antisense plasmids shown in Fig. 1A produced RNAs that contained the first 446 nt of the human PMCA1 mRNA and a simian virus 40 3′-untranslated region that included a polyadenylylation signal and an RNA splice site. This region of PMCA1 has no homology with the mammalian PMCA isoforms PMCA2, 3, and 4. However, the human and rat PMCA1 sequences are 94% identical over this region. The PMCA isoforms expressed in undifferentiated PC6 cells are PMCA1b, 4b, and some form of PMCA2 which has not been clearly established (unpublished observation).
Cells cotransfected with a plasmid containing the G418-resistance gene (pRSVneo) and with either the sense or antisense RNA producing constructs were selected for stable integration by resistance to G418. Two G418-resistant clones from the putative antisense (RSV9-2 and RSV9-9) and two from the sense (RSV14-2 and RSV14-4) transfectants were chosen for more thorough characterization. RT-PCR analysis showed that these four cell lines expressed the appropriate antisense or sense RNA transcribed from the RSV promoter of the transfected plasmid (data not shown).
The PMCA1 contents of the wild type, sense and antisense transfected PC6 cells were determined by specific immunoblotting assays. As shown in Fig. 1B, PMCA1 was undetectable with a specific anti-PMCA1 antibody in an immunoblot of extracts from PMCA1 antisense expressing cell lines (RSV9-2 and RSV9-9). However, identically prepared samples of PC6 and sense controls (RSV14-2 and RSV14-4) yielded bands of comparable intensities in the immunoblot at the expected molecular weight of ≈135 kDa.
Total PMCA content in cells was determined using a polyclonal antibody raised against a synthetic peptide corresponding to the calmodulin-binding domain which is identical in all isoforms of the PMCA examined to date. Total PMCA content was reduced in the two PMCA1 antisense cell lines (RSV9-2 and RSV9-9) by about 37% relative to the average of the control cell lines. The remaining 63% of the PMCA was presumably due to the PMCA2 and 4 isoforms expressed in these cells since PMCA1 was not detectable.
In vivo functional assays provided further evidence for detectable reduction of PMCA content in the antisense transfected PC6 cells. Fig. 2 shows assays of calcium fluxes in control and antisense transfected PC6 cells by aequorin bioluminescence. As can be seen in Fig 2 Upper, addition of bradykinin (at 80 sec), which causes release of calcium from internal stores via an IP3-dependent mechanism, led to a rapid rise in the cytosolic-free calcium which was indistinguishable in both rate of increase and amplitude in all cell lines. Removal of free calcium was also very rapid following peak response. However, while the sense-transfected (RSV14-2) and wild-type (PC6) cells showed identical removal rates, calcium removal was detectably slower in the antisense transfectant (RSV9-2). This is better illustrated by the plot of pCa as a function of time taken from the descending portions of the flux curves (Fig. 2 Lower). The apparent rates of calcium removal for the wild-type (•) and sense-transfected (▴) PC6 cells were estimated to be 4.0 μM/sec from these plots as compared with 2.8 μM/sec for the antisense transfected cells.
Figure 2.
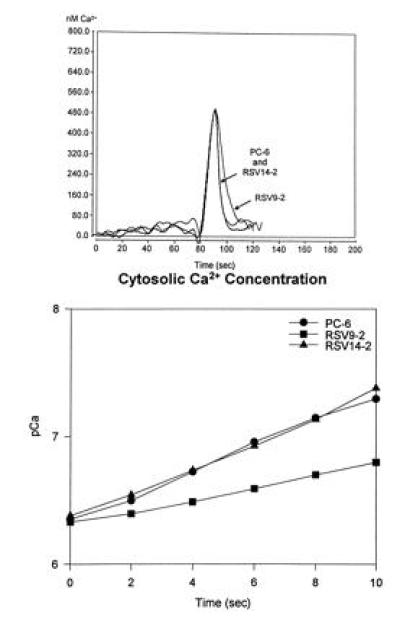
Bradykinin-induced calcium flux measurements in wild-type and transfected PC6 cells. Cells were scrape-loaded with aequorin and allowed to attach overnight to the surface of the Sykes–Moore chambers. The cells were then placed in a luminometer and changed into calcium- and serum-free medium; bradykinin (100 nM) was applied and the luminescence was monitored. (Upper) The calcium transients measured for each cell line. (Lower) A semilog plot of the rate of Ca2+ removal for each of the cell lines between 90 and 100 sec).
Effects of PMCA1 Antisense RNA on NGF-Induced Neurite Extension.
Owing to evidence linking neurite proliferation and calcium fluxes, we decided to examine NGF-dependent neurite outgrowth in control and PMCA1 antisense-transfected PC6 cells. The standard definition of a neurite in responding pheochromocytoma cells is a process of two cell body diameters in length or longer and possessing a well-defined growth cone (21). No gross differences were noted in the growth characteristics or morphology of the undifferentiated antisense and control cell lines and none produced significant neuritic processes as shown in Fig. 3A Left (−NGF). After treatment with NGF for 6 days (Fig. 3A Right, +NGF), the parental PC6 and sense transfected cell lines (RSV14-2, RSV14-4) produced numerous processes clearly identifiable as neurites by their length and the presence of growth cones. In contrast, the antisense transfectants, RSV9-2 and RSV9-9, produced far fewer processes that could be clearly identified as neurites. The histograms in Fig. 3B show the population of neurite lengths observed for these cell lines with or without NGF treatment. The antisense transfectant RSV9-2 produced almost no neurites. The other antisense transfectant, RSV9-9, did appear to produce some neurite-like processes in response to NGF. However, most of these were less than two cell diameters in length. The actual photograph of these cells (Fig. 3A) reveals that many of these processes were abnormal in shape and lacked defined growth cones. In contrast, the majority of the processes produced by control cell lines had growth cones and were of lengths greater than 2 cell diameters. Indeed, a significant number were greater than 9 cell diameters—far longer than any processes produced by the antisense cell lines.
Figure 3.
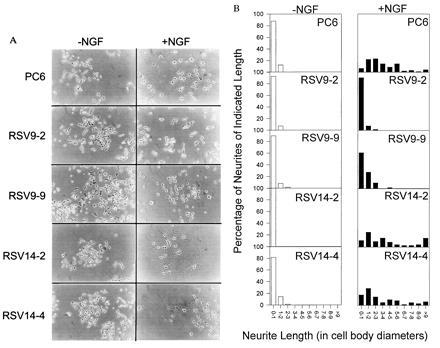
(A) NGF responsiveness of PC6 cells. Two PMCA1 antisense RNA expressing transfectants (RSV9-2, RSV9-9) and two PMCA1 sense RNA expressing control transfectants (RSV14-2, RSV14-4) were plated on standard tissue culture plastic and allowed to attach overnight. After attachment, fresh medium containing 100 ng/ml 2.5S murine NGF was added and the cells were allowed to continue in culture. The medium, with fresh NGF, was changed every 3 days. Cells were photographed on day 6. (B) Quantitation of neurite length in PMCA1 antisense expressing cells and controls. the length of neurites on cells in A was determined. Length was expressed in cell body diameters of the cell to which the neurite was attached. Length was only measured for neurites that remained in the frame of the picture and whose attachment to a cell could be clearly seen. For branched neurites only the longest branch was scored.
If one uses the dual criteria of at least 2 cell diameters in length and presence of defined growth cones to identify neurites, the percentage of cells producing neurites in response to NGF for each cell line was as follows: RSV9-2, 3.4%; RSV9-9, 17.2%; and RSV14-2, RSV14-4, and PC6, all 100%. Clearly, by these measures, the PMCA1 antisense transfected PC6 cells have an impaired ability to produce and/or extend neurites in response to NGF.
Determination of NGF Receptor Functionality.
A number of criteria were used to demonstrate that the NGF signaling pathway was intact in the PMCA1 antisense transfected cell lines used here. Initially, NGF-dependent tyrosine phosphorylation of proteins in the NGF signaling pathway was examined. Stimulation of NGF receptor has been shown to lead to phosphorylation of the 42-kDa erk-1 and 40-kDa erk-2 proteins (24). Western blots of NGF-stimulated or unstimulated cells probed with an antiphosphotyrosine antibody, showed that induction of phosphorylation of two proteins with these relative molecular weights (Fig. 4A; arrowheads) occurred in antisense cell lines (Upper) in response to NGF. Reprobing of the blot with specific antibodies showed these bands to be erk1 and erk2 (Lower).
Figure 4.
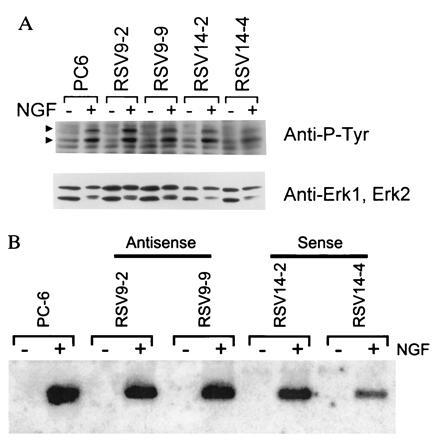
(A) NGF-mediated protein tyrosine phosphorylation. Cells were treated for 5 min with 100 ng/ml 2.5S murine NGF or vehicle (PBS), lysed, resolved on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with an antiphosphotyrosine antibody (RC20). The NGF-induced P-Tyr containing bands indicated by arrowheads correspond to erk1 (upper arrowhead) and erk2 (lower arrowhead) based on relative molecular weights and reprobing with erk1- and 2-specific antibodies (Lower). (B) NGF-mediated NGFI-A gene induction. Cells were treated with 100 ng/ml 2.5S murine NGF or vehicle (PBS) for 1 hr. The RNA was harvested, resolved on a formaldehyde agarose gel, blotted to a nitrocellulose membrane, and probed with a radiolabeled NGFI-A cDNA. An apparent decrease in material going from left to right on the autoradiogram is actually due to uneven transfer of the gel as can be judged by a concomitant decrease in background signal. The efficiency of transfer does not affect the conclusions drawn from the data.
To further assess NGF receptor function, we assayed induction of transcription of the immediate early gene NGFI-A (25). Fig. 4B shows a Northern blot of NGFI-A mRNA from the two antisense RNA producing cell lines, the two sense control lines and wild type PC6 cells. The production of NGFI-A mRNA was still inducible to approximately the same levels with NGF in all cell lines, demonstrating that events many steps downstream from NGF binding to its receptor still function in the antisense cell lines. In addition, this latter point is significant since expression of this gene can be controlled by calcium-dependent enzymes acting through CREB (7, 8), and there was no background expression in the absence of NGF in the antisense transfectants. Thus, NGFI-A transcription could be stimulated by elevated basal calcium levels resulting from the lack of PMCA1.
DISCUSSION
Calcium may act through any of several different pathways to effect the process of neurite extension. It has been shown to be involved in the expression of the NGF-induced immediate early genes NGFI-A, NGFI-B, and c-fos through stimulation of Ca2+/Celmodulin-dependent (CaM) kinases and calcineurin which affect the phosphorylation state of cAMP response element binding protein (CREB) (7, 8). Further, induction of CREB-dependent transcription apparently can be controlled solely by changes in calcium entering at the plasma membrane (8). Neurite extension (postinitiation) is also regulated by calcium through the actions of the cell adhesion molecules L1 (5), neural cell adhesion molecule (NCAM), and N-cadherin (3). These three cell adhesion molecules are associated with L- and N-type calcium channels that open when they bind homomeric molecules either on artificial substrates or on other cells (3, 5). The calcium influx associated with these cell adhesion molecules is essential for neurite extension based on the fact that buffering of cytosolic calcium with the calcium chelator, 1,2 bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA), removal of extracellular calcium or antagonism of L- and N-type calcium channels all result in a loss of neurite growth (3, 5). Calcium also affects the stability of microtubules in developed neurites since elevation of calcium concentrations by addition of hydrogen peroxide (26) or direct application of the calcium ionophore A23187 (4) leads to microtubule disassembly and neurite collapse. Calcium levels also affect lamellepodial and filopodial movement in hippocampal neurons, presumably by affecting microfilament polymerization/depolymerization (4).
Here we report that expression of the PMCA1 isoform of the PMCAs is also involved in normal neurite extension. Whether the loss of neurite extension in cells lacking PMCA1 results directly from its absence or indirectly by some unknown mechanism, it is clear that early events in NGF-mediated differentiation are not detectably altered. Fig. 4A shows that NGF-dependence of tyrosine phosphorylation of the second messenger associated proteins erk1 and erk2 (24) is the same in antisense and control cell lines. Additional downstream events required for NGF-dependent transcription of the NGF-inducible immediate early gene, NGFI-A, are also intact (Fig. 4B). Resting calcium levels do not appear to be drastically altered in cells when PMCA1 expression is blocked (Fig. 2), nor are there any obvious morphological changes (Fig. 3A) or changes in growth rate (data not shown).
PC6 cells in which PMCA1 expression is blocked do not appear to attach as efficiently to substrates as control cell lines (P.C.B., J.E.S., and T.C.V., unpublished data). Previous studies have indicated that integrins are directly involved in attachment of pheochromocytoma cells and are required for developing processes to the surrounding extracellular matrix (27, 28). PC12 cells, the parental line from which PC6 was derived, use two major integrins dimers, α1,β1 and α3,β1 (29). α1,β1 integrin binds primarily to collagens and α3,β1 integrins are involved in binding to laminin (27). Turner et al. (28) have shown that α1 integrin is required for NGF-dependent extension of neurites. Preliminary studies in our laboratory indicate that expression of α1 integrin is greatly reduced in PMCA1 antisense cells (P.C.B., J.E.S., and T.C.V., unpublished data). Therefore, loss of neurite extension associated with loss of PMCA1 may well be due to the decreased attachment efficiency of cells to the extracellular matrix.
Marks et al. (30) have shown that attachment to the extracellular matrix proteins, fibronectin or vitronectin, make neutrophils more sensitive to cytosolic calcium concentrations. They concluded that calcium transients are necessary for modulating affinity of integrins for their substrates and loss of these calcium transients prevents release of the cell from the substrate. This conclusion is consistent with recent work showing that the calcium-binding protein, calreticulin, stabilizes the high affinity state by binding to the KXGFFKR sequence of α integrins (31). PMCA1 could be involved in formation of these calcium transients.
The role of PM Ca2+-ATPase in neurite production seen here may reflect processes underlying formation of preprogrammed neural circuits in development. The production of afferents from one brain region to another is temporally and spatially programmed. Also, directed axonal movement in development is dependent on contact with specific cells along a path (guidepost cells). The growth cone–guidepost cell interactions are mediated by several classes of molecules (NCAM, N-cadherins, L1, and integrins) that are coupled to Ca2+ channels which allow calcium entry when correct coupling with a corresponding molecule on another cell is made (3, 5). We have previously shown the expression of specific isoforms of the PMCAs are developmentally regulated in different regions of the rat brain (12) and in myocytes (13). It is interesting to speculate that the expression of specific PMCA isoforms at programmed times may provide components necessary for regulation of movement and/or specific neurite proliferation during development of the nervous system.
Acknowledgments
We thank Ralph Bradshaw, Thomas Soderling, Jon Ivins, Kate Ivins, Tavner Delcamp, Simona Raffioni, Roland Mullins, and Molly Peck for many helpful discussions and Charlotte Randall for technical assistance. We thank Ralph Bradshaw and Thomas Soderling for the gifts of NGFI-A cDNA probes and Ernesto Carafoli for the gift of PMCA1 antibodies. This work was supported by National Institutes of Health Grant NS21868 (T.C.V.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: NGF, nerve growth factor; RT-PCR, reverse transcriptase-coupled PCR; PMCA, plasma membrane Ca2+-ATPase; RSV, Rouse sarcoma virus.
References
- 1.Anglister L, Farber I C, Shahar A, Grinvald A. Dev Biol. 1982;94:351–365. doi: 10.1016/0012-1606(82)90353-0. [DOI] [PubMed] [Google Scholar]
- 2.Streit J, Lux H D. J Neurosci. 1989;9:4190–4199. doi: 10.1523/JNEUROSCI.09-12-04190.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doherty P, Ashton S V, Moore S E, Walsh F S. Neuron. 1991;6:247–258. doi: 10.1016/0896-6273(91)90360-c. [DOI] [PubMed] [Google Scholar]
- 4.Mattson M P. Exp Gerontol. 1992;27:29–49. doi: 10.1016/0531-5565(92)90027-w. [DOI] [PubMed] [Google Scholar]
- 5.Williams E J, Doherty P, Turner G, Reid R A, Hemperly J J, Walsh F S. J Cell Biol. 1992;119:883–892. doi: 10.1083/jcb.119.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon F, Magendantz M. J Cell Biol. 1981;89:157–161. doi: 10.1083/jcb.89.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enslen H, Soderling T R. J Biol Chem. 1994;269:20872–20877. [PubMed] [Google Scholar]
- 8.Deisseroth K, Bito H, Tsien R W. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 9.Carafoli E, Guerini D. Trends Cardiovasc Med. 1993;3:177–184. doi: 10.1016/1050-1738(93)90003-O. [DOI] [PubMed] [Google Scholar]
- 10.Brandt P, Neve R L, Kammesheidt A, Rhoads R E, Vanaman T C. J Biol Chem. 1992;267:4376–4385. [PubMed] [Google Scholar]
- 11.Keeton T P, Burk S E, Shull G E. J Biol Chem. 1993;268:2740–2748. [PubMed] [Google Scholar]
- 12.Brandt P, Neve R L. J Neurochem. 1992;59:1566–1569. doi: 10.1111/j.1471-4159.1992.tb08476.x. [DOI] [PubMed] [Google Scholar]
- 13.Brandt P, Vanaman T C. J Neurochem. 1994;62:799–802. doi: 10.1046/j.1471-4159.1994.62020799.x. [DOI] [PubMed] [Google Scholar]
- 14.Pittman R N, Wang S, DiBenedetto A J, Mills J C. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauffer T P, Guerini D, Carafoli E. J Biol Chem. 1995;270:12184–12190. doi: 10.1074/jbc.270.20.12184. [DOI] [PubMed] [Google Scholar]
- 16.Niggli V, Penniston J, Carafoli E. J Biol Chem. 1979;254:9955–9958. [PubMed] [Google Scholar]
- 17.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 59–78. [Google Scholar]
- 18.James P, Maeda M, Fischer R, Verma A K, Krebs J, Penniston J T, Carafoli E. J Biol Chem. 1989;263:2905–2910. [PubMed] [Google Scholar]
- 19.Brandt P, Zurini M, Neve R L, Rhoads R E, Vanaman T C. Proc Natl Acad Sci USA. 1988;85:2914–2918. doi: 10.1073/pnas.85.9.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newcomb T G, Mullins R D, Sisken J E. Cell Calcium. 1993;14:539–549. doi: 10.1016/0143-4160(93)90075-h. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y Y, Bradshaw R A. J Cell Physiol. 1995;164:522–532. doi: 10.1002/jcp.1041640310. [DOI] [PubMed] [Google Scholar]
- 22.Ausubel F M. In: Current Protocols in Molecular Biology. Ausubel F M, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. New York: Wiley; 1987. pp. 4.9.1–4.9.12. [Google Scholar]
- 23.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen T T, Scimeca J-C, Filloux C, Peraldi P, Carpentier J-L, Van Obberghen E. J Biol Chem. 1993;268:9803–9810. [PubMed] [Google Scholar]
- 25.Milbrandt J. Science. 1987;238:797–799. doi: 10.1126/science.3672127. [DOI] [PubMed] [Google Scholar]
- 26.Hinshaw D B, Miller M T, Omann G M, Beals T F, Hyslop P A. Brain Res. 1993;615:13–26. doi: 10.1016/0006-8993(93)91110-e. [DOI] [PubMed] [Google Scholar]
- 27.Reichardt L F, Tomaselli K J. Annu Rev Neurosci. 1991;14:531–570. doi: 10.1146/annurev.ne.14.030191.002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner D C, Flier L A, Carbonetto S. J Neurosci. 1989;9:3287–3296. doi: 10.1523/JNEUROSCI.09-09-03287.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arregui C O, Carbonetto S, McKerracher L. J Neurosci. 1994;14:6967–6977. doi: 10.1523/JNEUROSCI.14-11-06967.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marks P W, Hendey B, Maxfield F R. J Cell Biol. 1991;112:149–158. doi: 10.1083/jcb.112.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppolino M, Leung-Hagesteijn C, Dedhar S, Wilkins J. J Biol Chem. 1995;270:23132–23138. doi: 10.1074/jbc.270.39.23132. [DOI] [PubMed] [Google Scholar]