

Abstract
The mitogen-activated protein kinase cascade of the Saccharomyces cerevisiae pheromone response pathway is organized on the Ste5 protein, which binds each of the kinases of the cascade prior to signaling. In this study, a structure–function analysis of Ste5 deletion mutants uncovered new functional domains of the Ste5 protein and revealed that Ste5 dimerizes during the course of normal signal transduction. Dimerization, mediated by two regions in the N-terminal half of Ste5, was first suggested by intragenic complementation between pairs of nonfunctional Ste5 mutants and was confirmed by using the two-hybrid system. Coimmunoprecipitation of differently tagged forms of Ste5 from cells in which the pathway has been activated by Ste5 overexpression further confirmed dimerization. A precise correlation between the biological activity of various Ste5 fragments and dimerization suggests that dimerization is essential for Ste5 function.
Mitogen-activated protein (MAP) kinase (MAPK) cascades, consisting of three evolutionarily conserved protein kinases operating in sequence, mediate responses to a wide variety of extracellular signals (1, 2). A well-defined MAPK cascade, consisting of the Ste11 (MAPKKK), Ste7 (MAPKK), and Fus3 or Kss1 (MAPK) kinases, is central to the pheromone response pathway in haploid cells of the yeast Saccharomyces cerevisiae (3, 4, 5). These kinases bind to the Ste5 protein (6, 7, 8), an essential element of the pathway, which acts as a scaffold to organize interactions between the kinases and prevent cross-talk between this cascade and other yeast MAPK modules (9). STE5 encodes a protein of 917 amino acids, with an acidic C terminus, a cysteine-rich N-terminal domain similar to a Lim-type zinc finger, and a small region of homology to Far1 (5, 10, 11, 12). While homologues of Ste5 have not yet been isolated, its essential role in the mating pathway and its demonstrated interactions with the evolutionarily conserved MAPK cascade suggest that Ste5 may have general relevance to signal transduction via these cascades.
We undertook to identify the essential regions of Ste5 by testing a series of deletion mutants for biological activity. The unexpected results of these experiments suggested that Ste5 acts as a dimer to transmit the pheromone signal. Dimerization was confirmed by using the two-hybrid system and by co-immunoprecipitation. Dimerization may be part of the mechanism by which Ste5 organizes and regulates signal transduction through its associated MAP kinase cascade.
MATERIALS AND METHODS
Yeast Strains.
Strains are described in Table 1. SMβ-2 is the progeny of a cross between strains βWT (isogenic to group I strains) and SM1414 (10). Genomic disruptions of mating pathway genes were created by standard procedures (14, 15) and were confirmed by testing for sterility and for complementation by the relevant wild-type gene. ste5 disruptions were created by using plasmids p2-1KS-C::HIS3 (10) and pKS-5bl. pKS-5bl contains the XbaI–XhoI fragment of STE5 in pBluescript KS (−) (Stratagene), with the 3.8-kb BglII-BamHI hisG-URA3-hisG fragment (15) replacing the KpnI–BglII fragment of STE5. Other disruptions were created by using the following plasmids: pBSIIste11::ADE2 and pUC119ste7::ADE2 for ste11::ADE2 and ste7::ADE2 (7); pJB225 for fus3::HIS3 (16); and p4PUC::HIS3 for ste4::HIS3 (10). The GAL1-lacZ reporter gene was inserted at the URA3 locus by using pRY121-2 μ− (13). Yeast transformations were done by a modification of the lithium acetate procedure (17).
Table 1.
Yeast strains
| Group | Strain | Genotype | Source/ref. |
|---|---|---|---|
| I | β5Δ | MATa ste5::HIS3 ura3-52 lys2 leu2 trp1-289 met− GAL+ | 10 |
| β5bl | MATa ste5::hisG ura3-52 lys2 leu2 trp1 his3Δ-200 met− GAL+ | This work | |
| II | SMβ-2 | MATa ste5-1 leu2 his3 ura3 trp1 met− | This work |
| SMβ2-5Δ | MATa ste5::HIS3 leu2 ura3 trp1 met− | This work | |
| III | SFY526 | MATa URA3::GAL1-lacZ his3-200 ade2-101 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | 13 |
| YM5d-121 | MATa ste5::hisG URA3::GAL1-lacZ his3-200 ade2-101 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | This work | |
| YM5d11a-121 | MATa ste5::hisG ste11::ADE2 URA3::GAL1-lacZ his3-200 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | This work | |
| YM5d7a-121 | MATa ste5::hisG ste7::ADE2 URA3::GAL1-lacZ his3-200 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | This work | |
| YM5d3h-121 | MATa ste5::hisG fus3::HIS3 URA3::GAL1-lacZ ade2-101 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | This work | |
| SFY 526-4Δ | MATa ste4::HIS3 URA3::GAL1-lacZ his3-200 ade2-101 lys2-801 trp1-901 leu2-3,112 can1 gal4-52 gal80-538 | This work | |
| IV | 310 | MATα lys1 | Lab stock |
Roman numerals indicate groups of isogenic strains.
Mating Assays.
Mating was assayed by a standard plate mating assay (18). To test for complementation of ste5ts, patches representing independent transformants were replica-plated to fresh SD plates (18), which were incubated at 23°C. Plates were shifted to the mating temperature (23 or 34°C) for 2 hr, replica plated to YPD plates preheated to the mating temperature, overlaid with a lawn of 310 cells, and allowed to mate overnight at the same temperature. The mixture was then replica-plated to SD without amino acids and incubated at 30°C to select for diploids.
Correction of the STE5 Sequence.
Errors in the STE5 sequence as previously reported (GenBank accession no. L01620L01620) (10) were corrected at position 1557, where the sequence ACCCTGGC was revised to ACCCCTGGGGC, and at position 1586, where the sequence CAACTCTATCCT was revised to CAAACTCTATCT, making the predicted open reading frame of 917 amino acids identical to that reported by others (accession nos. L07865L07865 and L23856L23856). Throughout this paper, numbering of Ste5 residues is according to the corrected sequence.
Ste5 Deletion Mutant Plasmids.
In-frame deletions of the Ste5 open reading frame were created in p2-1PN (10), using available restriction sites. The mutant products created were as follows: SteΔT25-R138, the KpnI site was ligated to the AvaI site with the oligomer TCGGGTAC; Ste5ΔS176-C239, the BsmI sites were ligated together with the oligomers TCTAGATG and TCTAGAAG; Ste5ΔC239-R335, the NsiI site was ligated to the EcoRI site with the oligomer AATTCTGCA; Ste5ΔG199-S505, the MscI site was ligated to the EcoRV site; Ste5ΔA468-L637, the HindIII–HindIII fragment was removed; Ste5ΔV586-D746, the HincII–HincII fragment (2328–2805) was removed; Ste5ΔN696-I858, the SspI–SspI fragment (2658–3141) was removed; Ste5ΔV586-I858, the HincII–SspI fragment (2328–3141) was removed; Ste5-M1-L754, the SpeI–NheI fragment was removed; Ste5-M1-V586, the SalI–XhoI fragment was removed; Ste5ΔT25-R138ΔV586-I858 contains both of the deletions from Ste5ΔT25-R138 and Ste5ΔV586-I858.
Two-Hybrid Plasmids.
The DNA-binding domain (DBD) and activation domain (AD) plasmids are derivatives of pMA424 (19) and pGAD2F (20), respectively. To express DBD fusion proteins with the TRP1 selectible marker, the EcoRV–EcoRV fragment of pMA424, including the ADH1 promoter, the Gal4 DBD, cloning sites, and the ADH1 terminator, was inserted into the PvuII sites of pRS424 (21). A unique BamHI site was created at the Ste5 T25 codon or the R138 codon by inserting the oligomer GGATCCGTAC into the KpnI site of p2-1PN or p2-1PNΔKpn-Ava, yielding p2-1PNK>B or p2-1PNΔKAK>B, respectively. For DBD fusion plasmids, The BamHI–SalI (encoding T25-D587) or BamHI–XhoI (T25-S900) fragments of p2-1PNK>B, or the BamHI-SalI fragment of p2-1PNΔKAK>B (R138-D587) or the BamHI-SalI fragment of p2-1PNKB>ΔBSM (T25-D587ΔS176-236) was inserted into the BamHI and SalI sites of pMA424, and the EcoRI–SalI fragment of Ste5 (I336-D587) was cloned into the blunted EcoRI and SalI sites of pMA424. Residues T25-V586, T25-I917, and T25-I504 were fused to the Gal4 AD by cloning the BamHI–HincII, BamHI-SmaI, or BamHI–EcoRV fragments of p2-1PNK>B into the BamHI and PvuII sites of pGAD2F, and the BamHI–BglII fragment (T25-D485) was inserted into the BamHI site of pGAD2F.
Myc- and Glutathione S-Transferase (GST)-Tagged Expression Vectors.
Expression vectors pGA2013 and pGA1913, respectively encoding Myc-tagged Ste5 and Ste11 under the control of the GAL1/10 promoter, were provided by G. Ammerer (Institut für Biochemie und Molekulare Zellbiologie der Universität Wien). To create Gst-Ste5, the BamHI–SmaI fragment of p2-1PNK>B was inserted into the BglII site and the filled-in EcoRI site of pGEX2K-L (modified from pGEX2TK (Pharmacia)), creating pGEX2KL-Ste5, and the EcoNI (filled-in)–NheI fragment of pGEX2KL-Ste5 was inserted into the EcoRV and XbaI sites of YcpIF7 (22) to make pF7G5, encoding GST-Ste5 (residues T25-I917) under the control of the GAL1 promoter.
ste5ts.
ste5-1 (ste5ts) was cloned by gap repair (14) of p2-1PN, following digestion of this plasmid with KpnI and XhoI and transformation into strain SM1414 (10). Plasmids repaired by recombination with genomic ste5-1 were recovered (23), and the XbaI–NheI ste5-1 fragment was inserted into pRS414 (24).
Immunoprecipitations.
Cells were grown in raffinose, induced with galactose for 3.5 hr, and collected as described (25). Lysis was by mixing with glass beads in 3 pellet volumes of NLB buffer [50 mM Tris·HCl, pH 7.4/50 mM NaCl/10% (vol/vol) glycerol/1 mM dithiothreitol/0.1% Triton X-100/10 mM NaF/50 mM β-glycerophosphate/5 μM zinc acetate (omitted where indicated)], supplemented with protease inhibitors: 10 μg/ml aprotinin, 1 mM benzamidine, 1 mM phenylmethanesulfonyl fluoride (PMSF), 5 μg/ml pepstatin A, and 25 μg/ml leupeptin. Lysates were cleared twice by centrifugation (15,000 × g) for 10 min at 4°C and stored at −70°C. Antibody was prebound for 1 hr at 4°C to protein G beads (Pharmacia) in modified NLB (without NaCl and glycerol). Lysates were diluted in ice-cold modified NLB supplemented with 1% BSA and protease inhibitors, were adjusted to a final NaCl concentration of 15 mM, and then cleared by centrifugation at 4°C (15,000 × g) for 10 min. The supernatant was added to a suspension of the antibody-bound protein G beads and was incubated for 2 hr at 4°C with end-over-end mixing. The beads were washed four times with cold modified NLB, resuspended in 30 μl of SDS/PAGE sample buffer, and boiled for 5 min prior to electrophoresis.
Antibodies.
Anti-Myc monoclonal antibody 9E10 was purchased from Babco (Emeryville, CA). Affinity-purified anti-Ste5 (residues I336–A468) rabbit antibodies were prepared by using standard procedures (26). Control monoclonal antibodies against insulin-like growth factor 1 receptor (IGF-1R) (27) were prepared by standard procedures.
β-Galactosidase Assay.
Cells were grown in selective medium to OD600 of 0.4–0.8 and were lysed by Vortex mixing with acid-washed glass beads in 0.1 M Hepes, pH 8/20% glycerol/1 mM dithiothreitol/2 mM phenylmethanesulfonyl fluoride. β-Galactosidase activity was assayed as described (28). Accumulation of product was measured as A405, and results are expressed as nmol of o-nitrophenol produced per min per mg of protein.
RESULTS
A series of Ste5 deletion mutants was tested for biological activity, as measured by complementation of mating of isogenic strains carrying either a genomic disruption (ste5Δ) or a temperature-sensitive (ste5ts) allele of STE5 (Fig. 1A and B). Some of the mutants failed to complement mating in either strain, indicating a loss of function. The Ste5ΔT25-R138 mutant rescued mating in both strains, identifying an N-terminal region dispensable for Ste5 function. While a point mutation in this region results in an activated allele (29), the T25–R138 deletion is not an activating mutation, as cells carrying this allele showed low basal activity of a FUS1::lacZ reporter gene (0.18 unit) and normal activation by α-factor (1.2 units), as compared with cells carrying full-length Ste5 (0.64 unit basal activity and 2.0 units upon stimulation with α-factor). As Ste5 fusion proteins beginning at T25 also complement ste5Δ (ref. 6 and data not shown), these results indicate that the first 137 residues of Ste5 are not required for mating.
Figure 1.
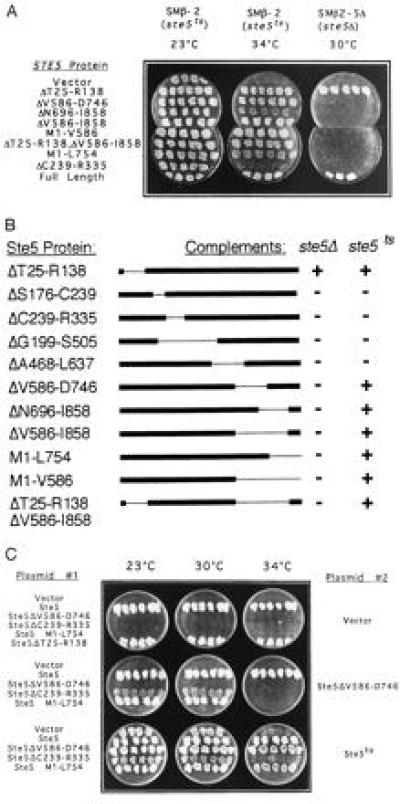
Complementation of ste5Δ and ste5ts by STE5 deletion mutants. (A) Mating assay of strains SMβ-2 and SMβ2-5Δ carrying representative STE5 deletion mutants on a 2 μ plasmid. Mating was assayed by a standard plate mating assay. Products of the mating reaction were replica plated to minimal media to select for diploids, shown here. (B) Results of mating assays of all of the Ste5 deletion mutants. Assays were performed as in A. (C) Mating assay of the ste5Δ strain β5Δ carrying pairs of ste5 mutant alleles. Group 1 plasmids are a subset of the plasmids assayed in A and B. Group 2 plasmids are based on the centromeric vector pRS414 (24) into which the indicated STE5 alleles were inserted.
Unexpectedly, mutants that have undergone C-terminal deletions beginning at V586 or further downstream complemented mating of the ste5ts strain, but not the ste5Δ strain (Fig. 1 A and B). Affinity-purified anti-Ste5 antibodies directed at residues 336–468 of Ste5 were used to verify expression of each of these mutants in the ste5Δ background (data not shown). In addition to mating assays, the inactivity of the C-terminal mutants in the ste5Δ background was verified by assaying for typical responses to α-mating pheromone: cell cycle arrest, morphological changes, and FUS1::lacZ transcriptional activation. In all assays ste5Δ cells expressing Ste5ΔV586-D746 were completely unresponsive (data not shown).
Rescue of ste5ts by mutant alleles which are intrinsically inactive suggests that two defective alleles of STE5 can cooperate to activate the pheromone response pathway. Notably, this phenomenon, which initially suggested the possibility of Ste5 dimerization, was observed in the course of a biological assay of the normal physiological function of Ste5: mating. Furthermore, the Ste5 mutant alleles were transcribed from the Ste5 promoter and only mildly overexpressed from a multicopy plasmid. To further verify that Ste5 dimerization occurs under physiological conditions, we expressed Ste5ΔV586-D746 from a centromeric plasmid, and we found it to complement ste5ts equally well (data not shown).
To establish the generality of this phenomenon, we searched for additional pairs of ste5 mutants that complement each other. These were identified by expressing Ste5ΔV586-D746 from a centromeric plasmid in a ste5Δ strain together with each of the deletion mutants shown in Fig. 1B. Two of the mutants, Ste5ΔC239-R335 and Ste5-M1-L754, complemented Ste5ΔV586-D746, though neither mutant complements ste5Δ (Fig. 1C). Complementation of Ste5ΔV586-D746 was more prominent at lower temperatures (Fig. 1C), suggesting that the protein–protein interactions required for signal transduction may be destabilized by the deletion mutations, especially at higher temperatures.
Each of the pairs of mutants found to complement ste5Δ was tested for α-factor-induced transcription of a FUS1::lacZ reporter gene. In all cases, pairs of mutants that complement mating of ste5Δ induced FUS1::lacZ, while each mutant alone was inactive (data not shown). Since induction of FUS1 transcription is an immediate consequence of activation of the pheromone signal transduction pathway (30), intragenic complementation between pairs of ste5 mutants must occur at the level of signal transduction.
Intragenic complementation between intrinsically inactive ste5 mutants, occurring at the level of signal transduction, strongly suggests that Ste5 functions as a dimer. Since Ste5 is a scaffold for the MAP kinase cascade (6, 7, 8), deletion mutants unable to bind one of the elements of the cascade are expected to be unable to transmit the signal. However, dimerization of two different mutants, each of which binds some but not all members of the cascade, can reconstruct a functional signal transduction module. Indeed, in the complementing pair of Ste5ΔC239-R335 and Ste5ΔV586-D746, Fig. 2A shows that the first mutant precisely lacks the domain required to bind Fus3/Kss1, while the second mutant may be defective in Ste7 binding, which requires residues C-terminal to V586 (6). Thus the second step of the kinase cascade, in which Ste7 phosphorylates Fus3 (or Kss1), can take place only in the context of a dimer.
Figure 2.

Summary of Ste5 domains. (A) The region of Ste5 required for mating and the Ste5 dimerization domain as defined in this work. For comparison, location of the Lim-like domain, the putative leucine zipper, and the Far1 homology domain are indicated, as well as the binding domains of Ste5, determined by others (6, 31). The Ste7 binding domain is poorly defined. The C-terminal region (thick line) is necessary for Ste7 binding but may not be sufficient, since the entire region shown (thick and thin lines) is the smallest fragment that bound Ste7 (6). (B) Sequence of the Ste5 LIM-like domain. The LIM consensus sequence is shown (32) as well as a LIM-like consensus derived from a comparison of Ste5, Far1, Pep5, and PS43 (12). (C) Sequence of the Ste5 putative leucine zipper. Characteristic hydrophobic residues at positions a and d of each heptad are indicated (33).
This interpretation suggests that each of the complementing mutants should contain the dimerization domain, which must therefore be located within Ste5 residues 138–239 and/or 335–586 (Fig. 2A). The first of these regions includes a LIM-like domain (Fig. 2B), a type of double zinc finger (32), known to mediate protein–protein interactions and to form dimers (34, 35). The second region includes a domain with the characteristic spacing of hydrophobic residues found in leucine zippers (Fig. 2C), another motif involved in dimerization (33).
To test the dimerization hypothesis and further identify the domains involved, we tested for Ste5–Ste5 interactions by using the two-hybrid system (36). Fusion proteins containing Ste5 N-terminal residues T25–V586 or R138–D587 dimerized in this system (Table 2). Further shortening of the Ste5 fragment from either the N terminus or the C terminus prevented dimerization (Table 2). Thus, the shortest continuous fragment found to dimerize in the two-hybrid system precisely overlaps the two dimerization domains predicted by the genetic data (Fig. 2A).
Table 2.
Domain analysis of Ste5–Ste5 two-hybrid interactions







| Strain | DBD-Ste5 protein | AD-Ste5
protein
|
|||
|---|---|---|---|---|---|
| AD alone | T25–V586 * | T25–D485 † | T25–I504 † | ||
| ste5Δ | DBD alone | 0.01 | 0.01 | 0.02 | 0.38 |
| * | T25–D587 | 4.2 | 43.8 | 4.4 | 4.1 |
| * | R138–D587 | 7.2 | 20.2 | ||
| † | I336–D587 | 0.24 | 0.16 | ||
| † | T25–D587 | 0.65 | 14.5 | ||
| Δ177–239 | |||||
Two-hybrid interactions were analyzed in isogenic strains carrying the plasmids shown. Cells were lysed and β-galactosidase activity was assayed; results are expressed as nmol of o-nitrophenol produced per min per mg of protein. For each strain, a mixture of five independent transformants was assayed twice with similar results, and a representative experiment is shown. Constructs that rescued mating of the ste5ts strain SMβ-2 at 34°C are marked by ∗. Those that did not are marked by †.
Both of the predicted dimerization domains are required for efficient dimerization in this system. Deletion of as few as 82 amino acids from the predicted C-terminal dimerization domain abolishes dimerization; however, this domain alone is not sufficient (compare T25–V586 to T25–I504 and I336–D587 in Table 2). By contrast, deletion of the Lim-like domain (residues 177–239) only slightly reduces the level of dimerization, suggesting that the contribution of the N-terminal dimerization domain may be by means of residues 138–176.
To assess the biological relevance of dimerization, each of the Ste5 fusion proteins assayed in the two-hybrid system was tested for the ability to rescue mating of the ste5ts strain SMβ-2 (Table 2). As noted before, N-terminal residues 1–137 were found to be dispensable for both mating and dimerization. By contrast, deletion of residues 505–586 prevented both dimerization and rescue of mating ste5ts. Indeed, each of the constructs that rescued mating of ste5ts also dimerized, strongly suggesting that dimerization of Ste5 is required for its activity in the mating pathway.
Repetition of the two-hybrid experiments in strains disrupted for each of the kinases of the MAPK cascade or for the G-protein β-subunit, Ste4 showed the Ste5–Ste5 interaction to be independent of Ste11, Ste7, Fus3, and Ste4 (Table 3). These findings suggest that the Ste5–Ste5 interaction is direct and not mediated by one of the other proteins known to bind to Ste5. Indeed, the ste7 disruption revealed an interaction between two nearly full-length Ste5 fusion proteins not detected in the wild-type background. The construct T25–D587 fails to dimerize with T25–I917 in any of the systems, while the background is very high (Table 3). We currently have no explanation of this finding.
Table 3.
Ste5 two-hybrid interactions in STE+ and ste− strains











| Strain | DBD-Ste5 protein | AD-Ste5
protein
|
||
|---|---|---|---|---|
| AD alone | T25–V586 | T25–I917 | ||
| STE+ | DBD alone | 0 | 0.1 | 0.2 |
| T25–D587 | 3.8 | 79.0 | 4.1 | |
| T25–S900 | 0.1 | 8.6 | 0.1 | |
| ste5Δ ste11Δ | ||||
| T25–D587 | 4.9 | 43.8 | 5.7 | |
| T25–S900 | 1.0 | 5.2 | 0.8 | |
| ste5Δ ste7Δ | ||||
| DBD alone | 0.08 | 0.1 | 0.15 | |
| T25–D587 | 5.0 | 38.3 | 1.1 | |
| T25–S900 | 0.01 | 1.2 | 5.7 | |
| ste5Δ fus3Δ | ||||
| *T25–D587 | 2.5 | 11.3 | 2.8 | |
| ste5Δ | ||||
| *T25–D587 | 2.8 | 25.6 | 5.7 | |
| STE+ste4Δ | ||||
| *T25–D587 | 3.1 | 14.8 | 6.1 | |
See legend of Table 2. ∗ indicates use of DBD plasmids that encode fusion proteins identical to the pMA424 series but carry a different selectable marker.
Dimerization was further demonstrated by immunoprecipitation. Two forms of Ste5, one bearing a Myc epitope tag (Ste5M) and the other a GST-Ste5 fusion protein, were expressed in the same cell under the control of the inducible GAL1/10 promoter. Fig. 3A shows that both forms of Ste5 are efficiently expressed and can be distinguished by their molecular weight. Immunoprecipitation of Ste5M with anti-Myc monoclonal antibodies resulted in coimmunoprecipitation of a fraction of the GST-Ste5 co-expressed in the same cell (Fig. 3B). This represents a specific interaction between the two Ste5 proteins, since it was dependent on the presence of the Ste5M protein and on the anti-Myc antiserum (Fig. 3B). The Ste5 dimers could be disrupted by incubation with EGTA or with the transition metal chelator 1,10-phenanthroline (Fig. 3C), supporting involvement of the Lim-like zinc finger in Ste5 dimerization in this system. This result contrasts with our findings in the two-hybrid system and may therefore reflect a general change in the Ste5 conformation caused by removing zinc from the Lim-like structure.
Figure 3.
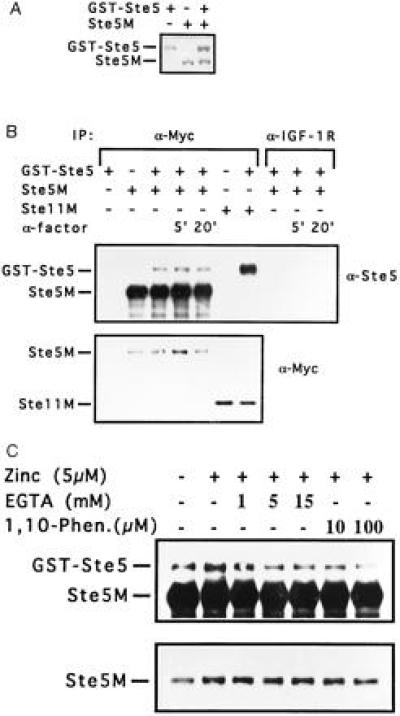
Coimmunoprecipitation of GST-Ste5 with Ste5M and with Ste11M. GST-Ste5, Ste5M, and Ste11M were expressed under control of the GAL1/10 promoter in a ste5Δ strain. Where indicated, 10 μg/ml α-factor was added for the last 5 or 20 min of growth. Cells were lysed as described in the text. (A) A 17-μg sample of each lysate was separated by SDS/PAGE (7% acrylamide) and blotted with affinity-purified anti-Ste5 antibodies. (B) A 300-μg sample of each lysate was immunoprecipitated with anti-Myc monoclonal antibody or with anti-IGF-1R monoclonal antibody in a final volume of 220 μl. Immune complexes were separated by electrophoresis on a 4–10% gradient gel and blotted. Ste5M and GST-Ste5 were detected with anti-Ste5 antibodies (Upper). The blot was erased and reprobed with anti-Myc antibodies to detect Ste5M and Ste11M (Lower). (C) Cells were lysed in NLB buffer without (left lane) or with (six right-hand lanes) 5 μM zinc acetate. A 300-μg sample of each lysate was immunoprecipitated as in B, except that prior to the addition of anti-Myc-bound protein G beads, lysates were preincubated at 4°C for 1 hr in the presence of chelators at the concentrations indicated. Immunoprecipitation was in a final volume of 132 μl. Ste5M and GST-Ste5 were detected with anti-Ste5 antibodies (Upper). A shorter exposure allows comparison of the amount of Ste5M in each lane (Lower).
The level of dimerization was not significantly affected by treatment of the cells with α-factor. Small fluctuations in the amount of GST-Ste5 coprecipitated (Fig. 3B Upper) match the fluctuations in the amount of Ste5M directly precipitated (Fig. 3B Lower). However, the high level of Ste5 expression achieved by overexpression under the GAL1/10 promoter is sufficient to induce activation of the mating pathway, as revealed by morphological changes of the cells. Therefore, this system cannot easily distinguish between constitutive dimerization and pheromone-induced dimerization.
The Ste5M–Ste5GST interaction is considerably weaker than a Ste11M–Ste5GST interaction which can also be detected in this system (Fig. 3B). This may be due to precipitation of mostly Ste5M–Ste5M dimers, which may be more stable than Ste5M–Ste5GST heterodimers. However, it is possible that the low level of dimerization detected reflects the normal in vivo situation. Support for this view may be found in the observation that disruption of STE7 was necessary to reveal interactions between full-length Ste5 molecules in the two-hybrid system, suggesting that Ste5 dimerization is normally a transient occurrence, which may be trapped by blocking the signal transduction pathway in certain ways.
DISCUSSION
Taken together, the experiments presented in this paper suggest a model in which Ste5 dimerizes during the course of signal transduction. Three independent sets of data support this conclusion: intragenic complementation between pairs of nonfunctional STE5 mutants, Ste5–Ste5 interactions in the two-hybrid system, and coimmunoprecipitation of two differently tagged forms of Ste5.
The observations suggest that dimerization is a transient occurrence, resulting in a weak signal in the coimmunoprecipitation assay. By contrast, dimerization is clearly revealed in genetic complementation assays as well as in the two-hybrid system. Both these systems have the advantage of high sensitivity for detection of transient interactions. Furthermore, the genetic assay reveals interactions which occur under physiological conditions during the course of a normal mating reaction. The striking correlation between the ability of various Ste5 fragments to functionally cooperate with the ste5ts-encoded protein and their ability to dimerize suggests that dimerization is essential for the function of Ste5.
The structure–function analysis of Ste5 revealed three previously unidentified domains. The T25–R138 deletion identified an N-terminal domain which is dispensable for Ste5 function but may have a regulatory role, as an activating mutation has been mapped to this domain (29). This finding implies that the Ste4 binding domain, recently identified in residues 1–214 of Ste5 (31), may actually be located within the smaller fragment of residues 138–214. The second domain revealed is the dimerization domain, which is predicted to be in residues 138–176 and/or 335–586 of Ste5. The two-hybrid system revealed that both of these domains are required for efficient dimerization. Finally, analysis of the complementing pair of mutants, Ste5 M1-L754 and Ste5ΔV586-D746, suggests the presence of an additional functional domain in the Ste5 C terminus. While both mutants are expected to bind Ste11 and Fus3/Kss1, the region required for Ste7 binding is known only to include residues C-terminal to 586. Since the two deletions span this region, at least one of the mutants must be defective in binding Ste7, while the other mutant must bind Ste7 to allow reconstitution of the MAPK cascade. This second mutant must be defective in yet another function of the Ste5 C terminus, such as binding to Ste20 or Bem1 (37). This complementing pair therefore defines a functional region of Ste5 located in its C terminus.
By analogy with the epidermal growth factor receptor, one might suggest that dimerization of Ste5 is necessary to facilitate activation of the kinases of the cascade by means of transphosphorylation. However, further characterization of the three-dimensional structure of Ste5 and the stoichiometry of the complexes it forms will be necessary to determine whether each of the kinases of the cascade binds simultaneously to one molecule of Ste5, or whether structural constraints dictate that a complete cascade can be formed only in the context of a Ste5 dimer.
The observation that disruption of STE7 allows interaction between full-length Ste5 molecules in the two-hybrid system (Table 3) suggests that Ste5 dimerization weakens Ste7 binding. Thus, it is possible that during the transient Ste5 dimerization Ste7 is ejected from the signal transduction complex, perhaps together with Fus3 to which it is tightly bound (6, 38) to phosphorylate downstream substrates. This leaves the remaining Ste5-Ste11 unit to interact with new Ste7 and Fus3 molecules, thereby amplifying the signal. The possible catalytic role of Ste5 may explain its low level of expression. A more definitive test of this speculative model awaits analysis of mutants defective in dimerization but not in other functions of Ste5.
Acknowledgments
We thank G. Ammerer and J. Balog (Institut für Biochemie und Molekulare Zellbiologie der Universität Wien) for their helpful correspondence and for providing plasmids pGA2013 and pGA1913. We gratefully acknowledge receipt of strain SFY526 and plasmid pRY121-2 μ− from S. Fields (State University of New York at Stony Brook) plasmids pBSIIste11::ADE2 and pUC119ste7::ADE2 from M. Wigler (Cold Spring Harbor Laboratories, Cold Spring Harbor, NY), and plasmid pJB225 from J. Brill and G. Fink (Whitehead Institute, Cambridge, MA).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: MAP, mitogen-activated protein; MAPK, MAP kinase; MAPKK, MAPK kinase (etc.); DBD, DNA-binding domain; AD, activation domain; GST, glutathione S-transferase; IGF-1R, insulin-like growth factor 1 receptor.
References
- 1.Marshall C J. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 2.Herskowitz I. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- 3.Schultz J, Ferguson B, Sprague G F J. Curr Opin Genet Dev. 1995;5:31–37. doi: 10.1016/s0959-437x(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 4.Kurjan J. Annu Rev Genet. 1993;27:147–179. doi: 10.1146/annurev.ge.27.120193.001051. [DOI] [PubMed] [Google Scholar]
- 5.Sprague G F, Jr, Thorner J W. In: The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression. Jones E W, Pringle J R, Broach J R, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 657–744. [Google Scholar]
- 6.Choi K-Y, Satterberg B, Lyons D M, Elion E A. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- 7.Marcus S, Polverino A, Barr M, Wigler M. Proc Natl Acad Sci USA. 1994;91:7762–7766. doi: 10.1073/pnas.91.16.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Printen J A, Sprague G F., Jr Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yashar B, Irie K, Printen J A, Stevenson B J, Sprague G F, Jr, Matsumoto K, Errede B. Mol Cell Biol. 1995;15:6545–6553. doi: 10.1128/mcb.15.12.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perlman P, Yablonski D, Simchen S, Levitzki L. Proc Natl Acad Sci USA. 1993;90:5474–5478. doi: 10.1073/pnas.90.12.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukai Y, Harashima S, Oshima Y. Mol Cell Biol. 1993;13:2050–2060. doi: 10.1128/mcb.13.4.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leberer E, Dignard D, Harcus D, Hougan L, Whiteway M, Thomaas D Y. Mol Gen Genet. 1993;241:241–254. doi: 10.1007/BF00284675. [DOI] [PubMed] [Google Scholar]
- 13.Bartel P, Chien C-T, Sternglanz R, Fields S. BioTechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- 14.Rothstein R. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 15.Alani E, Cao L, Kleckner N. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elion E A, Brill J A, Fink G R. Proc Natl Acad Sci USA. 1991;88:9392–9396. doi: 10.1073/pnas.88.21.9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gietz D, St. Jean A, Woods R A, Schiestl R H. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sprague G F., Jr Methods Enzymol. 1991;194:77–93. doi: 10.1016/0076-6879(91)94008-z. [DOI] [PubMed] [Google Scholar]
- 19.Ma J, Ptashne M. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- 20.Chien C-T, Bartel P L, Sternglanz R, Fields S. Proc Natl Acad Sci USA. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christianson T W, Sikorski R S, Dante M, Shero J H, Hieter P. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 22.Foreman P K, Davis R W. Gene. 1994;144:63–68. doi: 10.1016/0378-1119(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 23.Robzyk K, Kassir Y. Nucleic Acids Res. 1992;20:3790. doi: 10.1093/nar/20.14.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kranz J E, Satterberg B, Elion E. Genes Dev. 1994;8:313–327. doi: 10.1101/gad.8.3.313. [DOI] [PubMed] [Google Scholar]
- 26.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 27.Soos M A, Field C E, Lammers R, Ullrich A, Zhang B, Roth R A, Andersen A S, Kjeldsen T, Siddle K. J Biol Chem. 1992;267:12955–12963. [PubMed] [Google Scholar]
- 28.Guarente L, Ptashne M. Proc Natl Acad Sci USA. 1981;78:2199–2203. doi: 10.1073/pnas.78.4.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasson M S, Blinder D, Thorner J, Jenness D D. Mol Cell Biol. 1994;14:1054–1065. doi: 10.1128/mcb.14.2.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolan J W, Fields S. Genes Dev. 1990;4:492–502. doi: 10.1101/gad.4.4.492. [DOI] [PubMed] [Google Scholar]
- 31.Whiteway M S, Wu C, Leeuw T, Clark K, Fourest-Lieuvin A, Thomas D Y, Leverer E. Science. 1995;269:1572–1575. doi: 10.1126/science.7667635. [DOI] [PubMed] [Google Scholar]
- 32.Perez-Alvarado G C, Miles C, Michelsen J W, Louis H A, Winge D R, Beckerle M C, Summers M F. Nat Struct Biol. 1994;1:388–398. doi: 10.1038/nsb0694-388. [DOI] [PubMed] [Google Scholar]
- 33.Adamson J G, Zhou N E, Hodges R S. Curr Opin Biotechnol. 1993;4:428–437. doi: 10.1016/0958-1669(93)90008-k. [DOI] [PubMed] [Google Scholar]
- 34.Schmeichel K L, Beckerle M C. Cell. 1994;79:211–219. doi: 10.1016/0092-8674(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 35.Feuerstein R, Wang X, Song D, Cooke N E, Liebhaber S A. Proc Natl Acad Sci USA. 1994;91:10655–10659. doi: 10.1073/pnas.91.22.10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fields S, Song O-K. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 37.Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas D Y, Leberer E. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- 38.Bardwell L, Cook J G, Chang E C, Coins B R, Thorner J. Mol Cell Biol. 1996;16:3637–3650. doi: 10.1128/mcb.16.7.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]