

Abstract
The aim of this study was to examine whether short- and long-term gene transfer of Ca2+ handling proteins restore left ventricular (LV) mechanoenergetics in aortic banding-induced failing hearts. Aortic banded rats received recombinant adenoviruses carrying sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) (Banding+SERCA), parvalbumin (Banding+Parv) or β-galactosidase (Banding+βgal), or an adeno-associated virus carrying SERCA2a (Banding+AAV.SERCA) by a catheter-based technique. LV mechanoenergetic function was measured in cross-circulated hearts. “Banding”, “Banding+βgal” and “Banding+saline” groups showed lower end-systolic pressure at 0.1 ml intraballoon water (ESP0.1), higher end-diastolic pressure at 0.1 ml intraballoon water (EDP0.1) and slower LV relaxation rate, compared with “Normal” and “Sham”. However, “Banding+SERCA” and “Banding+Parv” showed high ESP0.1, low EDP0.1, and fast LV relaxation rate. In “Banding”, “Banding+βgal” and “Banding+saline”, slope of relation between cardiac oxygen consumption and systolic pressure-volume area, O2 cost of total mechanical energy, was twice higher than normal value, whereas slope in “Baning+SERCA” and “Banding+Parv” was similar to normal value. Furthermore, O2 cost of LV contractility in the 3 control banding groups was ∼3 times higher than normal value, whereas O2 cost of contractility in “Banding+SERCA”, “Banding+AAV.SERCA” and “Banding+Parv” was as low as normal value. Thus, high O2 osts of total mechanical energy and of LV contractility in failing hearts indicate energy wasting both in chemomechanical energy transduction and in calcium handling. Improved calcium handling by both short- and long-term overexpression of SERCA2a and parvalbumin transforms the inefficient energy utilization into a more efficient state. Therefore enhancement of calcium handling either by resequestration into the SR or by intracellular buffering improves not only mechanical but energetic function in failing hearts.
Keywords: gene therapy, heart failure, energetic function, SERCA2a, parvalbumin
1. Introduction
Calcium handling is central to the process of excitation-contraction (E-C) coupling. Sarcoplasmic reticulum (SR), which releases Ca2+ during contraction by a trigger of Ca2+ entry and takes it up during relaxation by SR-ATPase (SERCA2a) pump, plays a major role in controlling the synchronized Ca2+ handling in myocardial cells [1]. Removal of Ca2+ from the cytoplasm is governed mainly by SERCA2a, of which the activity is regulated by phospholamban, and to a lesser extent by Na+-Ca2+ exchanger (NaCaX). SERCA2a protein/mRNA expression and activity were decreased in human heart failure and in animal models of heart failure [2,3]. This SERCA2a reduction results in abnormal Ca2+ handling, which causes a prolongation of Ca2+ transient, an increase in diastolic intracellular Ca2+, a decrease in systolic intracellular Ca2+ and a reduced SR Ca2+ content. Therefore, abnormal Ca2+ handling by the SERCA2a reduction contributes to the systolic and diastolic dysfunction in failing hearts [4,5].
Our group [6-11] has previously shown that overexpression of SERCA2a, ablation of phospholamban, or overexpression of parvalbumin by adenoviral gene transfer modify intracellular Ca2+ handling and modulate physiological mechanical performance in normal, senescent, aortic banding-induced failing and ischemia/reperfusion-injured rat whole hearts. In addition, global cardiac gene transfer of SERCA2a improved survival and energetic state shown as phosphocreatine/ATP ratio in failing rat hearts induced by aortic banding [9], and reduced ventricular arrhythmias in the model rat of ischemia followed by reperfusion [10]. More recently, SERCA2a overexpression improved the energy utilization in special reference to myocardial oxygen consumption in diabetic failing hearts [12].
The aim of this study was to examine whether restoring calcium cycling by either re-sequestering the elevated intracellular calcium back into the SR (through SERCA2a gene transfer) or by buffering the elevated intracellular calcium (through parvalbumin gene transfer) would improve the left ventricular (LV) mechanical and energetic functions in the aortic banded rats, especially in terms of oxygen cost of LV contractility.
2. Materials and methods
2.1. Recombinant adenoviral vectors
Recombinant adenoviral vectors were used with cytomegalovirus-driven expression cassettes for SERCA2a, parvalbumin, or β-galactosidase with a second cassette in each adenovirus containing green fluorescent protein substituted for E1 by means of homologous recombination. Adenoviral SERCA2a (Ad.SERCA), adenoviral parvalbumin (Ad.Parv) and adenoviral β-galactosidase (Ad.βgal) had concentrations of 9 X 1010 pfu/ml, 10 X 1010 pfu/ml and 8 X 1010 pfu/ml, respectively, with a particle/pfu ratio of 10:1. Wild-type adenovirus contamination was excluded by the absence of PCR-detectable E1 sequences.
2.2. Recombinant adeno-associated virus (AAV) vectors
The AAV1 vector used in this study was manufactured using standard calcium phosphate transfection methods in 293 cells. Three plasmids were used, one containing helper functions from adenovirus, one containing the AAV rep2 and cap1 genes, and the third containing the vector genome. Vector production, harvest, purification and testing were as previously described [13].
2.3. Animals
All animal experiments in this study were performed with the approval of the Animal Care Committee of Massachusetts General Hospital and in accordance with the National Institute of Health′s Guide for the Care and Use of Laboratory Animals. Five-week-old male Wistar rats (Charles River, Mass; body weight (BW), 100 to 120 g) were anesthetized with intraperitoneal (i.p.) Ketamine-Xyla-Jet mixture and placed on a ventilator. An incision on the right side of chest was made, exposing the aortic root, and a tanatalum clip with an internal diameter of 0.58 mm (Weck Hemoclip) was placed on the ascending aorta. Animals in the sham group underwent a similar procedure without insertion of a clip. Twenty-four to twenty-six weeks later, about 90 % of the animals thus operated survived (aortic banding, 89%; sham-operation, 93%), and were randomized into 8 groups:
six normal control rats (Normal);
six uninfected sham-operated rats (Sham);
six uninfected aortic banded rats (Banding);
five aortic banded rats infected with Ad.βgal (Banding+βgal)
five aortic banded rats injected with saline (Banding+saline);
six aortic banded rats infected with Ad.SERCA (Banding+SERCA);
five aortic banded rats infected with Ad.Parv (Banding+Parv);
three aortic banded rats infected with AAV1 carrying SERCA2a (Banding+ AAV.SERCA).
2.4. Cardiac gene delivery
The in vivo cardiac gene transfer has been described previously in detail by our group [14]. Briefly, after the rats were anesthetized and a thoracotomy performed, a 22-guage catheter containing 200 μl of adenovirus and 50 μl adenosine (3 mg/ml) was advanced from the apex of the left ventricle to the aortic root. The aorta and main pulmonary artery were clamped for 30 seconds distal to the site of the catheter and the solution injected, then the chest was closed, and the animals were allowed to recover. In the group (4) - (8), 60-67% of the rats thus operated survived until LV mechanical and energetic studies (Banding+βgal, 60%; Banding+saline, 63%; Banding+SERCA, 62%; Banding+Parv, 64%; Banding+AAV.SERCA; 67%). The rats underwent LV mechanical and energetic studies 2-3 days after the injection of the adenovirus or saline and 45 days after the AAV injection.
2.5. LV mechanical and energetic studies
Surgical preparations
The LV mechanical and energetic studies were performed on the excised, cross-circulated rat heart preparations. The surgical preparations have been described previously in detail [15]. Briefly, in each experiment, 2 male 500 to 650g Wistar rats (blood supplier and metabolic supporter) and 1 heart donor rat were anesthetized with pentobarbital sodium (50 mg/kg, ip) and intubated and heparinized (1000 U, iv). In the beating heart, cross-circulated and maintained at 37°C, a thin latex balloon, which was connected to a pressure transducer for measuring LV pressure (LVP), was inserted into the LV. Systolic unstressed volume (V0=0.08 ml for normal or sham-operated hearts and V0=0.06 ml for aortic banded hearts) should be determined as the volume at which peak isovolumic pressure was zero. Heart rate was maintained constant at 300 beats/min by right atrium electrical pacing. Systemic arterial blood pressure of the support rat served as coronary perfusion pressure (diastolic-systolic, 80-120 mmHg; mean, about 90 mmHg). Arterial pH, Po2, and Pco2 of the support rat and perfused blood were maintained within their physiological ranges by supplemental oxygen and sodium bicarbonate throughout the experiment.
Calculation for oxygen consumption
Cardiac VO2 was obtained as the product of coronary flow and arteriovenous O2 content difference. The right ventricular (RV) component of total VO2, which is considered constant irrespective of LV volume, was calculated by multiplying biventricular VO2 under LV volume-unloading with the weight ratio of RV/RV+LV. The RV VO2 was subtracted from the total VO2 to yield LV VO2.
Experimental protocol
LVP, LV VO2 and systolic pressure-volume area (PVA) data were obtained at five different LV volumes, changed with a step of 0.025 ml, without any inotropic interventions (control volume run). After control volume run, Ca2+ inotropism run was performed at a midrange LV volume (mLVV) by intracoronary infusion of 1% CaCl2 solution. In some experiments, dobutamine (78 μM) inotropism run was also performed after Ca2+ inotropism run. The steady state of heart was reached 2-3 min after change of LV volume and 4 min after change of the infusion rate. Cardiac arrest was induced by intracoronary infusion of 1M KCl (12 ml/h) to obtain VO2 for basal metabolism. In each steady state, the data sampling, performed at 500 Hz for 2 s, was repeated 3 times at intervals of 0.5 min.
2.6. Data analysis
PVA calculation
The best-fit end-systolic/diastolic pressure-volume relations (ESPVR/EDPVR) were obtained by fitting the data with the exponential functions [12]. PVA was defined as the pressure-volume area circumscribed by the curvilinear ESVPR, EDPVR and the systolic portion of the ventricular pressure-volume trajectory.
VO2 for Ca2+ handling in E-C coupling during inotropic run
During Ca2+/dobutamine inotropic run at a mLVV, ESP-volume relations at different infusion rates were obtained as a best-fit exponential function curve. We calculated PVA at mLVV (PVAmLVV) by integrating each ESPVR from V0 up to mLVV. We then obtained the two composite VO2-PVAmLVV data points. Next, the lines including each VO2-PVAmLVV data point were drawn in parallel to control VO2-PVA relation. VO2-intercept value (PVA-independent VO2) at each infusion rate was thus obtained. VO2 for Ca2+ handling in E-C coupling was obtained by subtracting basal metabolic VO2 per beat, measured in KCl-induced arrest hearts, from PVA-independent VO2.
LV contractility
Equivalent maximal elastance (eEmax), an index for LV contractility, was obtained by calculating ESP-volume ratio of the specific virtual triangle, which is energetically equivalent to the real PVAmLVV [16].
Oxygen cost of LV contractility
The oxygen cost of LV contractility was obtained as the slope of the linear relation between VO2 for Ca2+ handling in E-C coupling and eEmax during Ca2+/dobutamine inotropism run. This slope is considered an index quantifying Ca2+ handling VO2 per unit LV contractility change.
Logistic time constant
To evaluate the LV relaxation rate, we analyzed LV isovolumic relaxation pressure-time curves at mLVV by using the logistic time constant (TL) derived from a logistic model.
2.7. Western blot for SERCA2a protein
Lysates from hearts were matched for protein concentration and then separated by SDS-PAGE and transferred to nitrocellulose membranes. Blots were incubated with SERCA2a antibodies (Affinity Bioreagents, CA) followed by detection with enhanced chemi-luminescence.
2.8. Statistics
Data are presented as mean ± SD. Multiple comparisons were performed by ANOVA with STATVIEW (Abacus Concepts, Barkeley, CA). Statistical significance was accepted at the level of P<0.05.
3. Results
3.1. Characterization of animals
There was no statistical difference in BW among the 7 groups (Table 1). LV/BW ratio and LV+RV/BW ratio, as well as LV weight and LV+RV weight, in 5 aortic banding groups were significantly higher than those of “Normal” and “Sham” groups, while RV/BW ratio, as well as RV weight, was not different among 7 groups (Table 1). Thus, the rats, which underwent an aortic banding for 24-26 weeks, had a significant increase in LV mass normalized to BW, and a cross-section of the aortic banded hearts showed apparent LV concentric hypertrophy (data not shown).
Table 1.
Morphometric analyses
| Group | n | BW (g) | LV (g) | RV (g) | LV+RV (g) | LV/BW (X10-3) | RV/BW (X10-3) | LV+RV/BW (X10-3) |
|---|---|---|---|---|---|---|---|---|
| Normal | 6 | 598 ± 34 | 1.014 ± 0.076 | 0.288 ± 0.022 | 1.301 ± 0.084 | 1.69 ± 0.08 | 0.48 ± 0.05 | 2.18 ± 0.11 |
| Sham | 6 | 627 ± 53 | 1.091 ± 0.115 | 0.297 ± 0.049 | 1.388 ± 0.159 | 1.74 ± 0.06 | 0.47 ± 0.05 | 2.21 ± 0.10 |
| Banding | 6 | 614 ± 53 | 1.351 ± 0.127 a,b | 0.302 ± 0.025 | 1.653 ± 0.143 a | 2.21 ± 0.24 a,b | 0.50 ± 0.07 | 2.71 ± 0.30a,b |
| Banding + βgal | 5 | 607 ± 65 | 1.312 ± 0.176 a,b | 0.306 ± 0.042 | 1.618 ± 0.207 a | 2.16 ± 0.14 a,b | 0.51 ± 0.05 | 2.66 ± 0.16 a,b |
| Banding + saline | 5 | 648 ± 59 | 1.488 ± 0.164 a,b | 0.312 ± 0.02 | 11.799 ± 0.160 a,b | 2.29 ± 0.12 a,b | 0.49 ± 0.07 | 2.78 ± 0.13 a,b |
| Banding + SERCA | 6 | 580 ± 46 | 1.317 ± 0.138 a,b | 0.325 ± 0.067 | 1.642 ± 0.195 a | 2.28 ± 0.28 a,b | 0.57 ± 0.15 | 2.85 ± 0.43 a,b |
| Banding + Parv | 5 | 554 ± 52 | 1.277 ± 0.115 a,b | 0.288 ± 0.023 | 1.565 ± 0.135 a | 2.32 ± 0.24 a,b | 0.52 ± 0.05 | 2.84 ± 0.29 a,b |
All data are shown as mean ± SD. BW, body weight; LV, left ventricle; RV, right ventricle; n, number of hearts.
P < 0.05 compared to “Normal” group.
P < 0.05 compared to “Sham” group.
3.2. LV mechanics
Figure 1A shows representative control ESVPRs and EDPVRs in “Sham”, “Banding” and “Banding+SERCA” hearts. Curvilinear ESPVRs and EDPVRs of “Banding+βgal” and “Banding+saline” hearts were similar to those of “Banding” hearts (data not shown). ESPVR of “Banding+SERCA” and “Banding+Parv” hearts was shifted upward compared with ESPVR of “Banding” hearts. Summarized data of LV mechanics are shown in Table 2. The volume intercepts of ESPVR and EDPVR, V0 and Vu, in 5 aortic banding groups were significantly smaller than those of “Normal” and “Sham” groups, resulting from LV concentric hypertrophy. In “Banding” hearts, the end-systolic pressure observed at 0.1 ml of intraballoon water volume (ESP0.1), was lower (130 ±27 mmHg), and end-diastolic pressure observed at 0.1 ml of intraballoon water volume (EDP0.1), was higher (22.1 ±6.2 mmHg), compared with “Normal” hearts (ESP0.1,188 ±15 mmHg; EDP0.1, 9.8 ±4.0 mmHg). However, in “Banding+SERCA” group, like in “Banding+Parv” group, ESP0.1 was increased over 200 mmHg and EDP0.1 was decreased to 12.4 ±2.7 mmHg, although ESP0.1 in “Banding+βgal” and “Banding+saline” groups was as low as that of “Banding” group. “Banding+βgal” group showed a higher EDP0.1, but without significant difference, compared with “Banding+SERCA” group. Two of “Banding+βgal” hearts had lower EDP0.1 (12.3 and 12.6 mmHg), shorter logistic time constant, TL (13.0 and 14.2 msec) and higher eEmax at mLVV (3538 and 3005 mmHg·ml-1·g), compared with the rest of “Banding+βgal” hearts. Thus, in the two hearts, the systolic/diastolic cardiac function was not much deteriorated. The systolic/diastolic function was dependent on the levels of SERCA2a protein expression or SERCA2a activity [17,18]. In this study, therefore, the degree of cardiac dysfunction appears somewhat different among the aortic-banded animals because of the different levels of down-regulated SERCA2a expression. In all groups, moreover, TL was obtained from LV isovolumic relaxation pressure-time curves at mLVV. TL in “Banding” group was significantly longer than that of “Normal” and “Sham” groups (p<0.05). In “Banding+SERCA” and “Banding+Parv” groups, however, TL was decreased to TL level observed in “Normal” and “Sham” groups, although TL in “Banding+βgal” and “Banding+saline” groups remained as long as that of “Banding” group.
Fig. 1.

(A) End-systolic volume relation (closed symbols) and end-diastolic pressure-volume relation (open symbols) in “Sham” (■and □), “Banding” (▲ and △) and “Banding+SERCA” (● and ○) hearts. Both relations were obtained by control volume-loading runs where balloon water was changed from 0 to 0.1 ml by 0.025 ml. (B) Comparison of logistic time constants among 7 groups. Logistic time constants were obtained from a best-fit logistic curve of pressure-time curve during isovolumic relaxation at midrange LV volume (0.05 ml of balloon water volume). *P<0.05 compared to “Normal” and “Sham” groups. #P<0.05 compared to “Banding”, “Banding+βgal” and “Banding+saline” groups. n=6 in “Normal”, n=6 in “Sham”, n=6 in “Banding”, n=5 in “Banding+βgal”, n=5 in “Banding+saline”, n=6 in “Banding+SERCA”, n=5 in “Banding+Parv”. (C) Linear relations between myocardial oxygen consumption per beat (VO2) and systolic pressure-volume area (PVA) in “Sham” (■), “Banding” (▲) and “Banding+SERCA” (●) hearts.
Table 2.
Variables of LV mechanics
| ESPVR |
EDPVR |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group | n | A (mmHg) | B (1/ml) | V0 (ml/g) | ESP0.1 (mmHg) | A′ (mmHg) | B′ (1/ml) | Vu (ml/g) | EDP0.1 (mmHg) |
| Normal | 6 | 221±20 | 13.2±5.4 | 0.079±0.007 | 188±15 | 0.35±0.40 | 28.1±11.2 | 0.079±0.007 | 9.8±4.0 |
| Sham | 6 | 191±20 | 45.9±21.6 | 0.074±0.008 | 188±15 | 1.44±1.07 | 28.0±6.5 | 0.074±0.008 | 13.4±4.7 |
| Banding | 6 | 177±45 | 25.3±17.4 | 0.045±0.005a,b | 130±27a,b | 0.93±0.35 | 43.4±5.5 | 0.045±0.005a,b | 22.1±6.2a |
| Banding +βgal | 5 | 132±24a,b | 57.0±26.8a | 0.047±0.007a,b | 128±27a,b | 2.12±2.79 | 34.0±12.6 | 0.047±0.007a,b | 15.1±3.0 |
| Banding + saline | 5 | 155±22a | 27.3±16.3 | 0.040±0.005a,b | 118±19a,b | 2.45±1.74 | 37.4±7.4 | 0.040±0.005a,b | 24.8±9.8a,b |
| Banding + SERCA | 6 | 206±46d | 65.6±30.1a | 0.046±0.005a,b | 210±46c,d,e | 2.45±3.56 | 33.6±12.2 | 0.046±0.005a,b | 12.4±2.7c,e |
| Banding + Parv | 5 | 238±39d,e | 47.6±18.3 | 0.047±0.004a,b | 232±31c,d,e | 1.83±1.13 | 33.7±10.9 | 0.047±0.004a,b | 16.7±3.8 |
All data are shown as mean ± SD. ESPVR, end-systolic pressure-volume relation; A and B are parameters in the equation ESP=A{1-exp[-B(V-V0)]};
V0, volume intercept of end-systolic pressure-volume relation;
ESP0.1, end-systolic pressure observed at maximum left ventricular volume (0.1 ml of balloon water volume);
EDPVR, end-diastolic pressure-volume relation; A′ and B′ are parameters in the equation EDP=A′{exp[B′(V-Vu)]-1};
Vu, volume intercept of end-diastolic pressure-volume relation;
EDP0.1, end-diastolic pressure observed at maximum left ventricular volume (0.1 ml of balloon water volume); n, number of hearts.
P < 0.05 compared to “Normal” group.
P < 0.05 compared to “Sham” group.
P < 0.05 compared to “Banding” group.
P < 0.05 compared to “Banding +βgal” group.
P < 0.05 compared to “Banding + saline” group.
3.3. LV energetics; VO2-PVA relations
Figure 1C shows representative control VO2-PVA relations in “Sham”, “Banding” and “Banding+SERCA” hearts. “Banding” heart showed a linear VO2-PVA relation with steeper slope compared with “Sham” and “Banding+SERCA” hearts. Summarized data of LV energetics are shown in Table 3. There was no significant difference in the VO2 intercept of VO2 -PVA relation among 7 groups. On the other hand, the slope in “Banding” group was about twice as high as that of “Normal” and “Sham” groups. However, the slope in “Banding+SERCA” and “Banding+Parv” groups was similar to the slope found in “Normal” and “Sham” groups, although the slope in “Banding+βgal” and “Banding+saline” groups remained high. In five aortic banding groups, moreover, the minute VO2 for basal metabolism and for Ca2+ handling in E-C coupling, which are both components of the VO2 intercept of VO2 -PVA relation, were not significantly different from those of “Normal” and “Sham” groups.
Table 3.
Variables of LV energetics
| VO2-PVA relation |
VO2 per minute (μl O2 · min-1 · g-1) |
||||
|---|---|---|---|---|---|
| Group | n | Slope (X10-2μl O2 · mmHg-1 ml-1) | VO2-intercept (μl O2· beat-1 · g-1) | Basal Metabolism | E-C Coupling |
| Normal | 6 | 1.13±0.29 | 0.303±0.043 | 29.0±3.2 | 60.3±13.4 |
| Sham | 6 | 1.26±0.24 | 0.348±0.026 | 29.6±3.4 | 71.8±5.3 |
| Banding | 6 | 2.61±0.82a.b | 0.364±0.040 | 33.8±3.1 | 72.4±11.8 |
| Banding + βgal | 5 | 2.19±0.25a,b | 0.310±0.044 | 35.1±3.4 | 56.7±13.5 |
| Banding + saline | 5 | 2.11±0.40a,b | 0.304±0.036 | 28.6±3.3 | 62.7±11.8 |
| Banding + SERCA | 6 | 1.09±0.23c,d,e | 0.377±0.064 | 33.4±2.7 | 78.6±17.9 |
| Banding + Parv | 5 | 1.37±0.34c,d,e | 0.350±0.032 | 32.0±4.1 | 69.4±7.1 |
All data are shown as mean ± SD. VO2, myocardial oxygen consumption per beat; PVA, systolic pressure-volume area;
E-C, excitation-contraction; n, number of hearts. VO2 per minute for basal metabolism was measured in the hearts under KCl-induced arrest. VO2 per minute for E-C coupling was obtained by subtracting VO2 per minute for basal metabolism from VO2 per minute for mechanically unloaded (i.e., free of balloon water) contraction.
P < 0.05 compared to “Normal” group.
P < 0.05 compared to “Sham” group.
P < 0.05 compared to “Banding” group.
P < 0.05 compared to “Banding + βgal” group.
P < 0.05 compared to “Banding + saline” group.
3.4. ESPmLVV in response to calcium
Table 4 shows changes in end-systolic pressure observed at a mLVV (ESPmLVV) in response to Ca2+ infusion. ESPmLVV prior to Ca2+ infusion in “Banding+SERCA” and “Banding+Parv” groups were significantly higher than those in “Banding”, “Banding+βgal” and “Banding+saline” groups. ESPmLVV was gradually increased as the infusion rate of Ca2+ solution was increased step-wise from 2 to 6 ml/h. In “Banding+SERCA” and “Banding+Parv” groups, the maximal increase in ESPmLVV in response to Ca2+ solution (6 ml/h) were similar to those in “Normal” and “Sham” groups, and were much higher than those in “Banding”, “Banding+βgal” and “Banding+saline” groups. In some experiments, we infused dobutamine solution into the coronary perfusion tubing at 2-4 ml/h of infusion rate after the Ca2+ infusion. The changes in ESPmLVV in response to dobutamine were similar to those to Ca2+ in all groups (data not shown).
Table 4.
ESP at midrange left ventricular volume in response to Ca 2+ infusion
| Group | n | ESPmLVV before Ca2+ infusion (mmHg) | ESPmLVV during Ca2+ infusion (mmHg) | Increase in ESPmLVV(mmHg) |
|---|---|---|---|---|
| Normal | 6 | 114.2 ± 26.0 | 168.2 ± 24.8 | 54.0 ± 18.1 |
| Sham | 6 | 144.5 ± 22.7 | 199.8 ± 21.7 | 55.3 ± 12.2 |
| Banding | 6 | 86.1 ± 26.7 | 98.8 ± 28.9a,b | 12.7 ± 4.6a,b |
| Banding + βgal | 5 | 94.6 ± 26.8 | 115.0 ± 34.8a,b | 20.3 ± 9.8a,b |
| Banding + saline | 5 | 91.1 ± 21.5 | 114.7 ± 18.6b | 23.6 ± 7.1a,b |
| Banding + SERCA | 6 | 154.1 ± 58.2c,d,e | 198.8 ± 62.6c,d,e | 44.7 ± 14.7c,d,e |
| Banding + Parv | 5 | 170.6 ± 26.1c,d,e | 231.8 ± 34.1a,c,d,e | 61.2 ± 11.2c,d,e |
All data are shown as mean ± SD.
ESPmLVV, end-systolic pressure observed at midrange left ventricular volume (0.05 ml of balloon volume); n, number of hearts.
ESPmLVV during Ca2+ infusion was obtained in a steady state of hearts after intracoronary infusion of 1% CaCl2 at a 6 ml/h infusion rate for at least 4 minutes.
P < 0.05 compared to “Normal” group.
P < 0.05 compared to “Sham” group.
P < 0.05 compared to “Banding” group.
P < 0.05 compared to “Banding + βgal” group.
P < 0.05 compared to “Banding + saline” group.
3.5. LV contractility
The eEmax at mLVV, an index of LV contractility, in “Banding+SERCA” and “Banding+Parv” groups was significantly higher than that in “Normal”, “Sham” and 3 other control aortic banding groups (Fig. 2). In the preliminary experiments, 3 aortic banded rats underwent LV mechanical and energetic studies 45 days after recombinant AAV.SERCA transfer. As shown in Fig. 2, “Banding+AAV.SERCA” group showed the high LV contractility similar to that found in “Banding+SERCA” and “Banding+Parv” groups. On the other hand, “Banding”, “Banding+βgal” and “Banding+saline” groups showed the lower LV contractility.
Fig. 2.

Comparison of eEmax at midrange LV volume, an index of LV contractility, among all groups. *P<0.05 compared to “Normal”, “Sham”, “Banding”, “Banding+βgal” and “Banding+saline” groups. n=6 in “Normal”, n=6 in “Sham”, n=6 in “Banding”, n=5 in “Banding+βgal”, n=5 in “Banding+saline”, n=6 in “Banding+SERCA”, n=3 in “Banding+AAV.SERCA”, n=5 in “Banding+Parv”.
3.6. LV energetcs; oxygen cost of LV contractility
Figure 3A shows representative relations between VO2 for Ca2+ handling in E-C coupling and eEmax at mLVV during Ca2+ inotropism run in “Sham”, “Banding” and “Banding+SERCA” hearts. These 3 distinct linear relations had the different slopes, which mean the different oxygen costs of LV contractility. In some experiments, the intracoronary dobutamine infusion was performed after the Ca2+ infusion in the same heart preparation. There was a fairly good correlation between the oxygen costs of LV contractility in response to Ca2+ and to dobutamine (Fig. 3B). The oxygen cost of LV contractility for Ca2+ in “Banding” group was about 3 times as high as that in “Normal” and “Sham” groups (Fig. 3C). In “Banding+SERCA”, “Banding+AAV.SERCA” and “Banding+Parv” groups, the oxygen costs of LV contractility were as low as those in “Normal” and “Sham” groups, but in “Banding+βgal and ”Banding+saline“ groups the oxygen costs remained as high as that of ”Banding“ group.
Fig. 3.
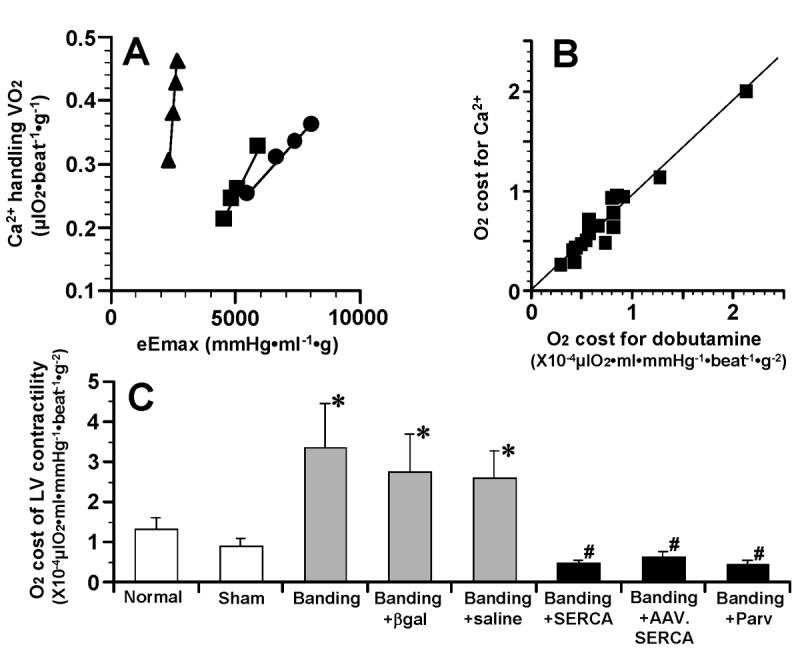
(A) Linear relations between Ca2+ handling VO2 and eEmax at midrange LV volume during Ca2+ inotropic run in “Sham” (■), “Banding” (▲) and “Banding+SERCA” (●) hearts. Slope of these linear relations means oxygen cost of LV contractility. (B) Correlation between O2 costs for Ca2+ and for dobutamine. In 17 heart preparations, dobutamine (78 μM) inotropic run was performed after Ca2+ inotropic run. An equation of regression line is f(x) = 0.93x + 1.8X10-6, r = 0.96. (C) Comparison of oxygen costs of LV contractility among all groups. *P<0.05 compared to “Normal” and “Sham” groups. #P<0.05 compared to “Banding”, “Banding+βgal” and “Banding+saline” groups. n=6 in “Normal”, n=6 in “Sham”, n=6 in “Banding”, n=5 in “Banding+βgal”, n=5 in “Banding+saline”, n=6 in “Banding+SERCA”, n=3 in “Banding+AAV.SERCA”, n=5 in “Banding+Parv”.
3.7. SERCA2a & parvalbumin protein expression
We examined the protein expression of SERCA2a in the heart preparations used for the analysis of mechanical and energetic function. As shown in Figure 4, there is a decrease in SERCA2a expression in the failing hearts compared to sham. The overexpression of SERCA2a either by adenovirus or AAV1 restores SERCA2a protein levels in the failing hearts. Gene transfer of parvalbumin induces a robust expression in the hearts infected.
Fig. 4.

Immunoblots of SERCA2a and parvalbumin from left ventricular tissues of various groups.
4. Discussion
In this study, using a unique technique for measuring LV mechanoenergetics in the cross-circulated heart preparations, we found that cardiac gene transfer of Ca2+ handling proteins (SERCA2a and parvalbumin) can restore not only mechanical but also energetic function in pressure overload-induced failing hearts.
4.1. Mechanical function and gene transfer
In the failing rats, we found a downward shift of ESPVR, a decreased ESP0.1 and deceased eEmax at mLVV compared with “Normal” and “Sham” rats. Furthermore, the logistic time constant of isovolumic relaxation and EDP0.1 were found to be increased in these failing rats. Thus the aortic banded rats used in this study showed both LV systolic and diastolic dysfunction. The central mechanisms for systolic dysfunction in this rodent model of heart failure have been reported to be: decreased SERCA2a expression [17], increased sarcolemmal NaCaX expression [19], and the transition of cardiac myosin isoform components from V1 with higher ATPase activity to V3 with lower ATPase activity [20]. The decreased SERCA2a and/or the increased NaCaX can reduce the Ca2+ content in the SR and limit the Ca2+ release from the SR, leading to diastolic increases in calcium and general intracellular calcium overload [19]. V1 is known to be associated with an increased shortening velocity of the cardiac fibers and V3 with a slower shortening velocity [21], although in aortic banded rats, the improved contractile function by clenbuterol, a selective β2-adrenergic agonist, was not related to changes in myosin isoform components [3]. On the other hand, the central mechanisms that cause diastolic dysfunction in this rodent model of heart failure are due to decreased SERCA2a expression and increase in cardiac stiffness due to enhanced collagen and fibrosis [3]. However, in this study the contractile dysfunction found in the “Banding” hearts (ESPVR, ESP0.1, EDP0.1, and time constant of isovolumic relaxation) could be completely corrected by SERCA2a gene transfer, indicating that the decreased SERCA2a expression plays a key role in both systolic and diastolic dysfunction. Furthermore, similar correction of contractile dysfunction could be accomplished by parvalbumin gene transfer as reported in normal and aged rats [11,22]. In senescent rat cardiomyocytes, introduction of parvalbumin, that functions as a Ca2+ sink due to its Ca2+ affinity and enhances relaxation in skeletal muscle, increased both the rate of Ca2+ transient decay and the rate of myocyte re-lengthening [23]. This in vitro study provides the cellular mechanism for improved diastolic function by parvalbumin gene transfer in the present in vivo study. On the other hand, the possible mechanism for improved systolic function by the parvalbumin gene transfer is as follows; the parvalbumin-induced decline in intracellular free Ca2+ during relaxation attenuates the intracellular Ca2+ overload and may improve the impaired Ca2+ handling in the present failing hearts. Finally, the prominent eEmax at mLVV could be induced by the SERCA2a or parvalbumin gene transfer in the aortic banded hearts. The high LV contractility, as indicated by high ESP0.1 and high eEmax at mLVV, seems due mainly to both the hypertrophied LV free wall and the enhanced Ca2+ handling by overexpression of the SERCA2a or parvalbumin.
In the failing hearts, the increase in ESPmLVV in response to Ca2+ infusion was much lower than that of “Normal” and “Sham” hearts. This blunted inotropic response to Ca2+ is most likely caused by multiple factors including abnormal Ca2+ handling, deficient production of cyclic AMP [24], along with a decrease in energy reserve via a deficient creatine kinase reaction [25]. Both SERCA2a and parvalbumin gene transfer was capable of completely reversing the inotropic response to Ca2+ (i.e., contractile reserve), indicating that in the aortic banded hearts the abnormal Ca2+ handling is a central cause of the lowered contractile reserve and regardless how calcium is decreased intracellularly, there is a beneficial response.
4.2. LV energetic function and gene transfer
In the failing hearts, the slope of the VO2-PVA relation, which is an index of the O2 cost of PVA, i.e., the O2 cost of total mechanical energy of LV contraction [26], was about twice as high as that of “Normal” and “Sham” hearts. A similar steeper slope of the VO2-PVA relation was reported in hyperthyroid rabbit hearts which actually have increased V1/V3 ratio [27]. In contrast, in hypothyroid [15] and diabetic [12] rat hearts with decreased V1/V3 ratio, the slope remained unchanged. Thus the inconsistent relationship between the slope and the V1/V3 ratio suggests that a change in myosin isoform components, i.e., a change in myosin ATPase activity, does not directly affect the contractile efficiency, which is the reciprocal of slope of the VO2-PVA relation [26]. The contractile efficiency reflects the chemo-mechanical energy transduction efficiency of the contractile machinery. Therefore, in the aortic banded hearts, the high O2 cost of total mechanical energy, shown by the steeper slope of the VO2-PVA relation, indicates that energy wasting in the chemomechanical energy transduction is present. In the pressure overload-induced hypertrophied myocardium, the slope of relation between VO2 and isometric developed tension was increased [28,29], and the increased VO2 was due to the increased nonphosphorylating (state 4) mitochondrial respiration linked to Ca2+ transport [28], while in volume overload-induced hypertrophied myocardium, the slope was not different from that of normal myocardium [30]. Therefore, It seems likely that in the aortic banded hearts, the increased slope of the VO2-PVA relation is attributable to the increased VO2 induced by the increased nonphosphorylating mitochondrial respiration. This high slope could be completely improved by gene transfer of SERCA2a or parvalbumin. We have speculated on one possible mechanism for this; the overexpression of SERCA2a or parvalbumin restores the increased cytosolic free Ca2+ o normal levels and improves the enhanced Ca2+ transport to mitochondria, leading to the normal nonphosphorylating respiration of mitochondria. On the other hand, the VO2-intercept (i.e., PVA-independent VO2) of the VO2-PVA relation, that reflects VO2 for non-mechanical works consisting of Ca2+ handling during E-C coupling and basal metabolism [26], did not differ among all the groups studied. Furthermore, there was no significant difference in minute VO2 for Ca2+ handling and basal metabolism among all the groups, showing that these minute VO2 are affected neither by cardiac hypertrophy nor by adenoviral gene transfer. In addition, the unchanged VO2 for Ca2+ handling found in the aortic banded hearts suggests that SERCA2a, albeit down-regulated, can exert its function normally because of a lower level of cytosolic free Ca2+ during diastole in steady state of mechanically unloaded and Ca2+ unloaded hearts.
The most important finding of this study is that the O2 cost of LV contractility, defined as the slope of relation between Ca2+ handling VO2 and eEmax, was increased in the failing hearts, and this increased O2 cost could be restored to normal levels by short-term and long-term gene transfer of SERCA2a or parvalbumin. The O2 cost of LV contractility is thought to indicate the energy cost of Ca2+ handling during E-C coupling in failing hearts where the Ca2+ responsiveness of myofilaments remains unchanged [31]. There seems three possible mechanisms for the increased O2 cost of LV contractility, which means the energy wasting in Ca2+ handling, in the aortic banded hearts. First, the molar coupling ratio of calcium to ATP in SERCA2a was decreased due to increased Ca2+ permeability of the SR membrane [32]. Such SERCA2a dysfunction would need more ATP to lower cytosolic Ca2+ during relaxation. Second, abnormal Ca2+ leak through ryanodine receptor (RyR), caused by RyR conformational change due to a partial loss of RyR-bound FK506-binding protein, was found in a canine model of heart failure [33]. This Ca2+ leak via RyR will elevate basal cytosolic Ca2+ levels during diastole, leading to the futile Ca2+ handling. However, the above-mentioned mechanisms cannot fully explain the increased O2 cost of LV contractility because of the down-regulated SERCA2a expression in our aortic banded hearts. Finally, cardiac NaCaX expression was reported to be up-regulated in heart failure [19,34,35]. Although NaCaX consumes no ATP to remove cytosolic Ca2+ in exchange with influx Na+ on the basis of a stoichiometry of 3Na+:1Ca2+, the influx Na+ will be pumped out by Na+-K+-ATPase with a stoichiometry of 3Na+:2K+:1ATP, resulting in the net stoichimetry of 1Ca2+:1ATP [36]. On the other hand, SERCA2a removes cytosolic Ca2+ on the basis of a stoichiometry of 2Ca2+:1ATP. Therefore, the Ca2+ extrusion via NaCaX leads to twice increase in energy expenditure compared with the Ca2+ uptake by SERCA2a. Thus it seems most likely that the energy wasting in Ca2+ handling is induced mainly by the enhanced NaCaX activity. Nevertheless, overexpression of SERCA2a or parvalbumin was capable of restoring the high O2 cost of LV contractility to normal levels in the aortic banded hearts. The enhanced expression of SERCA2a or parvalbumin appears to induce the decrease in the cytosolic free Ca2+ level during diastole and thereby the decrease in Ca2+ extrusion via NaCaX, leading to the decrease in the energy cost of Ca2+ handling, namely, the decrease in the O2 cost of LV contractility.
4.3. Therapeutic implications
In patients with congestive heart failure (CHF), inotropic agents improve contractility and hemodynamics in the short term. However, they increase VO2, and subsequently induce an energetic imbalance, followed by arrhythmias and worsening survival. Therefore, long-term inotropic interventions have been shown to increase morbidity and mortality in patients with CHF [37]. In this study, gene transfer of SERCA2a or parvalbumin was found to improve the energy wasting both in chemomechanical energy transduction and in Ca2+ handling during E-C coupling in pressure-overload failing hearts. In a previous study [9], our group had already shown improvement in mortality and energy reserve (measured by NMR) in the same model of heart failure following short-term gene transfer of SERCA2a. Therefore, targeting Ca2+ handling in CHF may provide great advantage of efficient energy utilization over standard inotropic agents.
4.4. Limitations of the study and future directions
In the present failing hearts, correction of the energetic dysfunction by SERCA2a overexpression was accomplished, in a long-term transgene expression system of AAV1-based vectors as well as in a short-term transgene expression system of adenoviral vectors. To further confirm this finding, large animal studies are also required to verify these provocative results, because the contribution of SERCA2a to the lowering of cytosolic Ca2+ levels varies between rodents and large mammals. In humans, about 75% of the Ca2+ is removed by SERCA2a, whereas in rodents 92% is removed by SERCA2a [1]. However, overexpression of SERCA2a was capable of improving contractile function in failing human cardiomyocytes [38]. Therefore, SERCA2a overexpression is expected to be useful for improving LV energetic function in failing human hearts. Taken together, large animal studies using AAV vectors are indispensable for leading to clinical trials of gene therapy for CHF.
4.5. Conclusions
High O2 costs of total mechanical energy and of LV contractility in the aortic banded hearts indicate the energy wasting both in chemomechanical energy transduction and in Ca2+ handling during E-C coupling. The gene transfer of Ca2+ handling proteins (SERCA2a and parvalbumin) was capable of correcting both the high O2 costs. Thus the gene transfer of the Ca2+ handling proteins transforms the inefficient energy utilization into a more efficient state, and improves not only mechanical but also energetic function in pressure-overload failing hearts. Gene transfer of Ca2+ handling proteins may be a potential therapeutic strategy with the benefit of efficient energy utilization.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health: R01 HL078691, HL057263, HL071763, HL080498, & HL083156, and a Leducq Transatlantic Network (R.J.H.), K08 HL069842 (F.d.M.), and K01 HL076659 (D.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- [2].Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–84. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- [3].Wong K, Boheler KR, Petrou M, Yacoub MH. Pharmacological modulation of pressure-overload cardiac hypertrophy. Changes in ventricular function, extracellular matrix, and gene expression. Circulation. 1997;96:2239–46. doi: 10.1161/01.cir.96.7.2239. [DOI] [PubMed] [Google Scholar]
- [4].Gwathmey JK, Morgan JP. Altered calcium handling in experimental pressure-overload hypertrophy in the ferret. Circ Res. 1985;57:836–43. doi: 10.1161/01.res.57.6.836. [DOI] [PubMed] [Google Scholar]
- [5].Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–6. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- [6].Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee K-H, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998;95:5251–6. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97:793–8. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schmidt U, del Monte F, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A, et al. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca2+-ATPase. Circulation. 2000;101:790–6. doi: 10.1161/01.cir.101.7.790. [DOI] [PubMed] [Google Scholar]
- [9].del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation. 2001;104:1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101:5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schmidt U, Zhu X, Lebeche D, Huq F, Guerrero JL, Hajjar RJ. In vivo gene transfer of parvalbumin improves diastolic function in aged rat hearts. Cardiovasc Res. 2005;66:318–23. doi: 10.1016/j.cardiores.2004.06.028. [DOI] [PubMed] [Google Scholar]
- [12].Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang LF, et al. Mechanical and metabolic rescue in a type II diabetes model of cardiomyopathy by targeted gene transfer. Mol Therapy. 2006;13:987–96. doi: 10.1016/j.ymthe.2006.01.002. [DOI] [PubMed] [Google Scholar]
- [13].Sandalon Z, Bruckheimer EM, Lustig KH, Rogers L, Peluso R, Burstein H. Secretion of a TNFR:Fc fusion protein following pulmonary administration of pseudotyped adeno-associated virus vectors. J Virol. 2004;78:12355–65. doi: 10.1128/JVI.78.22.12355-12365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hajjar RJ, del Monte F, Matsui T, Rosenzweig A. Prospects for gene therapy for heart failure. Circ Res. 2000;86:616–21. doi: 10.1161/01.res.86.6.616. [DOI] [PubMed] [Google Scholar]
- [15].Ohga Y, Sakata S, Takenaka C, Abe T, Tsuji T, Taniguchi S, et al. Cardiac dysfunction in terms of left ventricular mechanical work and energetics in hypothyroid rats. Am J Physiol. 2002;283:H631–41. doi: 10.1152/ajpheart.00046.2002. [DOI] [PubMed] [Google Scholar]
- [16].Sakata S, Ohga Y, Abe T, Tabayashi N, Kobayashi S, Tsuji T, et al. No dependency of a new index for oxygen cost of left ventricular contractility on heart rates in the blood-perfused excised rat heart. Jpn J Physiol. 2001;51:177–85. doi: 10.2170/jjphysiol.51.177. [DOI] [PubMed] [Google Scholar]
- [17].Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, et al. Relationship between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and non-failing human myocardium. Circ Res. 1994;75:434–42. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- [18].Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–37. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- [19].Pogwizd SM, Bers DM. Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci. 2002;976:454–65. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- [20].Lompré AM, Schwartz K, d′Albis A, Lacombe G, Thiem NV, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature. 1979;282:105–7. doi: 10.1038/282105a0. [DOI] [PubMed] [Google Scholar]
- [21].Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance: plasticity of the myocardium generated through protein isoform switches. J Clin Invest. 1989;84:1693–700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Szatkowski ML, Westfall MV, Gomez CA, Wahr PA, Michele DE, DelloRusso C, et al. In vivo acceleration of heart relaxation performance by parvalbumin gene delivery. J Clin Invest. 2001;107:191–7. doi: 10.1172/JCI9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huq F, Lebeche D, Iyer V, Liao R, Hajjar RJ. Gene transfer of parvalbumin improves diastolic dysfunction in senescent myocytes. Circulation. 2004;109:2780–5. doi: 10.1161/01.CIR.0000131764.62242.96. [DOI] [PubMed] [Google Scholar]
- [24].Perreault CL, Shannon RP, Komamura K, Vatner SF, Morgan JP. Abnormalities in intracellular calcium regulation and contractile function in myocardium from dogs with pacing-induced heart failure. J Clin Invest. 1992;89:932–8. doi: 10.1172/JCI115674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liao R, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS. Decreased energy reserve in an animal model of dilated cardiomyopathy; relationship to contractile performance. Circ Res. 1996;78:893–902. doi: 10.1161/01.res.78.5.893. [DOI] [PubMed] [Google Scholar]
- [26].Suga H. Ventricular energetics. Physiol Rev. 1990;70:247–77. doi: 10.1152/physrev.1990.70.2.247. [DOI] [PubMed] [Google Scholar]
- [27].Goto Y, Slinker BK, LeWinter MM. Decreased contractile efficiency and increased nonmechanical energy cost in hyperthyroid rabbit heart. Relation between O2 consumption and systolic pressure-volume area or force-time integral. Circ Res. 1990;66:999–1011. doi: 10.1161/01.res.66.4.999. [DOI] [PubMed] [Google Scholar]
- [28].Cooper G, Satava RM, Harrison CE, Coleman HN. Mechanism for the abnormal energetics of pressure-induced hypertrophy of cat myocardium. Circ Res. 1973;33:213–23. doi: 10.1161/01.res.33.2.213. [DOI] [PubMed] [Google Scholar]
- [29].Gunning JF, Coleman HN. Myocardial oxygen consumption during experimental hypertrophy and congestive heart failure. J Mol Cell Cardiol. 1973;5:25–38. doi: 10.1016/0022-2828(73)90033-3. [DOI] [PubMed] [Google Scholar]
- [30].Cooper G, Puga FJ, Zujko KJ, Harrison CE, Coleman HN. Normal myocardial function and energetics in volume-overload hypertrophy in the cat. Circ Res. 1973;32:140–8. doi: 10.1161/01.res.32.2.140. [DOI] [PubMed] [Google Scholar]
- [31].Hajjar RJ, Schwinger RHG, Schmidt U, Kim CS, Lebeche D, Doye AA, et al. Myofilament calcium regulation in human myocardium. Circulation. 2000;101:1679–85. doi: 10.1161/01.cir.101.14.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Krause S, Hess ML. Characterization of cardiac sarcoplasmic reticulum dysfuntion during short-term, normothermic, global ischemia. Circ Res. 1984;55:176–84. doi: 10.1161/01.res.55.2.176. [DOI] [PubMed] [Google Scholar]
- [33].Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–6. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- [34].Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, et al. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–8. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- [35].Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–19. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- [36].Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–81. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- [37].Stevenson LW. Clinical use of inotropic therapy for heart failure: looking backward or forward? Part II: chronic inotropic therapy. Circulation. 2003;108:492–7. doi: 10.1161/01.CIR.0000078349.43742.8A. [DOI] [PubMed] [Google Scholar]
- [38].del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–11. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]