

SUMMARY
Telomere maintenance is critical for cancer progression. To examine mechanisms of tumor suppression induced by short telomeres we crossed mice deficient for the RNA component of telomerase, mTR-/-, with Eμ-Myc transgenic mice; an established model of Burkitt’s lymphoma. Short telomeres suppressed tumor formation in Eμ-myc transgenic animals. Expression of bcl2 blocked apoptosis in tumor cells but, surprisingly, mice with short telomeres were still resistant to tumor formation. Staining for markers of cellular senescence showed that pre-tumor cells induced senescence in response to short telomeres. Loss of p53 abrogated the short telomere response. This study providesin vivo evidence for the existence of a p53 mediated senescence mechanism in response to short telomeres that suppresses tumorigenesis.
SIGNIFICANCE
Telomerase inhibition has potential therapeutic benefits for cancer treatment. Experimental systems indicate that short telomeres impair tumor formation by inducing apoptosis. Therefore blocking apoptosis was expected to block the tumor suppressive effect of short telomeres. However, we found that despite efficient inhibition of apoptosis, short telomeres retained potent tumor suppressive signaling in Eμ-myc induced lymphomas. This response required p53 and showed hallmarks of cellular senescence. Thus, telomerase inhibition can engage multiple p53-dependent tumor-suppressor pathways thus may have therapeutic potential for tumors that have lost specific p53 effector functions but retain wildtype p53.
INTRODUCTION
Burkitt’s lymphoma is a highly aggressive, rapidly growing and lethal cancer. Tumors that arise are uniformly associated with translocations that activate the c-myc oncogene. A transgenic mouse model of Burkitt’s lymphoma in which c-myc is expressed in B cells (Adams et al., 1985) provides an excellent model to understand the biology of lymphoma and to test mechanisms that may limit tumor growth. Understanding the pathways that interrupt tumor growth will allow the development of specific therapies for lymphoma.
Cells contain two major intrinsic pathways of tumor suppression, apoptosis and senescence, that can be activated by multiple stimuli (Lowe et al., 2004). Telomerase inhibition was proposed as a potential cancer therapy when telomere shortening was first described in human cells (Harley et al., 1990). Telomerase is critical for telomere length maintenance; when telomerase is absent or blocked, progressive telomere shortening occurs with each cell division. In primary cultures of human cells, telomere shortening results in dysfunctional telomeres which trigger a DNA damage response that ultimately leads to an irreversible state of cellular senescence (Bodnar et al., 1998;d’Adda di Fagagna et al., 2003). In mouse cells, short telomeres also initiate a DNA damage response (Hao et al., 2004), however, in response, these cells do not enter senescence (Blasco et al., 1997;Parrinello et al., 2003).In vivo short telomeres induce apoptosis in multiple highly proliferative tissues including testis germ cells and lymphocytes (Hemann et al., 2001a;Lee et al., 1998). Senescent cells were detected in a subset of hepatocytes that were induced to proliferate via partial hepatectomy in mice with dysfunctional telomeres (Lechel et al., 2005;Satyanarayana et al., 2003). However, senescence was not observed in quiescent hepatocytes in animals with acute telomere dysfunction due to telomere uncapping (Denchi et al., 2006). Strong evidence has accumulated that short telomeres indeed limit tumor growth. Crosses of mTR-/- mice to tumor prone models demonstrate that the short telomere response significantly limits tumor formation (Gonzalez-Suarez et al., 2000;Greenberg et al., 1999;Qi et al., 2003;Qi et al., 2005;Rudolph et al., 2001;Wong et al., 2003). In these experiments, decreased tumor formation correlated with an increase in DNA damage, chromosome instability, and apoptosis. However, senescence was not reported in these studies. Taken together, these observations raise the question whether short telomere induced senescence may act as a tumor suppressor mechanism.
The p53 tumor suppressor protein is the major mediator of the DNA damage response, apoptosis, and senescence. Consistent with this role, p53 deficiency abrogates many of the cellular responses to short telomeres (Chin et al., 1999). In addition, short telomeres stimulated the formation of tumors in p53 deficient mice and caused the occurrence of tumor types that are not normally associated with this tumor prone mouse model (Artandi et al., 2000). The loss of p53 confers multiple advantages to uncontrolled proliferation, including loss of the DNA damage checkpoint, genome stability, senescence, and apoptosis (Vogelstein et al., 2000). It is therefore unclear which p53 effector functions are important for tumor suppression in response to short telomeres.
In the work presented here we demonstrate that short telomeres suppress tumorigenesis in a mouse model of Burkitt’s lymphoma and examine the requirement of apoptosis in mediating this response. We found that, surprisingly, inhibition of apoptosis did not abrogate the reduction in lymphoma formation in response to short telomeres. After long latency, micro-lymphomas that did appear displayed multiple markers of senescence. Genetic evidence showed that p53 was absolutely required for tumor suppression. We conclude that short telomeres activate a p53 dependent cellular senescence pathway limits tumor formation in vivo.
RESULTS
The Eμ-myc transgenic mouse is an established model of Burkitt’s lymphoma. Overexpression of the Myc oncogene in B cells leads to B cell lymphoma with a median onset of four to six months (Adams et al., 1985). To assess the impact of short telomeres on lymphomagenesis we generated Eμ-myc transgenic mice that were deficient for the RNA component of telomerase (Blasco et al., 1997) (Eμ-myc; mTR-/-). To obtain Eμ-myc transgenic mice with short telomeres we intercrossed Eμ-myc; mTR-/- mice for six successive generations in the absence of telomerase. This breeding strategy allows for the comparison of Eμ-myc transgenic mice that are wildtype for telomerase, Myc mTR+/+; those that lack telomerase but have long telomeres, Myc mTR-/- G1; and those that lack telomerase but have short telomeres Myc mTR-/-G5/6.
To determine the effect of short telomeres on tumor formation we monitored cohorts of Myc mTR+/+, Myc mTR-/- G1, and Myc mTR-/- G5/6 mice for lymphoma onset. Consistent with previous studies (Adams et al., 1985), Myc mTR+/+ and Myc mTR-/-G1 animals developed fully penetrant B-cell lymphoma and the latency of lymphoma onset was indistinguishable between the two cohorts (Fig 1A). In contrast, only 7 of 25 Myc mTR-/- G5/6 animals developed lymphoma over the course of the study (P < 10–4,Fig 1A). As expected, telomere length was significantly shorter in Myc mTR-/- G5/6 tumors compared to Myc mTR+/+ and Myc mTR-/- G1 tumors (Fig. 1B). Further, these tumors displayed a high frequency of chromosome end-to-end fusions and non-reciprocal translocations (Fig1. C,D). In contrast Myc mTR+/+ and Myc mTR-/- G1 tumors showed no non-reciprocal translocations and only one independent, but clonal, end-to-end chromosome fusion. These observations indicate that short telomeres induce extensive genomic instability and are a potent inhibitor of tumorigenesis in Eμ-Myc transgenic mice.
Figure 1.
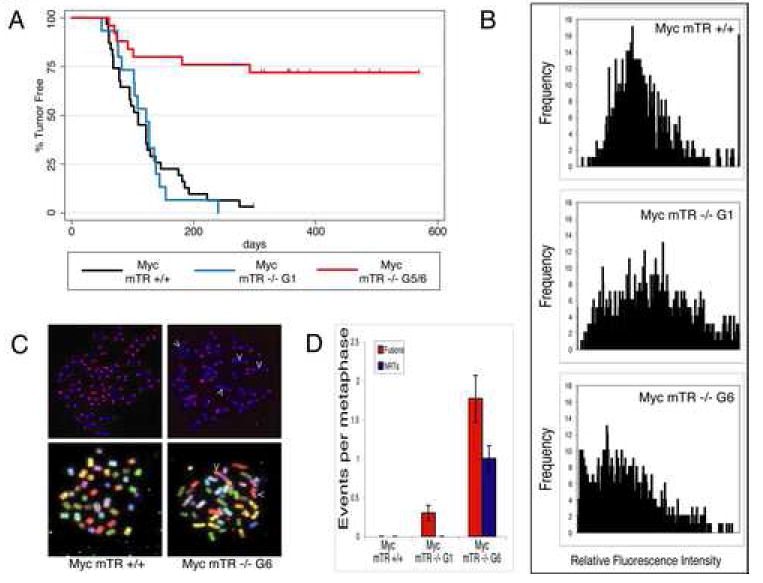
Short telomeres decrease penetrence of tumor formation and initiate genomic instability. A) Kaplan-Meier survival analysis showing lymphoma onset in Myc mTR+/+ (black, n= 31), Myc mTR -/- G1 (blue, n= 18) and Myc mTR -/- G5/6 (red, n=25) cohorts. B) Quantitative fluorescencein situ hybridization (Q-FISH) of tumor cell metaphase spreads from Myc mTR +/+ (top), Myc mTR -/- G1 (middle), and Myc mTR -/- G6 (bottom). Ten metaphases each from three separate lymphomas are represented for each genotype. C) Representative Q-FISH (top) and spectral karyotypes (bottom) for Myc mTR +/+ (left) and Myc mTR -/- G6 (right) tumors. Arrow heads indicate end-to-end chromosome fusions and non-reciprocal translocations in Q-FISH and SKY images respectively. D) Quantitation of chromosome abnormalities. The number of chromosome end-to-end fusions (red bars) and non-reciprocal translocations (blue bars) per metaphase are shown (10 metaphases per tumor n=3). Bars indicate standard error.
Previous studies in telomerase deficient mice have suggested that the apoptotic pathway is critical for short telomere tumor suppression (Gonzalez-Suarez et al., 2000;Greenberg et al., 1999;Qi et al., 2003;Qi et al., 2005;Rudolph et al., 2001;Wong et al., 2003). To directly test the requirement of apoptosis for short telomere mediated tumor suppression, we disabled apoptotic signaling by expressing the Bcl-2 oncogene using an adoptive transfer protocol (Fig. 2A). We cultured bone marrow, enriched for hematopoietic stem cells, from young, non-lymphomic Myc mTR+/+, Myc mTR-/-G1, and Myc mTR-/- G5/6 animals. Whole bone marrow was infected with a murine stem cell retrovirus (MSCV) that expresses Bcl-2 and EGFP from a bicistronic message (Schmitt et al., 2002). Adoptive transfer of Bcl-2 expressing Eμ-myc bone marrow into lethally irradiated syngeneic recipient animals results in rapid lymphoma onset approximately six weeks post transplant (Schmitt et al., 2002). Disabling apoptosis results in a rapid onset of tumors during Eμ-myc lymphomagenesis (Schmitt et al., 2002). As expected, transplantation of Bcl-2 positive HSCs derived from Myc mTR+/+ and Myc mTR-/- G1 animals resulted in rapid tumor onset in all recipient animals in both genotypes (8 of 8 Bcl-2; Myc mTR+/+ and 7 of 7 Bcl-2; Myc mTR-/- G1 mice were lymphoma bearing by 42 days post transplantFig. 2B). These animals harbored highly aggressive tumors displaying complete effacement of the cervical lymph nodes and infiltration of tumor cells into adjacent salivary gland. Surprisingly, animals transplanted with Bcl-2 positive Myc mTR -/- G5/6 bone marrow failed to develop palpable tumors for more than100 days post transplant (0 of 3 Myc mTR-/-G5 (129 days) and 0 of 5 Myc mTR-/- G6 (100 days),Fig. 2B). Histological evaluation of lymphatic tissues from these animals showed small encapsulated tumor masses in the cervical lymph nodes with little infiltration into salivary gland (Fig. 5A). To confirm that Bcl-2 was blocking apoptosis in the tumor cells in these animals, we used a terminal UTP nick end labeling (TUNEL) protocol to examine the extent of apoptosis in lymphomas from Bcl-2 expressing mice. Apoptotic levels were significantly reduced in Bcl-2 expressing lymphomas compared to spontaneously arising Eμ-myc lymphomas that do not express Bcl-2 (Fig. 2C). Importantly, the amount of apoptotic cells was similar in all Bcl-2 positive lymphomas tested (t-test; p=0.6 for Myc mTR +/+ to Myc mTR -/- G6 comparison). These data indicate that short telomeres effectively abolished tumor progression even when apoptosis was blocked.
Figure 2.
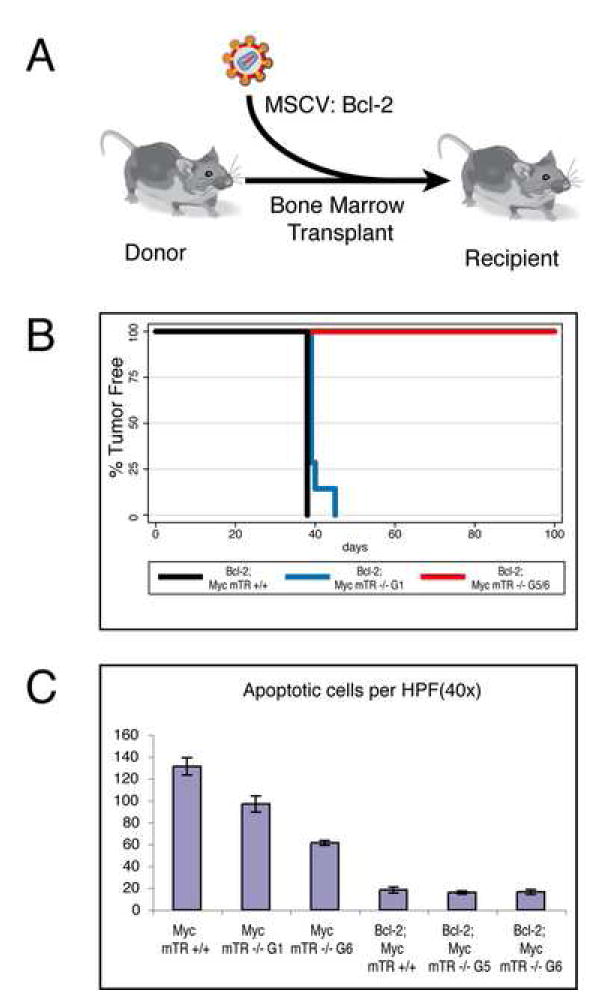
Short telomeres suppress lymphoma formation despite abrogation of the apoptotic program. A) Experimental bone marrow transplant scheme. Bone marrow from donors of defined genotypes was infected with Bcl-2 expressing murine stem cell virus prior to transplantation into lethally irradiated wildtype recipient animals. Iconography reproduced by kind permission of New Science Press fromImmunity (DeFranco et al., 2007).B) Kaplan-Meier survival analysis of Myc mTR +/+ (black, n=8), Myc mTR -/-G1 (blue, n=7), and Myc mTR -/- G5/6 (red, n=3(G5), n=5 (G6)) cohorts. C) TUNEL analysis of tumor masses from Bcl-2; Myc mTR+/+, Bcl-2; Myc mTR-/- G5, and Bcl-2 Myc mTR-/- G6. Bars indicate standard error.
Figure 5.
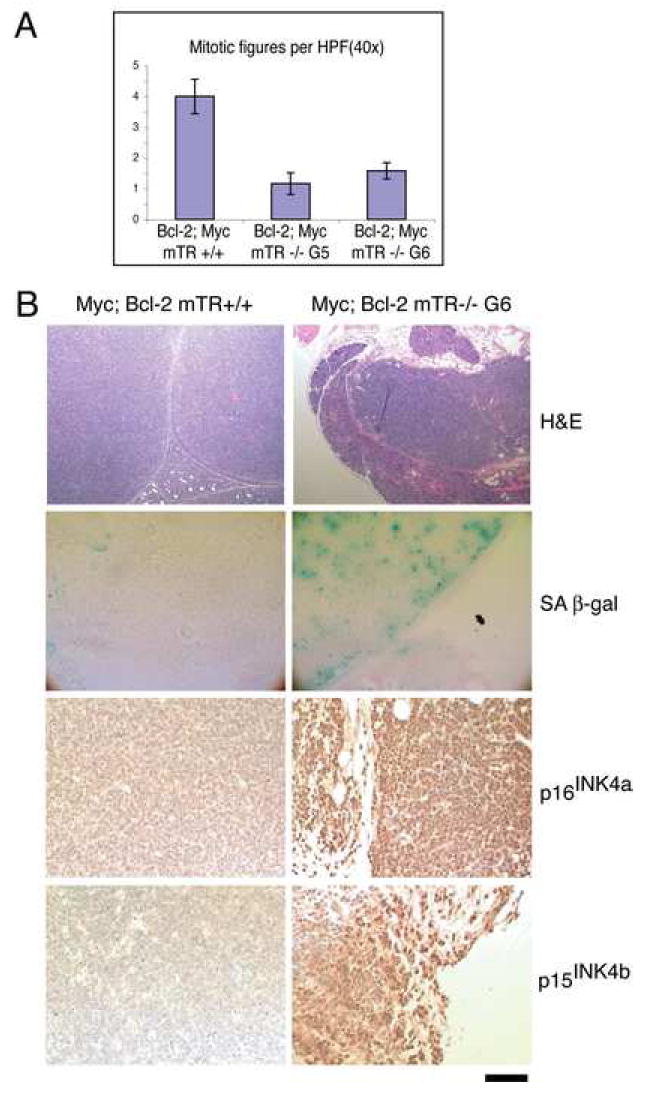
Micro-lymphomas from Bcl-2; Myc mTR-/- G5/6 animals are senescent. A) Mitotic Indices for Bcl-2; Myc mTR+/+, Bcl-2; Myc mTR-/- G5, and Bcl-2; Myc mTR-/- G6. Bars indicate standard error. B) Histological analysis of Bcl-2; Myc mTR+/+ and Bcl-2; Myc mTR-/- G6 lymphomas. Top) H&E stain showing complete effacement of cervical lymph node in Bcl-2; Myc mTR +/+ and small encapsulated micro-lymphoma surrounded by salivary glands in Bcl-2; Myc mTR-/- G6. Senescence associated β-gal activity, p16INK4a and p15INK4b immuno-stain specifically associated with Bcl-2; mTR -/- G6 micro-lymphomas. Scale bar represents 250 μm for low power hematoxylin and eosin (H&E) stained images (top panels) and 100 μm for all other images.
Decreased tumor formation in the absence of apoptosis suggested another mechanism is limiting tumor growth. p53 regulates multiple pathways that act to suppress tumor formation including apoptosis and senescence (Lowe et al., 2004;Vogelstein et al., 2000). To genetically test whether p53 is required for the short telomere induced suppression of lymphoma, we crossed mice doubly heterozygous for mTR and p53 (mTR+/- p53+/-) to Myc mTR-/- G6 mice (Fig. 3A). This cross results in intergenerational G7 (iG7) littermates that all have half long and half short telomeres and are either telomerase positive (mTR+/-) or telomerase negative (mTR-/-). The iG7-/- mice have short telomeres that mimic mTR-/- G6 mice (Feldser et al., 2006; Hemann et al., 2001b;Qi et al., 2003). Eμ-myc transgenic animals that are heterozygous for p53 (p53+/-) invariably develop p53 deficient lymphomas due to loss of the remaining wildtype p53 allele (LOH) (Schmitt et al., 1999). As expected, Myc iG7 mTR+/- p53+/- mice formed tumors rapidly. Interestingly, Myc iG7 mTR-/- p53+/- mice also formed tumors with similar rapid onset as their telomerase positive counterparts; all animals developed lymphoma by day 42 after birth (Fig. 3B). Despite similar rates of lymphoma onset, iG7 mTR-/- tumors had significantly shorter telomeres, high rates of chromosome end-to-end fusions and numerous non-reciprocal translocations, whereas their telomerase positive (mTR+/-) counterparts had none (Fig. 3D). These genetic experiments indicate that short telomere induced tumor-suppression requires the p53 tumor-suppressor pathway and that loss of p53 abrogates the cellular response to telomere dysfunction.
Figure 3.
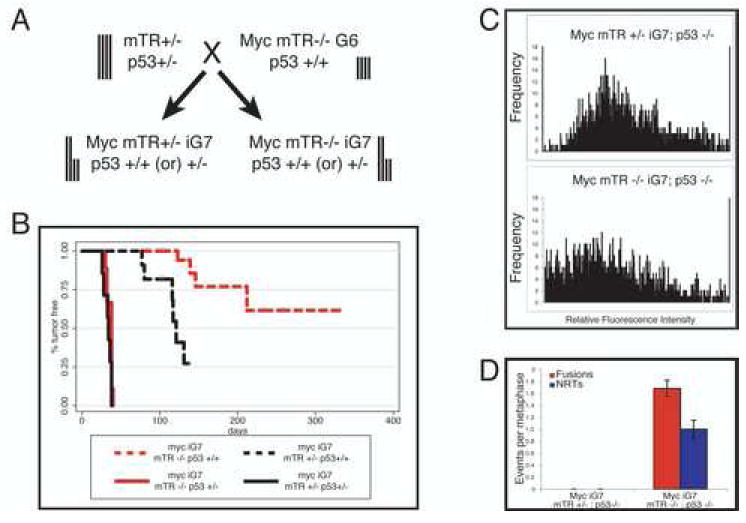
Short telomeres block tumor formation in a p53 dependent manner. A) Intergenerational breeding scheme. Vertical bars represent telomere lengths. B) Kaplan-Meier survival analysis of Myc iG7 mTR +/- p53 +/+ (black dashed, n=11) Myc iG7 mTR -/- p53 +/+ (red dashed, n=20), Myc iG7 mTR +/- p53 +/-(black solid, n=7), and Myc iG7 mTR -/- p53 +/- (red, solid n=9) cohorts. Myc p53+/- animals invariably develop Myc p53-/- tumors at around 40 days. Short telomeres promote chromosomal abnormalities in p53 deficient lymphomas. C) Q-FISH analysis of metaphase spreads from Myc mTR +/- p53 -/- (top) and Myc mTR -/-p53 -/- (bottom) lymphomas. Ten metaphases each from three separate lymphomas are represented for each genotype. D) Quantitation of chromosome abnormalities. The number of chromosome end-to-end fusions (red bars) and non-reciprocal translocations (blue bars) per metaphase are shown. (10 metaphases per tumor n=3). Bars indicate standard error.
In addition to these genetic data, several lines of evidence suggest that loss of p53 is critical for tumor development in the presence of short telomeres. First, tumors that are derived from Myc mTR-/- G5/6 animals (Fig 1) are unable to elicit a G1 checkpoint in response to γ-IR and these tumors also do not initiate apoptosis after γ-IR treatment (Fig 4A,B and D). Second, immunoblot analysis showed that in these tumors p53 was either undetectable regardless of γ-IR treatment or was grossly over-expressed and uninducible (Fig 4C). Finally, p19ARF was over-expressed in all tumors from Myc G5/6 animals (6 out of 6 lymphomas tested χ2 test p=0.01 compared to Myc mTR+/+). These observations are consistent with p53 inactivation in the tumors; either by deletion or by point mutation which impairs p53 degradation (Eischen et al., 1999). This implies that for tumors to grow in the presence of short telomeres the p53 pathway must be inactivated.
Figure 4.
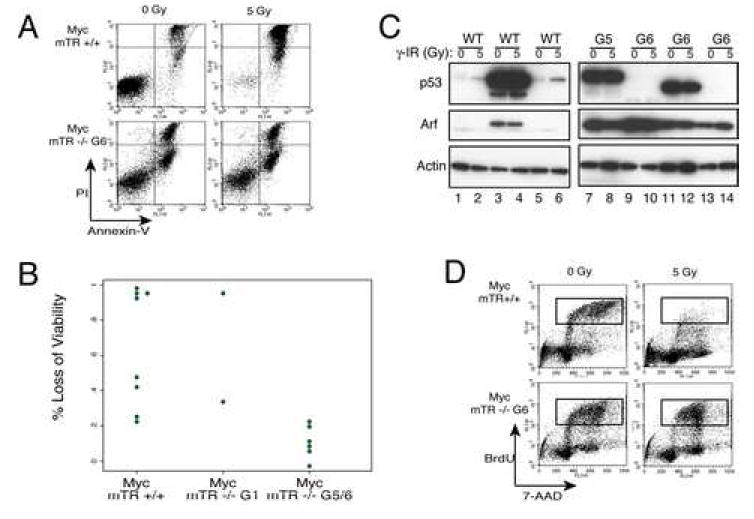
A) Loss of apoptotic induction in tumor cells derived from Myc mTR-/- G5/6 mice. Six hours after 0 or 5 Gy γ-IR cells were stained with Annexin-V FLUOS (FL-1) and propidium iodide (FL-3) and processed by flow cytometry. Representative tumors from Myc mTR +/+ (top) and Myc mTR -/- G6 (bottom) are shown. Sensitivity to γ-IR is shown by loss of viable cells (lower left quadrant) after irradiation. B) Summary of the percent of viable cells, as described in A, for all tumors analyzed. C) Immunoblot of non-irradiated or irradiated (5 Gy γ-IR) tumor cell lysates for p53, Arf, and Actin. Representative tumors from Myc mTR +/+ (left panel) and Myc mTR -/- G5/6 (right panel). Normal p53 induction shown in lanes 1-2, and 5-6, overexpression of mutant form in lanes 3-4, 7-8, and 11-12. Complete loss of p53 protein is shown in lanes 9-10, and 13-14. Arf expression is seen only in samples with suspected p53 mutations (Eischen et al., 1999). D) Radio-resistant DNA synthesis in lymphomas with p53 mutations. Cells were treated with 5 Gy γ-IR after 1 hour BrdU was added to culture medium then six hours post irradiation cells were fixed and stained with anti-BrdU antibody and subjected to FACS analysis (FL-3 = 7-AAD signal and FL-1 = anti-BrdU FLUOS) the boxed area indicates cells in S-phase.
The requirement for p53 but not apoptosis implies that p53 mediated cellular senescence may be responsible for tumor suppression. To determine whether short telomeres could be blocking tumor progression by initiating senescence, we analyzed micro-lymphomas from Bcl-2; Myc mTR-/- G5/6 animals (Fig 2) for proliferation defects and the presence of senescence markers. We scored mitotic indices from tissue sections derived from Bcl-2; Myc mTR+/+, and Bcl-2; Myc mTR-/- G5/6 lymphomas. Compared to Bcl-2;Myc mTR+/+ sections, Bcl-2; Myc mTR-/- G5/6 sections had significantly fewer mitotic figures per high power field (Fig. 5A p<0.0001) indicating that short telomeres negatively affect cellular proliferation. Next Bcl-2; Myc lymphomas were analyzed for the presence of markers that are specifically upregulated in senescent tissues (Collado and Serrano, 2006). Bcl-2 expressing Myc mTR-/- G5/6 micro-lymphomas stained positive for SA-βgal, and displayed both p16INK4a and p15INK4b immunohistochemical staining. In contrast, Bcl-2 expressing Myc mTR+/+ lymphomas did not stain for any of these markers ( Fig. 5 B). These staining patterns indicate that Bcl-2 expressing Myc mTR-/- G5/6 micro-lymphomas were indeed senescent. Taken together with the failure of these lymphomas to progress, these data indicate that short telomeres can induce senescencein vivo to effectively abolish tumor progression.
DISCUSSION
Understanding cellular mechanisms that may limit tumor growth could provide new therapeutic approaches to cancer therapy. Telomere shortening, through the inhibition of telomerase, has been proposed as a potential cancer treatment. Here we show that short telomeres can block lymphoma induction in a mouse model of Burkitt’s lymphoma. Short telomeres have been shown to reduce tumor formation in several other mouse tumor models (Gonzalez-Suarez et al., 2000;Greenberg et al., 1999;Qi et al., 2003;Qi et al., 2005;Rudolph et al., 2001;Wong et al., 2003), however in all of those systems apoptosis was thought to be responsible for the decreased incidence of tumor formation. We found that blocking apoptosis still resulted in tumor suppression. p53 was required for the tumor reduction due to short telomeres, and direct evidence indicated a p53 mediated senescence program was activated in the small tumors that did form. These results indicate that short telomeres can mediate a p53 dependant senescence program that limits tumor formation in vivo.
Telomere mediated senescence in tumors may not have been detected in previous studies because it likely is initiated in only a few cells. Recently, cellular senescence induced by oncogene expression has been established as a tumor suppressor mechanismin vivo (Braig et al., 2005;Chen et al., 2005;Collado et al., 2005;Michaloglou et al., 2005). Activation of cellular senescence in these instances occurs in all cells in the initiating tumor mass, thus facilitating detection of senescence markers. In contrast, senescence induced by short telomeres may occur initially in only a few cells in which telomeres become critically short. This may impede the detection of short telomere mediated senescence during tumor formation.
Short telomeres can engage both the apoptosis and the senescence pathways. p53 is necessary to carry out both apoptotic and senescence pathways (Fig. 6). The requirement for the loss of p53 for the growth of the Eμ-myc lymphomas with short telomeres suggest both apoptosis and the senescence play roles in limiting tumor growth. If only one tumor suppressor pathway, apoptosis or senescence, were necessary for short telomere mediated tumor suppression we would have expected mutations in other components of theses pathways to also allow tumor formation in these mice. For example, if disruption of apoptosis were the only p53 effector function required for tumor formation in the presence of short telomeres, we would have expected over-expression of Bcl-2 to confer a selective growth advantage regardless of telomere length. Consistent with this, we speculate that loss of p16 pathway components in tumors that express Bcl-2 would be resistant to the tumor suppressive effects of short telomeres (Fig. 6).Tumor suppressor programs induced by p53 are context dependent. Restoration of p53 function in p53 null tumors induces tumor regression in multiple tumor types (Martins et al., 2006;Ventura et al., 2007;Xue et al., 2007). However, p53 restoration induces apoptosis in lymphomas (Martins et al., 2006;Ventura et al., 2007), and senescence in sarcomas(Ventura et al., 2007) and liver carcinomas(Xue et al., 2007). It is unclear whether p53-induced senescence or apoptosis correlates with cell type, oncogenic lesion, or both. We show here that in a tumor type that is predisposed to p53-mediated apoptosis, short telomeres can effectively redirect the p53 tumor suppressor response to induce senescence. It would be interesting if short telomeres could, likewise, alter p53-mediated senescence programs to induce apoptosis in tumor types predisposed to undergo senescence.
Figure 6.
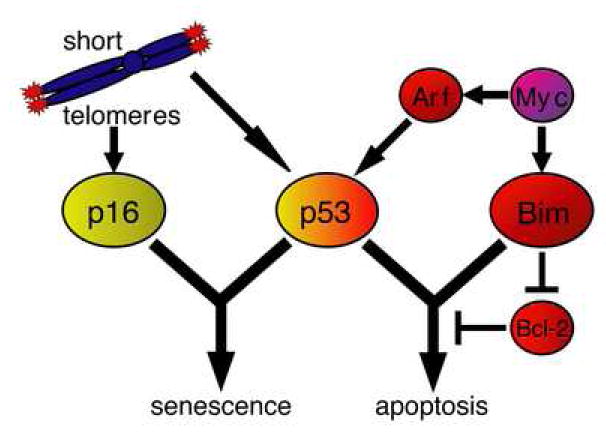
Tumor suppressor signaling downstream of short telomeres and myc. The myc oncogene negatively regulates its effects on cell proliferation by inducing both Arf and Bim tumor suppressors. Arf activity results in stabilization of the p53 protein, whereas Bim is directly transcribed by myc and induces apoptosis by inhibiting Bcl-2. Activation of both Arf and Bim is required for myc induced apoptosis. Apoptotic signaling can also be disrupted by over-expression of the downstream anti-apoptotic Bcl-2 oncogene. The short telomere response signals through both p16 and p53 tumor suppressor pathways, which carry out senescence and/or apoptotic tumor suppressor functions. The p53 tumor suppressor lies at the crossroads of senescence and apoptosis and is required for both programs.
Short telomeres and p53 deficiency have profound effects on tumor formation. First, short telomeres promote transformation of p53 deficient mouse embryo fibroblasts (Chin et al., 1999). Second, short telomeres contribute to mortality of tumor prone p53 null mice by increasing tumor formation and facilitating tumorigenesis in tissues not usually prone to tumor formation in this system (Artandi et al., 2000;Jacks et al., 1994). Here we used a mouse tumor model in which the initiating mutation, the Myc transgene, is unrelated to p53 and found that the growth of tumors with short telomeres requires p53 loss. This requirement for p53 has implications for the clinical application of telomerase inhibitors. First, the fact that short telomeres can induce both apoptosis and senescence to limit tumorigenesis indicates both of these pathways can be targeted in cancer therapy. However, the data also imply that telomerase inhibition will be most effective in tumors that have an intact p53 pathway. Alternatively telomerase inhibition may be combined with approaches that reinstate p53 function or other mechanisms to activate senescence or apoptosis programs in tumors. The lymphoma model described here thus provides an excellent system to understand the genetic requirements that will guide the development of more sophisticated approaches to targeting telomerase in cancer therapy.
Experimental Methods
Lymphoma Monitoring and Analysis
The Institutional Animal Care and Use Committee at The Johns Hopkins University approved all animal procedures. Eμ-myc transgenic animals were inspected twice weekly for lymph node enlargement by palpation and for overall signs of distress. Animals were considered lymphomic when tumor mass was easily palpated; no more than one centimeter in diameter. Lymphoma bearing animals were euthanized and tumor masses were dissociated through nylon mesh into single cell suspensions and cultured on mitomycin C treated 3T3 feeder layers in B-cell media (45% DMEM (Invitrogen), 45% IMDM (Invitrogen), 10% FBS (Hyclone), Penn/Strep/Glutamine (Invitrogen)).
Chromosome Analysis
Metaphase spreads were generated by arresting exponentially growing cultures with 0.5 μg/mL KaryoMax (Gibco) 2 hours before hypotonic swelling in 0.075 M KCl and fixation in 3:1 methanol:acetic acid. Fixed cells were dropped onto glass slides over a steaming water bath. Telomere length was determined by hybridization of methaphases with Cy3 (CCCTAA)3 PNA probes (PE Biosystems) as described (Lansdorp et al., 1996). Spectral karyotyping was performed on metaphases as described (Liyanage et al., 1996). Images were obtained using IP-Lab software on a Zeiss Axioscope microscope.
Immunoblot, Apoptosis and Cell Cycle Analysis
Exponentially growing lymphoma cell (2 × 106 cells) were grown in six-well plates. Cells were exposed to 0 or 5 Gy γ-IR from a cesium source (Gamma Cell) prior to analyses. For Immunoblot analysis: Cells were lysed in RIPA buffer containing protease inhibitors (Roche), denatured in SDS loading buffer, separated on a 4-12% gradient Tris-acetate gel (Novex), transferred to PVDF (MilliPore), and probed with anti-p53 (CM5 Novocastra), anti-Arf (Ab80 Abcam) and anti-actin (Sigma). For Apoptosis analysis: Cells were harvested, washed in PBS, then stained with Annexin-V FLUOS staining kit (Roche) per manufactures instruction. For BrdU incorporation: BrdU was added to culture medium one hour post irradiation and analysis was performed six hours after γ-IR. Cells were harvested and analyzed usingin situ cell proliferation kit (Roche) per manufacturers instructions.
Bone Marrow Transplantation Protocol and Retrovirus Production
Donor animals (6-10 week old on C57/B6J background) were primed with 5-FU (150 mg/kg) four days prior to bone marrow harvest. Marrow was harvested from tibia and femur by flushing with Hank’s balanced salt solution with a 23 gauge needle. After red cell lysis, cells were cultured in IMDM with 18% FBS (Hyclone) 4% WEHI-3 conditioned media, 10ng/mL IL-3, 10 ng/mL IL-6, and 100ng/mL mSCF (Fitzgerald). Ecotropic retrovirus was produced using Phoenix cells (G. Nolan, Stanford) by transfecting 12μg MSCV:Bcl-2 (Schmitt et al., 2002) into 2×106 cells with 20μL Lipofectamine 2000 (Invitrogen) overnight. Virus was collected 24 and 48 hours post transfection and used immediately to infect bone marrow cells four times by 90 minute spinfection (1000g). Recipient animals (C57/B6J, Jackson Labs) were lethally irradiated with 9 Gy total body γ-IR (Gamma Cell cesium source) and transplanted with 2.5×105 bone marrow cells.
In situ Apoptosis and Mitosis
Formalin fixed tissue sections were stained usingin situ cell death kit (Roche) per manufactures instructions and counterstained with DAPI. Mitotic cells were counted based on morphology and apoptotic cells identified by Cy-3 label after TUNEL reaction.
Histology and Immunohistochemistry
Tissues harvested from lymphoma bearing animals were fixed 4% neutral buffered formalin, then sectioned into 5μm thick sections for H&E and immunochemical stain. Prior to antibody hybridization, antigens were retrieved by boiling slides for 10 minutes in 10mM Citrate solution. Positive staining was routinely assessed by comparing serial sections exposed to either specific primary antibodies or antibody dilution buffer only (negative controls). All subsequent steps were identical. p16Ink4a antibody (F-12 Santa Cruz, 1:100) and p15INK4b (Ab-6 Lab Vision, 1:50) antibody were visualized with Vectastain Elite kit (Vector Labs). For SAβ-gal activity, tissues were snap frozen in OCT mounting media (Sakura) and 12 μm sections were stained as described (Dimri et al., 1995).
Acknowledgments
We would like to thank Scott Lowe and Michael Hemann for reagents, protocols and helpful discussions, David Huso for help with histology, and Michael Hemann, Stephen Desiderio and members of the Greider lab for critical reading of the manuscript. This work was supported by NIH grant P01CA16519 to C.W.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- DeFranco AL, Locksley RM, Robertson M. Immunity: The Immune Response in Infectious and Inflammatory Disease. London: New Science Press; 2007. [Google Scholar]
- Denchi E, Celli GB, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev. 2006;20:2648–2653. doi: 10.1101/gad.1453606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldser D, Strong MA, Greider CW. Ataxia telangiectasia mutated (Atm) is not required for telomerase-mediated elongation of short telomeres. Proc Natl Acad Sci U S A. 2006;103:2249–2251. doi: 10.1073/pnas.0511143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet. 2000;26:114–117. doi: 10.1038/79089. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Hao LY, Strong MA, Greider CW. Phosphorylation of H2AX at short telomeres in T cells and fibroblasts. J Biol Chem. 2004;279:45148–45154. doi: 10.1074/jbc.M403924200. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Rudolph KL, Strong MA, DePinho RA, Chin L, Greider CW. Telomere dysfunction triggers developmentally regulated germ cell apoptosis. Mol Biol Cell. 2001a;12:2023–2030. doi: 10.1091/mbc.12.7.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001b;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap AK, Tanke HJ. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- Lechel A, Satyanarayana A, Ju Z, Plentz RR, Schaetzlein S, Rudolph C, Wilkens L, Wiemann SU, Saretzki G, Malek NP, et al. The cellular level of telomere dysfunction determines induction of senescence or apoptosis in vivo. EMBO Rep. 2005;6:275–281. doi: 10.1038/sj.embor.7400352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat Genet. 1996;14:312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Strong MA, Karim BO, Armanios M, Huso DL, Greider CW. Short telomeres and ataxia-telangiectasia mutated deficiency cooperatively increase telomere dysfunction and suppress tumorigenesis. Cancer Res. 2003;63:8188–8196. [PubMed] [Google Scholar]
- Qi L, Strong MA, Karim BO, Huso DL, Greider CW. Telomere fusion to chromosome breaks reduces oncogenic translocations and tumour formation. Nat Cell Biol. 2005 doi: 10.1038/ncb1276. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KE, Wustefeld T, Blasco MA, Manns MP, Rudolph KL. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. Embo J. 2003;22:4003–4013. doi: 10.1093/emboj/cdg367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev. 1999;13:2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007 doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007 doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]