

Abstract
ECF-L is a novel autocrine stimulator of osteoclast (OCL) formation that enhances the effects of 1,25-(OH)2D3 and RANK ligand (RANKL) and is increased in inflammatory conditions such as rheumatoid arthritis. ECF-L acts at the later stages of OCL formation and does not increase RANKL expression. Thus, its mechanism of action is unclear. Therefore, RAW 264.7 cells and M-CSF-dependent murine bone marrow macrophage (MDBM) cells were treated with RANKL and/or with recombinant ECF-L expressed as a Fc fusion protein (ECF-L-Fc) to determine their effects on NF-κB, AP-1 and JNK activity, and on the expression of the adhesion molecules that have been implicated in OCL formation. These parameters were measured by semiquantitative and PCR and Western blot analysis. In addition, the role of ICAM-1 was further assessed by treating normal mouse marrow cultures with ECF-L-Fc and 10−10 M 1,25-(OH)2D3 in the presence or absence of a blocking ICAM-1 antibody or treating marrow cultures from ICAM-1 knockout mice with ECF-L and 1,25-(OH)2D3. ECF-L-Fc by itself only modestly increased NF-κB binding and JNK activity in RAW 264.7 cells, which was further enhanced by RANKL. In contrast, ECF-L-Fc increased LFA-1α and ICAM-1 mRNA levels 1.8-fold in mouse marrow cultures, and anti-ICAM-1 almost completely inhibited OCL formation induced by 10−10 M 1,25-(OH)2D3 and ECF-L. Furthermore, ECF-L did not increase OCL formation in marrow cultures from ICAM-1 knockout mice. Taken together, these results demonstrate that ECF-L enhances RANKL and 1,25-(OH)2D3-induced OCL formation by increasing adhesive interactions between OCL precursors through increased expression of ICAM-1 and LFA-1.
Keywords: ECF-L, Osteoclast, RANKL, ICAM-1, LFA-1
Introduction
We recently identified eosinophil chemotactic factor-L (ECF-L) as an OCL stimulatory factor produced by OCL [1]. ECF-L was originally purified as an eosinophil chemotactic factor from the culture supernatant of mouse splenocytes [2]. ECF-L is also chemotactic for T lymphocytes and bone marrow polymorphonuclear leukocytes in vitro. ECF-L is identical to Ym1 but not its isomer, Ym2 [3]. Ym1 is a macrophage protein that is transiently synthesized and secreted by activated macrophages during inflammation, such as rheumatoid arthritis [4]. Furthermore, prominent expression of Ym1 has been observed in early myeloid precursor cells in fetal liver, spleen and bone marrow [5].
We previously reported that ECF-L increases OCL formation in a dose-dependent manner in mouse bone marrow cultures in the presence of very low concentrations of 1,25-(OH)2D3 10−10 M or RANKL (10 ng/mg) [1]. Chemotactic assays showed that ECF-L increased migration of OCL precursors. Importantly, ECF-L does not induce RANKL, but a neutralizing antibody to ECF-L blocked RANKL or 1,25-(OH)2D3-induced OCL formation in mouse marrow cultures not treated with exogenous ECF-L. These results demonstrated that ECF-L is an important cofactor for RANKL-induced OCL formation. Mechanistic studies showed that recombinant ECF-L increased the average number of nuclei per OCL compared with cultures treated with control Fc protein and acted at the later differentiation/fusion stages of OCL formation. Taken together, these results support an important role for ECF-L in OCL formation. However, the molecular mechanisms responsible for the effects of ECF-L on osteoclastogenesis are unknown. Since ECF-L enhances the effects of RANKL and acts on the later stages of OCL formation, we investigated the effects of ECF-L on the RANK signaling pathway, including NF-κB and JNK, and on the expression of cell adhesion molecules implicated in OCL precursor fusion, ICAM-1 and LFA-1.
Materials and methods
Materials
Fetal calf serum (FCS) and tissue culture media were purchased from Life Technologies (Grand Island, NY). Restriction enzymes and Taq polymerase were purchased from Invitrogen (Carlsbad, CA). RANKL and 1,25-(OH)2D3 were generously provided for these experiments by Amgen (Seattle, WA) and Teijin (Tokyo, Japan) respectively. NF-κB and AP-1 oligonucleotides were purchased from Promega (Madison, WI) as were the gel shift assay kits. The JNK assay kit and c-Jun antibody were from Cell Signaling Technology (Beverly, MA). Goat polyclonal antibody to ICAM-1, bovine anti-goat IgG and polyclonal antibody to actin from Santa Cruz Biotechnology (Santa Cruz, CA) were used for Western blot analysis. Purified hamster anti-mouse ICAM-1 monoclonal antibody and hamster IgG (BD Bioscience, San Diego, CA) were used in mouse marrow cultures. ICAM-1 knockout mice that lack the entire coding region of ICAM-1 were generously provided by Dr. A. Beaudet (Baylor College of Medicine, Houston, TX). These ICAM-1 mutant mice have been used in other studies and shown to totally lack ICAM-1 expression [6].
Bone marrow cell cultures
The femurs and tibias of adult mice were aseptically removed and dissected free of adhering tissues. The bone ends were cut off with scissors, and the marrow cells were recovered by slowly flushing one end of the bone with α-Minimal Essential Medium (α-MEM) using a sterile 25 g needle fitted to a 1 ml syringe. The marrow cells were collected, washed with α-MEM and red blood cells removed by treatment with 0.727% NH4Cl–0.017% Tris–HCl (pH 7.2)–phosphate-buffered saline (PBS) solution. After washing, 5 × 106 cells/ml were cultured for 24 h in α-MEM containing 10% FCS, 100 IU/ml penicillin G, 100 μg/ml streptomycin and rhM-CSF (100 ng/ml). The cells were then washed vigorously with PBS twice to remove nonadherent cells. The adherent cells were harvested by pipetting with 0.02% EDTA in PBS and seeded at 3 × 105 cells/ml in a 10-cm dish. After a further 3-day culture in the same media (the second passage), marrow cells were recovered. There was approximately a 10-fold increase in the cell number recovered compared with the initial number of cells plated. These cells were used as M-CSF-dependent bone marrow macrophage (MDBM) cells for subsequent assays [7].
Production and purification of recombinant ECF-L
The production and purification of recombinant ECF-L (ECF-L-Fc) were carried out as described previously [1]. Briefly, a mouse ECF-L cDNA was generated by PCR, and the EcoRV-digested PCR product was fused with the Fc coding domain of human IgG1. The ECF-L-Fc construct was stably transfected into 293 cells, using a CaPO4 mammalian transfection kit (Stratagene, La Jolla, CA), according to the manufacturer’s protocol. ECF-L-Fc fusion protein was purified from 293 cells conditioned media by protein G affinity chromatography (Pierce, Rockford, IL). A mammalian expression vector pcDNA3 including the Fc coding domain of human IgG1 was also transfected into 293 cells and purified for use as control Fc protein.
NF-κB and AP-1 electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared as described previously [8]. EMSA was performed by incubating nuclear extracts prepared from control Fc and ECF-L-Fc treated RAW 264.7 cells stimulated with RANKL (20 ng/ml) for 30 min. Nuclear extracts were incubated with [γ-32P] ATP-labeled oligonucleotides representing AP1 mutant (5′ GATCCATCTGGGAATGTCAGGAGAGAAGG-3′) or MHC kB (5′-GAGGCT GGGGAT TCCCCATCTC-3′) binding sites. Electrophoresis was performed using a 4% nondenaturing polyacrylamide gel for supershift anti-p65 antibody F-6, and anti-Fos K-25 from Santa Cruz Biotechnology was used (Santa Cruz, CA).
JNK activity assays and Western blot analysis
JNK activity was measured in mouse marrow cells after stimulation with ECF-L-Fc and/or RANKL and was determined in cell extracts, using a JNK assay kit, according to the manufacturer’s protocol. Briefly, JNK was precipitated by incubating cell extracts overnight with c-Jun fusion protein beads. Beads were resuspended in kinase buffer supplemented with 100 μM ATP and incubated for 30 min at 30°C. The reaction was terminated with SDS sample buffer. Samples were subjected to SDS-PAGE and then transferred to nitrocellulose membranes. After blocking, the membranes were incubated with phospho-c-Jun (Ser63) antibody at 1:1000 dilution overnight. Horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma, St. Louis, MO) was used as a secondary antibody (1:10,000), and the blots were developed with an enhanced chemiluminescent system (Pierce, Rockford, IL) on Kodak X-ray films. The c-Jun antibody was used as an internal control.
RNA-based assays
Mouse bone marrow cells (1.2 × 107/well) and free stromal cells were cultured with ECF-L-Fc and/or 10−10 M 1,25-(OH)2D3 in 6-well plates for 1 day. Total RNA was extracted with RNA-BEE (Tel Test, Friendswood, TX) according to the manufacturer’s protocol. RT was carried out in an identical 20-μl reaction with Superscript II RNase H− (Invitrogen) or with MuLV reverse transcriptase (Roche Applied Science) using an oligo (dT) primer and 10 ng of RNA, according to the manufacturer’s protocol. The expression levels of mouse LFA-1 and ICAM-1 mRNA were determined by RT-PCR analysis. Specific primer sets were designed from published cDNA sequences: murine LFA-1α (sense: 5′-CAGTGAGGACCTCATCACAT-3′; antisense: 5′-GAGGTGTTC-CAAATGGAAGC-3′), LFA-1β (sense: 5′-GTCGGAAGGACAATAGTTCC-3′; antisense: 5′-TATCCAACAGCCTTCCGAGT-3′), murine ICAM-1 (sense: 5′-AACAGCAGTGACTCTGTGTC-3′; antisense: 5′-GGTGAAGTCTGT-CAAACAGG-3′), GAPDH (sense: 5′-ACCACAGTCCATGCCATCAC-3′; antisense: 5′-TCCACCACCCTGTTGCTGTA-3′). GAPDH was used as an internal control. The annealing temperatures for each PCR reaction were as follows: 58°C for LFA-1α and β, 55°C for ICAM-1 and 60°C for GAPDH. The identity of all RT-PCR products was confirmed by sequence analysis.
Western blot analysis and antibodies
Mouse bone marrow cells (2 × 107/well) were cultured with ECF-L-Fc and/or 10−10 M 1,25-(OH)2D3 in 6-well plates for 1 day. Control cultures were treated with a control Fc protein. Protein extraction was performed at 4°C. Cells were washed and lysed by adding 150 mM NaCl, 0.1% Nonidet P-40, 10 mM Tris–HCl, pH 7.8, with 1 mM EDTA, 2 mM Na3VO4, 10 mM NaF and 10 μg/ml aprotinin. Lysates were clarified by centrifugation at 5000×g for 20 min. Protein was measured by alkaline copper reduction with bicinchoninic acid (absorbance at 660 nm) (Pierce). Twenty micrograms of the cell lysates was separated on 8% polyacrylamide gels in Laemmli buffers and transferred electrophoretically onto polyvinylidene difluoride membranes. The membranes were incubated with an ICAM-1 antibody (1:1000) for 1 h. Horseradish peroxidase-conjugated bovine anti-goat IgG was used as a secondary antibody (1:2000). Antibody to actin was used for an internal control.
Immunoreactive proteins were visualized using enhanced chemiluminescent (Amersham Biosciences). Where indicated, blots were stripped for 20 min in 2% SDS, 100 mM 2-mercaptoethanol, 62.5 mM Tris–HCl, pH 6.7, at 50°C.
Murine OCL-like multinucleated cell formation assays
Mouse bone marrow cultures from C57Bl6 or ICAM-1 knockout mice were performed as previously described [9] to assess the role of ICAM-1 in OCL formation induced by ECF-L-Fc. In selected experiments, varying concentrations of anti-ICAM-1 or control IgG were added to C57Bl6 marrow cultures. Briefly, freshly isolated C57Bl6 or ICAM-1 knockout mouse bone marrow cells (106/ml in α-MEM containing 10% FCS/well) were cultured in 48 well plates for 6–7 days. At the end of culture period, the cells were fixed and stained for tartrate resistant acid phosphatase (TRACP), using a TRACP staining kit (A-367, Sigma) to identify OCL-like multinucleated cells. OCLs containing >3 nuclei/cell were counted with an inverted microscope.
Statistical analysis
All experiments were performed in quadruplicate, and the mean±SD for the number of OCLs formed was determined. The means of individual treatment groups were compared using Student’s t test, and the results were considered significantly different for p<0.05. All experiments were repeated at least three times to demonstrate the reproducibility of the results.
Results
Effects of ECF-L-Fc on the JNK activity and DNA binding of NF-κB and AP-1
Since RANKL signals through JNK and NF-κB, we determined if ECF-L also stimulated these signaling pathways to enhance osteoclastogenesis. RAW 264.7 cells, a murine monocytic cell line that forms OCL, were treated with ECF-L-Fc (200 ng/ml), control Fc (200 ng/ml) and/or RANKL (20 ng/ml) for 30 min to measure DNA binding activity for NF-κB and AP-1. As shown in Fig. 1, purified ECF-L-Fc protein modestly increased the DNA binding activity of NF-κB and AP-1 by 40% respectively. Under these conditions, low levels of RANKL by itself did not increase NF-κB binding to DNA, but RANKL and ECF-L-Fc showed an increase in NF-κB activity. JNK activity was also modestly increased in mouse bone marrow cells treated with ECF-L-Fc or RANKL. The effects of ECF-L and RANKL on JNK activity were additive (Fig. 2).
Fig. 1.
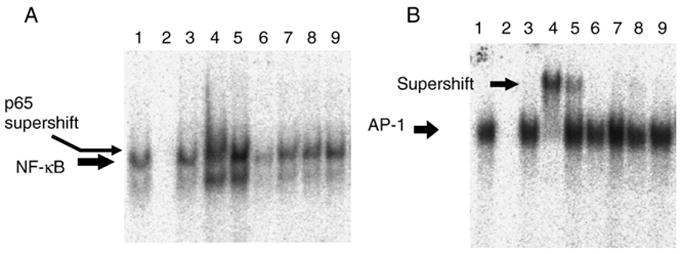
ECF-L-Fc increases the DNA binding of p65 NF-κB and AP-1 in RAW 264.7 cells. RAW 264.7 cells were treated with purified ECF-L-Fc (200 ng/ml) and/or RANKL (20 ng/ml) for 30 min. Control cultures were treated with a control Fc protein (200 ng/ml). Nuclear extracts (NE) were made and EMSA for (A) NF-κB and (B) AP-1 was performed. (A) A typical EMSA using a 32P-labeled NF-κB probe is shown and is representative of at least 3 independent experiments. Lanes 1–5 and 8, NE from RANKL treated cells; lane 2, 50× unlabeled NF-κB oligo; lane 3, 50× unlabeled mAP-1 oligo; lane 4, anti-p65 antibody (F-6, sc-8008); lane 5, non-specific mouse antibody; lane 6, NE from control cells; lane 7, NE from ECF-L treated cells; lane 9, NE from ECF-L plus RANKL treated cells. (B) A typical EMSA using a 32P-labeled AP-1 probe is shown and is representative of at least 3 independent experiments. Lanes 1–5 and 8, NE from RANKL treated cells; lane 2, 50× unlabeled AP-1 oligo; lane 3, 50× unlabeled mAP-1 oligo; lane 4, anti-Fos antibody (K-25, sc-253); lane 5, non-specific rabbit antibody; lane 6, NE from control cells; lane 7, NE from ECF-L treated cells; lane 9, NE from ECF-L plus RANKL treated cells.
Fig. 2.
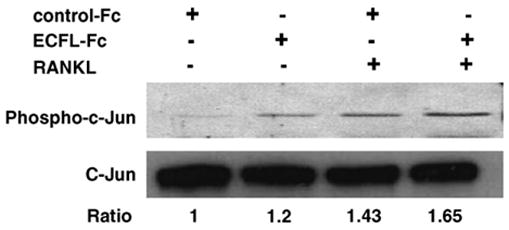
ECF-L-Fc increases JNK activity. Mouse marrow cells were treated with purified ECF-L (200 ng/ml) and/or RANKL (20 ng/ml) for 30 min, and cell extracts were analyzed by Western blot using phospho-c-Jun antibody. Control cultures were treated with a control Fc protein (200 ng/ml). ECF-L-Fc modestly increased JNK activity. RANKL-induced JNK activity was increased further by ECF-L-Fc. c-Jun was used as an internal control for the Western blots. A densitometry analysis was performed and reported as a ratio of phosphor-c-Jun to c-Jun. A similar pattern of results was seen in three independent experiments.
Effects of ECF-L-Fc on the expression of LFA-1 and ICAM-1 mRNA
Since ECF-L acts at the differentiation/fusion stage of OCL formation, we then examined the effects of ECF-L on the expression of the adhesion molecules LFA-1 and ICAM-1 that are involved in OCL precursor fusion. LFA-1α mRNA expression was increased 1.8-fold by the addition of ECF-L-Fc within 24 h. However, LFA-1β expression was not increased by ECF-L-Fc and/or 1,25-(OH)2D3. The mRNA expression levels of ICAM-1, the cognate receptor for LFA-1, were also increased after treatment with ECF-L-Fc alone and were not further increased with combined treatment with ECF-L-Fc and 1,25-(OH)2D3 (Fig. 3).
Fig. 3.

ECF-L increases (A) LFA-1α and ICAM-1 expression. Mouse marrow cells were treated with ECF-L-Fc (200 ng/ml) and/or 10−10 M 1,25-(OH)2D3 and RANKL for 24 h. Control cultures were treated with a control Fc protein (200 ng/ml). Total RNA was isolated and RT-PCR analysis for murine LFA-1α and β was performed. LFA-1α mRNA levels were increased by ECF-L-Fc. ICAM-1 mRNA levels were also increased by ECF-L-Fc. GAPDH was used as an internal control for the RT-PCR. A densitometric analysis was performed and reported as a ratio to GAPDH. (B) Real time PCR shown the expression of ICAM-1 using a Taqman probe similar pattern of results was seen in three independent experiments.
Role of ICAM-1 in ECF-L-Fc induced OCL formation
To further investigate the importance of the interaction between ICAM-1 and LFA-1 in ECF-L-induced osteoclastogenesis, we tested the effects of an anti-ICAM-1 antibody on OCL formation in marrow cultures treated with ECF-L-Fc and low concentrations of RANKL (10 ng/ml) (Fig. 4A), A representative picture of TRACP staining showed that OCL formation was stimulated 2-fold by ECF-L-Fc (200 ng/ml) in the presence of RANKL (10 ng/ml) compared to RANKL alone. This stimulatory effect of ECF-L-Fc was totally inhibited by the addition of an anti-ICAM-1 antibody (1.0 μg/ml) as seen in (Fig. 4B). The anti-ICAM-1 antibody was not toxic to the cultures and did not significantly inhibit OCL formation in marrow cultures treated with the combination of control Fc (0.1–1 ng/ml) and RANKL (10 ng/ml).
Fig. 4.
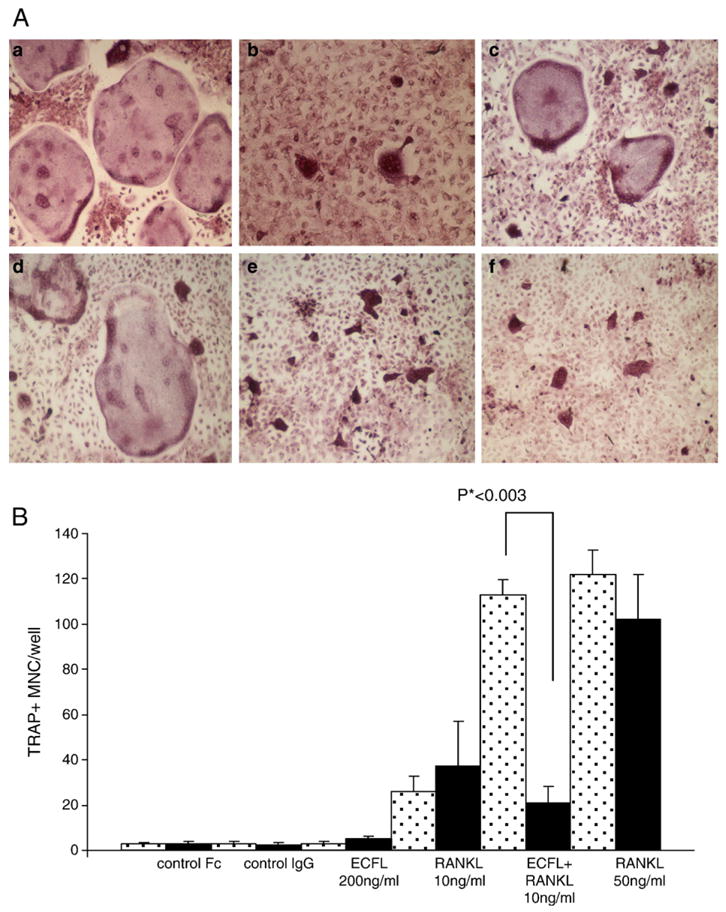
Anti-ICAM-1 inhibits ECF-L-induced OCL formation. (A) TRACP staining was performed and the number of TRACP+ multinucleated cells in each well was counted. MDBM cells were treated with RANKL 50 ng/ml (a), RANKL 10 ng/ml (b) and RANKL 10 ng/ml+ECF-L-Fc 200 ng/ml (c) and in the presence of the blocking anti-ICAM 1 antibody RANKL 50 ng/ml, (d) RANKL 10 ng/ml (e) and RANKL 10 ng/ml+ECF-Fc 200 ng/ml (f). (B) Anti-ICAM-1 inhibits ECF-L-induced OCL formation. Anti-ICAM-1 antibody was added to mouse bone marrow cultures stimulated with 200 ng/ml of ECF-L-Fc. Control cultures were treated with a control Fc protein (200 ng/ml) and control IgG protein. Anti-ICAM-1 antibody did not inhibit TRACP+ multinucleated cells induced by 50 ng/ml RANKL, but inhibited TRACP+ MNC formation induced by 10 ng/ml RANKL and 200 ng/ml of ECF-L-Fc. Results represent the mean±SD. A similar pattern of results was seen in three independent experiments. Stippled bars represent cultures treated with control IgG and solid bars are cultures treated with anti-ICAM-1 antibody.
Role of ICAM-1 in ECF-L-Fc induced OCL formation in the presence of 1,25-(OH)2D3 (10−10 M)
We tested the effects of an anti-ICAM-1 antibody on OCL formation in marrow cultures treated with ECF-L-Fc and low concentration of 1,25-(OH)2D3 (10−10 M). The stimulatory effect of ECF-L-Fc was totally inhibited by the addition of an anti-ICAM-1 antibody (1.0 μg/ml) as seen in Fig. 5.
Fig. 5.
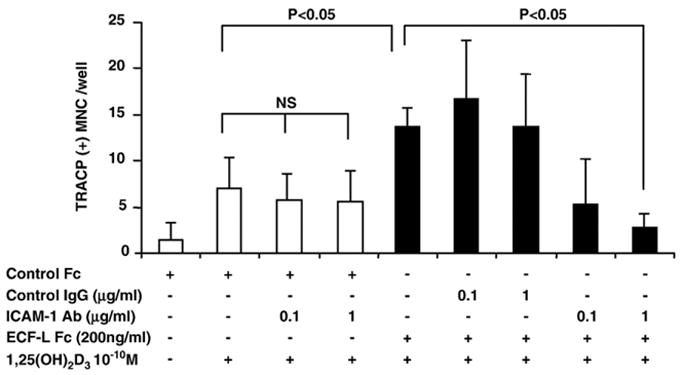
Anti-ICAM-1 inhibits ECF-L-induced OCL formation, treated by 10−10 M 1,25-(OH)2D3 and/or ECF-L-Fc (200 ng/ml). Anti-ICAM-1 antibody was added to mouse bone marrow cultures stimulated with 200 ng/ml of ECF-L-Fc and/or 10−10 M 1,25-(OH)2D3. Control cultures were treated with a control Fc protein (200 ng/ml) and control IgG protein. Anti-ICAM-1 antibody did not inhibit TRACP+ MNC induced by 10−10 M 1,25-(OH)2D3, but inhibited TRACP+ MNC formation induced by 10−10 M 1,25-(OH)2D3 and ECF-L-Fc. Results represent the mean±SD. A similar pattern of results was seen in three independent experiments. Open bars represent cultures not treated with ECF-L and solid bars represent cultures treated with ECF-L.
To further investigate the role of ICAM-1 in ECF-L stimulated osteoclastogenesis, OCL formation was assayed in cultures of marrow from ICAM-1 knockout mice. 1,25-(OH)2D3 (10−10 M) increased OCL formation in marrow cultures from ICAM-1 knockout mice, although OCL formation was lower than in control mouse cultures treated with 1,25-(OH)2D3. Importantly, OCL formation was not enhanced in cultures of ICAM-1 knockout marrow cells by addition of ECF-L-Fc. In contrast to ICAM-1 knockout mice (Fig. 6), OCL formation induced by 1,25-(OH)2D3 (10 −10 M) was further increased by addition of ECF-L-Fc in cultures from control mice.
Fig. 6.
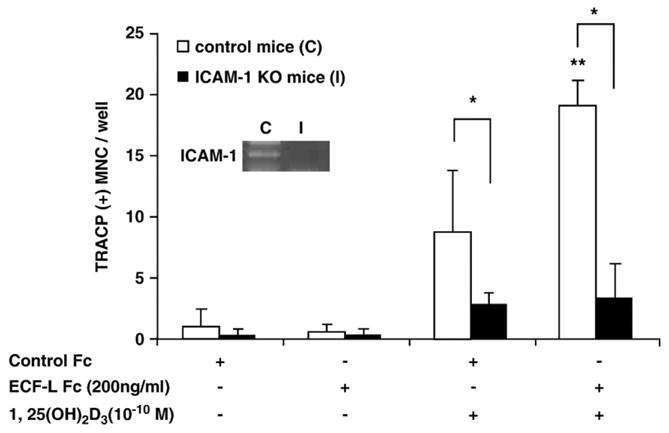
Osteoclast formation is not increased by ECF-L-Fc in ICAM-1 knockout mouse marrow cultures. Mouse marrow cells from control and ICAM-1 knockout mice were treated by 10−10 M 1,25-(OH)2D3 and/or ECF-L-Fc (200 ng/ml). Control cultures were treated with a control Fc protein (200 ng/ml). TRACP+ MNC formation was increased by 10−10 M 1,25-(OH)2D3 in control and ICAM-1 knockout mouse marrow cultures, but ECF-L-Fc significantly increased TRACP+ MNC formation in cultures from control mice in the presence 10−10 M 1,25-(OH)2D3 compared with ICAM-1 knockout mice. RT-PCR for ICAM-1 mRNA expression was performed with marrow cells from control and ICAM-1 mice (*p<0.05 between control and ICAM-1 knockout mice, **p<0.05 vs. 1,25-(OH)2D3 treated cells from control mice). A similar pattern of results was seen in two independent experiments.
Discussion
We previously identified ECF-L as a novel OCL-stimulating factor that enhanced OCL formation in the presence of low concentrations of 1,25-(OH)2D3 and RANKL and showed that ECF-L played an important role in RANKL-mediated osteoclastogenesis, even though ECF-L does not induce expression of RANKL. Therefore, we investigated the molecular mechanisms responsible for ECF-L effects on OCL formation.
Binding of RANKL to RANK results in activation of NF-κB and the protein kinase JNK, which increases transcription of AP-1 [10,11]. ECF-L significantly enhanced NF-κB, AP-1 binding activity and JNK kinase activity. ECF-L did not increase p44/42 or p38 MAPK activity (data not shown). However, these effects were modest and could not by themselves account for the effects of ECF-L on OCL formation.
Cell-to-cell contact is necessary for OCL formation since OCL form by cell fusion rather than endomitosis [12, 13]. Cell adhesion molecules such as ICAM-1 and E-cadherin have been reported to be involved in these cell-to-cell interactions [14, 15]. ICAM-1 is expressed on osteoblasts [16], OCL precursors and OCL [11] and ICAM-1 expression can be regulated by intracellular signaling through NF-κB and JNK [16]. ICAM-1 is one of the ligands for LFA-1 [17], and ICAM-1 and LFA-1 interactions have been reported to be involved in cell-to-cell contact between OCL precursors and osteoblastic/stromal cells and between OCL precursors [9,13]. ECF-L increased ICAM-1 and LFA-1α expression in nonadherent mouse marrow cells, at both the mRNA and protein level, but had no effect on LFA-1/ICAM-1 expression in ST2 stromal cells (data not shown). Furthermore, ECF-L did not increase β1 or β3 integrin expression (data not shown). Importantly, ECF-L also enhanced the capacity of 1,25-(OH)2D3 to further increase ICAM-1 expression, and ECF-L had no effect on OCL formation in marrow cultures from ICAM-1 knockout mice. Tanaka et al. reported that inflammatory cytokines, such as IL-1 and TNF-α1, increased bone resorption in rheumatoid arthritis by increasing expression of RANKL and ICAM-1 on osteoblasts [18] and that OCL precursors bind to osteoblasts through ICAM-1. Similarly, β1 integrins signaling through focal adhesion kinase increases ICAM-1 and RANKL expression on the surface of osteoblasts to increase OCL formation [19]. Furthermore, Harada et al. reported that blocking ICAM-1 binding to LFA-1 on OCL precursors inhibited OCL formation. Taken together, these findings strongly support an important role for ICAM-1 in OCL formation. Our results demonstrate that ECF-L, an autocrine factor produced by OCL, acts as a regulator of ICAM-1 expression on OCL precursors and enhances the effects of RANKL and 1,25-(OH)2D3 on OCL formation by increasing ICAM-1 on OCL precursors. ECF-L’s effects on ICAM-1 expression differ from those of TNF-α and IL-1 since IL-1 and TNF increase ICAM-1 expression on osteoblasts and not osteoclast precursors [18,19,20]. These results suggest that, in addition to TNF and IL-1, ECF-L, which is increased in inflammatory conditions, may play an important role in the increased bone loss, associated with rheumatoid arthritis and may be a novel therapeutic target for treatment of the bone destruction associated with rheumatoid arthritis.
Acknowledgments
This study was supported by R01-AR41336 from NIAMS. We thank Arthur Beaudet for providing ICAM-1 deficient mice with the support of NIH grant AI-32177.
References
- 1.Oba Y, Chung HY, Choi SJ, Roodman GD. Eosinophil chemotactic factor-L (ECF-L): a novel osteoclast stimulating factor. J Bone Miner Res. 2003;18:1332–41. doi: 10.1359/jbmr.2003.18.7.1332. [DOI] [PubMed] [Google Scholar]
- 2.Owhashi M, Arita H, Hayai N. Identification of a novel eosinophil chemotactic cytokine (ECF-L) as a chitinase family protein. J Biol Chem. 2000;275:1279–86. doi: 10.1074/jbc.275.2.1279. [DOI] [PubMed] [Google Scholar]
- 3.Webb DC, McKenzie ANJ, Foster PS. Expression of the Ym2 lectin-binding protein is dependent on interleukin-4 and IL-13 signal transduction. J Biol Chem. 2001;276:41969–76. doi: 10.1074/jbc.M106223200. [DOI] [PubMed] [Google Scholar]
- 4.Chang NA, Hung SI, Hwa KY, Kato I, Chen JE, Liu CH, et al. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. J Biol Chem. 2001;276:17497–506. doi: 10.1074/jbc.M010417200. [DOI] [PubMed] [Google Scholar]
- 5.Hung SI, Chang AC, Kato I, Chang NA. Transient expression of Ym1, a heparin-binding lectin, during developmental hematopoiesis and inflammation. J Leukocyte Biol. 2002;72:72–82. [PubMed] [Google Scholar]
- 6.Robker RL, Collins RG, Beaudet AL, Mersmann HJ, Smith CW. Leukocyte migration in adipose tissue of mice null for ICAM-1 and Mac-1 adhesion receptors. Obes Res. 2004;12:936–40. doi: 10.1038/oby.2004.114. [DOI] [PubMed] [Google Scholar]
- 7.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–88. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 8.Andrews NA, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tani-Ishii N, Penninger JM, Matsumzoto G, Teranaka T, Umemoto T. The role of LFA-1 in osteoclast development induced by co-cultures of mouse bone marrow cells and MC3T3-G2/PA 6 cells. J Periodontal Res. 2002;37:184–91. doi: 10.1034/j.1600-0765.2002.00610.x. [DOI] [PubMed] [Google Scholar]
- 10.Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96:3540–5. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–4. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- 12.Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13:66–80. doi: 10.1210/edrv-13-1-66. [DOI] [PubMed] [Google Scholar]
- 13.Harada H, Kukita T, Kukita A, Iwamoto Y, Iijima T. Involvement of lymphocyte function-associated antigen-1 and intercellular adhesion molecule-1 in osteoclastogenesis: a possible role in direct interaction between osteoclast precursors. Endocrinology. 1998;139:3967–75. doi: 10.1210/endo.139.9.6171. [DOI] [PubMed] [Google Scholar]
- 14.Kurachi K, Morita I, Murota S. Involvement of adhesion molecules LFA-1 and ICAM-1 in osteoclast development. Biochem Biophys Acta. 1993;1178:259–66. doi: 10.1016/0167-4889(93)90202-z. [DOI] [PubMed] [Google Scholar]
- 15.Mbalaviele G, Chen H, Boyce BF, Mundy GR, Yoneda T. The role of cadherin in the generation of multinucleated osteoclasts from mononuclear precursors in murine marrow. J Clin Invest. 1990;95:2757–65. doi: 10.1172/JCI117979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okada Y, Morimoto I, Ura K, Watanabe K, Eto S, Kumegawa M, et al. Cell-to-cell adhesion via intercellular adhesion molecule-1 and leukocyte function-associated antigen-1 pathway is involved in 1, 25(OH)2D3, PTH and IL-1α-induced osteoclast differentiation and bone resorption. Endocr J. 2002;49:483–95. doi: 10.1507/endocrj.49.483. [DOI] [PubMed] [Google Scholar]
- 17.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–34. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:325–8. doi: 10.2174/1568010054022015. [DOI] [PubMed] [Google Scholar]
- 19.Nakayamada S, Okada Y, Saito K, Tamura M, Tanaka Y. Beta1 integrin/focal adhesion kinase-mediated signaling induces intercellular adhesion molecule 1 and receptor activator of nuclear factor kappaB ligand on osteoblasts and osteoclast maturation. J Biol Chem. 2003;278:45368–74. doi: 10.1074/jbc.M308786200. [DOI] [PubMed] [Google Scholar]
- 20.Okada Y, Morimoto I, Ura K, Watanabe K, Eto S, Kumegawa M, et al. Cell-to-Cell adhesion via intercellular adhesion molecule-1 and leukocyte function-associated antigen-1 pathway is involved in 1alpha,25(OH)2D3, PTH and IL-1alpha-induced osteoclast differentiation and bone resorption. Endocr J. 2002;49:483–95. doi: 10.1507/endocrj.49.483. [DOI] [PubMed] [Google Scholar]