

1. Introduction
Large regions of eukaryotic genomes are comprised of repetitive DNA sequences, which are frequently concentrated in the pericentromeric and telomeric heterochromatin. Dipteran insect genomes, such as Drosophila melanogaster and Anopheles gambiae, are estimated to be 30-60% repetitive. Mammalian genomes are roughly 50% repetitive (Lander et al., 2001; Waterston et al., 2002), with at least 20% of genome content composed of long interspersed nuclear element retrotransposons (LINEs) alone (Hansen, 2003). The sequence composition and organization of repeats is diverse, and includes highly-repeated satellite sequences, tandem clusters of essential genes (e.g. ribosomal DNA), complete and incomplete transposable elements (TEs), and complex, interdigitated nests of TEs. TEs have been shown to have dramatic negative consequences on their host genomes by disrupting genes, generating recombinational instability (reviewed in Deininger et al., 2003), and altering transcriptional regulation (Thornburg et al., 2006). For example, the P transposable element has recently invaded the of D. melanogaster genome, spreading globally in the 20th century, and is responsible for the disruption of many genes (reviewed in Engels, 1983). This tendency has been widely exploited as a genetic tool to mutagenize the genes of this organism for experimental purposes (Spradling et al., 1999). Despite once being considered solely as a destructive force or ‘junk DNA’, we now know that some repetitive sequences can also have roles in genome architecture and inheritance, including centromere function (Grady et al., 1992), generation of exon diversity (Britten, 2006; Sorek et al., 2002; Sorek et al., 2004), and the regulation of genes (Bejerano et al., 2006). Recent evidence has even linked the RNA interference (RNAi) machinery with silencing of TEs, as a potential mechanism for maintaining genome stability (reviewed in Bernstein and Allis, 2005; Casacuberta and Pardue, 2006).
It is essential to identify and analyze repetitive regions in sequenced genomes in order to fully understand genome structure and function. Knowledge of repeated sequences improves gene annotations by identifying open reading frames (ORFs) from transposable elements that contaminate genes. For example, gene-finding programs such as GENSCAN (Burge and Karlin, 1997) and Genie (Reese et al., 1997) often produce gene predictions containing exons that are, in fact, unmasked TE ORFs embedded in introns. Similarly, interpretation of microarray datasets is complicated by the fact that many of the genes being measured are ORFs from transposable elements. Finally, phylogenetic comparison of repeat sequences and frequencies between related species can give insights into genome rearrangements and evolution (Price et al., 2004; Caspi and Pachter, 2006).
By far, D. melanogaster has the mostly completely assembled and thoroughly studied of genomes, where dedicated sequence finishing of the euchromatic and heterochromatic regions (reviewed in Celniker and Rubin, 2003), careful manual inspection of repeat clusters, and automated BAC fingerprinting analysis have been used to validate the sequence assembly. The identification and annotation of interspersed repeats in D. melanogaster has taken decades of work and predated the completion of the genome sequence. Indeed, often only one instance of a given type of TE can be found in the D. melanogaster genome (Kaminker et al., 2002). The detailed description of the TEs in D. melanogaster, the experimental determination of a boundary between euchromatin and heterochromatin regions by BAC-FISH (Hoskins et al., 2002), and the availability of over 20 megabases (Mb) of nearly-finished repeat-rich heterochromatin (Carlson J., 2006) makes this species ideal for studying the relationship between repeats and genome structure. Furthermore, genome assemblies for twelve other Dipteran species, including the mosquito, A. gambiae (Aultman et al., 2002), and eleven other Drosophilids (Table 1) have recently been generated (Agencourt, 2005; TIGR, 2005; WUSTL, 2005; BDGP, 2006), making in-depth comparative analysis of these genomes possible.
Table 1.
Genome sequences analyzed
| Genome Version used for PILERDF Input | Genome Version used for Repeat Analyses | Assembled Size of Genome used for Repeat Analyses (MB) | |
|---|---|---|---|
| D. melanogaster | BDGP Release 3 | BDGP Release 5 | 169 |
| A. gambiae | Genbank Release 1 April 2002 | Release MOZ2a | 288 |
| D. sechellia | CAF1 | CAF1 | 167 |
| D. simulans | Dsim DPGP syntenic assembly | CAF1 | 142 |
| D. yakuba | WUSTL Genome Sequencing Center. April 7 2004 (full, non-random arms only) | CAF1 | 169 |
| D. erecta | Agencourt Arachne Assembly August 1 2005 | CAF1 | 153 |
| D mojavensis | Agencourt Arachne Assembly July 21st, 2004 | CAF1 | 194 |
| D. ananassae | Salzberg TIGR group Celera Assembly July 15th, 2004 | CAF1 | 231 |
| D. virilis | Agencourt Arachne Assembly July 21st, 2004 | CAF1 | 206 |
| D. persilimis | CAF1 | CAF1 | 188 |
| D. pseudoobscura | Salzberg TIGR Group CABA assembly Aug 2004 | CAF1 | 153 |
| D. grimshawi | CAF1 | CAF1 | 200 |
| D. willistoni | CAF1 | CAF1 | 237 |
Although the cost of sequencing has been greatly reduced, most of these genomes are not as highly finished as D. melanogaster, complicating conclusions about large-scale genome structure and evolution. Recent comparative studies also suggest that TEs have only recently expanded in many of these lineages and that few, if any, TEs are common between even closely related species (Caspi and Pachter, 2006). Unfortunately, since TE sequences diverge quickly and often have regions where there is no requirement for sequence conservation, the use of DNA homology-based repeat identification often fails or under-predicts the true repeat content. While the identification of simple, low-complexity nucleotide repeats and longer 200-350bp tandem repeated arrays is relatively straightforward, interspersed elements, including TEs, have a more complex structure, often including terminal repeats, group-specific antigens (GAG), polymerase (POL), and envelope (ENV) genes, reverse transcriptases (RT), integrase proteins, and other genes. Retrotransposable elements, such as LINE and long-terminal-repeat (LTR) elements require RT, which is error-prone relative to cellular polymerases and makes these TEs more mutable than typical coding-sequences (Roberts et al., 1989). Meanwhile, DNA transposons have tandem-inverted-repeats (TIRs) used to transpose in the genome via a cut- and-paste mechanism. Yet other transposable elements do not have self-encoded transposition ORFs. For example, miniature TEs (MITEs) are non-autonomous elements found in several vertebrates (Hikosaka et al., 2000), plants (Casacuberta et al., 1998), and mosquitoes (Tu, 1997; Tu, 2001) whose propagation depends on the transposition machinery of other elements. Thus, TEs vary widely in length, sequence composition, coding sequence content, and mode of replication, complicating efforts to predict them de novo or to identify them outside of homology-based approaches. To address these issues, we have developed computational methods for generating new repeat libraries and annotating repetitive elements.
TEs insert themselves into the genome through a variety of mechanisms (reviewed in Kazazian, 2004). The PILER-DF program (Edgar and Myers, 2005) predicts interspersed elements by searching for three or more sequence regions that are globally alignable and surrounded by unique sequence, implying a sequence-specific copying mechanism that is the hallmark of TEs. These criteria are often not satisfied, for example if a TE is only present twice in a genome or is always found embedded in other repetitive sequence, and in such a case PILER-DF will fail to identify that TE. We applied the PILER-DF program to the D, melanogaster, A. gambiae, and eleven recently sequenced Drosophila genomes (Table 1) and identified a number of species-specific TE predictions. Here we present a repeat annotation pipeline and apply it to these PILER-DF predictions.
Although building libraries of species-specific transposable elements is an important step in describing the repeat landscape of genomes, these libraries are only a starting point. Programs such as RepeatMasker (Smit, 1996-2004) annotate repetitive sequence in genomes using DNA-based alignments to a known library of repeats. Since redundancy in the DNA code allows amino acid conservation even when the DNA sequence changes, one method for accomplishing more complete repeat annotation is to identify elements using protein homology. We therefore developed the RepeatRunner program, which is based on the Comparative Genomics Library (Yandell et al., 2006). RepeatRunner integrates DNA-based RepeatMasker output with BLASTX (Altschul et al., 1990) protein-based search results of a custom Genbank-TE (GB-TE) protein library (See Section 2.1). Here we describe our improved methodology for identifying and annotating species-specific repeats using PILER-DF and RepeatRunner. We show that the identification of repetitive DNA is greatly improved using PILER-DF libraries and RepeatRunner compared to RepeatMasker alone, and provide the masked genomes of 13 Dipteran species and their complete repeat annotations.
2. Materials & Methods
2.1 Datasets & Sequences
All datasets described in the text are available from ftp://ftp.dhgp.org/pub/DHGP/PILER-DF. The list of genomes and their respective assembly versions are listed in Table 1. In most cases PILER-DF predictions were made on early builds of newly sequenced genomes, while we performed our masking of repeats on the most current versions of the Drosophilid genome assemblies (AAA, 2006). We used Repbase version 10.07 (Jurka et al., 2005) for WU-BLAST (Altschul et al., 1990) based annotation (see Section 2.3). We also used a custom BDGP Drosophila TE library (Ashburner, 2005), which included several TEs from D. virilis and the newly described INE-1 element (Quesneville et al., 2005). We generated a Genbank-TE protein database (GB-TE, Supplemental File 1c) from Genbank release 143 by extracting only peptide sequences containing at least one of the following text strings anywhere in their annotations: “reverse transcriptase”, “transposon”, “repetitive element”, “RNA-directed DNA polymerase”, “pol protein”, “non-LTR retrotransposon”, “mobile element”, “retroelement”, “polyprotein”, “retrovirus”, “group-specific antigen (gag)”, or “polymerase (pol)”.
2.2 PILER-DF
The PILER-DF program was executed on PALS (Edgar and Myers, 2005) self-alignments of preliminary genome assemblies (Table 1). We found that the number and quality of PILER-DF predictions were not greatly affected by the genome assembly version used (data not shown). Centroid consensus sequences, defined as the individual repetitive element most related by DNA sequence identity to all other elements in the same family, were determined from raw PILER-DF output using a custom script. In order to reduce the redundancy of homologous predictions, centroid sequences were compared to one another using NCBI-BLASTN and any predictions with 90% or higher identity to any other PILER-DF prediction for a given species over 90% or more if its length were discarded. A FASTA file of all non-redundant PILER-DF centroids is provided in Supplemental File 1d.
2.3 Annotation of PILER-DF Predictions
An overview of our annotation process is shown in Figure 1. Briefly, non-redundant, centroid PILER-DF predictions (see Section 2.2) were compared to curated repeat libraries from Repbase and the BDGP Drosophila TE library using WU-BLAST. Predictions were named using a four-letter abbreviation of the genus and species of origin and a numerical “Superfamily.family” identifier for the element (e.g. Dmel.1.34). Members of a given family are globally alignable to each other, while members of a superfamily are locally but not globally alignable. We assigned each prediction to one of the classes shown in Table 2 by the following procedure: Each prediction was used to search Repbase, GB-TE and the BDGP Drosophila TE library using both WU-BLASTN and WU-TBLASTX. High-scoring-pairs (HSPs) with e-values less than 10-5 were considered significant. Each HSP was assigned to one or more classes as follows. If the HSP was a hit to GB-TE, we used a manually curated lookup table (Supplemental File 1b) that assigned one or more classes to each of the text strings used to create the database; for example, “reverse transcriptase” was mapped to “LINE” and “LTR” elements. In the case of Repbase and the BDGP TE library, the class was extracted directly from the sequence annotation. If 90% or more of the class assignments agreed then the majority assignment was accepted, otherwise the class was considered to be undetermined, but the highest scoring class was still recorded in Supplemental File 1a. Any prediction that was 99% or more subsumed by an element described in Repbase was automatically classified as a species-specific example of that TE. Finally, we searched for terminal repeats by aligning the first half of a prediction to the second. If a terminal repeat was found, we verified whether it was consistent with the assigned class, and in cases where the class was undetermined to this point, the presence of terminal inverted or long-terminal repeats alone was used to classify the prediction as either TIR or LTR.
Figure 1.
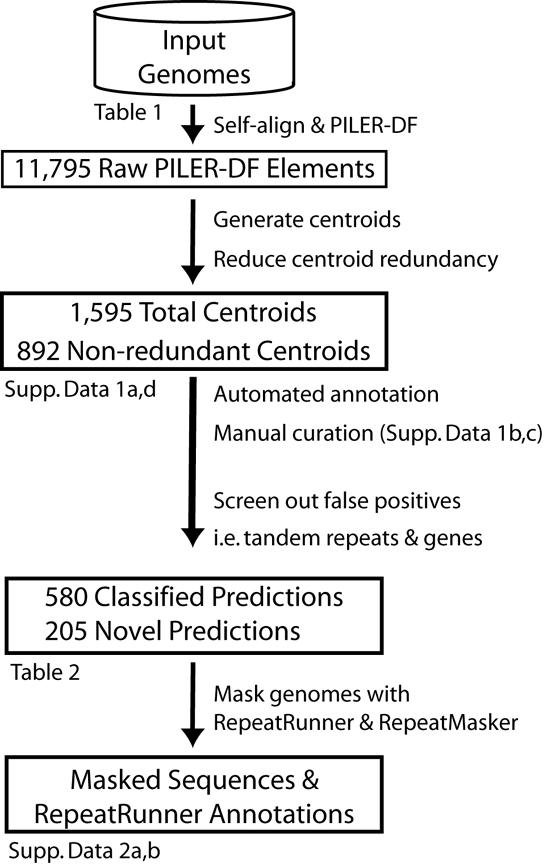
Repeat Prediction Pipeline
An outline of the process used to generate and annotate PILER-DF predictions (see Sections 2.2-2.3). Links to figures and supplemental data elaborating on specific steps in the dataflow are indicated.
Table 2.
PILER-DF Summary
| D. melanogaster | D. sechellia | D. simulans | D. yakuba | D. erecta | D. ananassae | D. persimilis | D. pseudoobscura | D. mojavensis | D. virilis | D. grimshawi | D. willistoni | A. gambiae | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LTR | 25 | 2 | 1 | 6 | 0 | 32 | 1 | 5 | 9 | 21 | 0 | 10 | 37 | 149 |
| LINE | 9 | 2 | 6 | 1 | 0 | 30 | 1 | 10 | 3 | 15 | 0 | 9 | 38 | 124 |
| TIR | 5 | 0 | 1 | 15 | 2 | 47 | 5 | 34 | 36 | 8 | 0 | 27 | 46 | 226 |
| FB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 2 |
| Helitron | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | 2 | 1 | 4 | 15 |
| SINE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 3 |
| Novel - LTR | 0 | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 |
| Novel - TIR | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 2 | 1 | 0 | 0 | 3 | 12 | 19 |
| Novel - Unknown | 0 | 1 | 0 | 4 | 2 | 61 | 1 | 22 | 10 | 7 | 2 | 31 | 64 | 205 |
| False + | 2 | 1 | 0 | 2 | 0 | 0 | 3 | 7 | 1 | 1 | 1 | 0 | 6 | 24 |
| Satellite | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 6 | 18 | 0 | 0 | 4 | 7 | 37 |
| TRF tandem repeat | 0 | 1 | 0 | 18 | 3 | 14 | 0 | 0 | 4 | 13 | 4 | 12 | 14 | 83 |
| TOTALS | 41 | 9 | 9 | 46 | 7 | 197 | 11 | 86 | 83 | 66 | 9 | 97 | 231 | 892 |
The number of PILER-DF predictions of a given ‘Class’ for 13 Dipteran species.
1 - ‘Novel-LTR’ and ‘Novel-TIR’ predictions had no significant sequence similarity to known TEs, but had detectable terminal repeats.
We identified two types of false positives: tandem repeats and protein-coding genes. The tandem repeat content of PILER-DF predictions was assessed using Tandem Repeat Finder (TRF, (Benson, 1999)). Any prediction containing more than 25% tandem repeats was flagged as a potential false positive and excluded from the repeat libraries used by RepeatRunner. All non-redundant centroids were compared to protein-coding gene sets from D. melanogaster (Release 4.2.1), D. pseudoobscura (Richards et al., 2005), and A. gambiae (Release MOZ2a) using WU-BLASTX. Predictions that aligned to D. melanogaster gene annotations with 90% or greater identity over 90% or more of their length were classified as false positives. HSPs with e-values less than 10-5 were visually inspected. All non-tandem repeat, non-false positive PILER-DF predictions were appended to the Repbase version 10.07 Drosophila and Anopheles repeat libraries to make a custom ‘Dipteran + PILER’ repeat library, which was used for subsequent RepeatMasker and RepeatRunner analyses.
2.4 Masking of Genomes
We used the standalone program RepeatMasker (Smit, 1996-2004) and the CGL program RepeatRunner (Yandell, 2006) to identify repetitive sequences for this study. RepeatRunner uses the RepeatMasker v2.4 program to identify repetitive regions by nucleotide identity and WU-BLASTX to identify regions by 3-frame translated similarity to a protein database, which in our case was GB-TE. For comparative purposes we analyzed genomes from the Comparative Annotation Freeze 1 (CAF1, (AAA, 2006), D. melanogaster Release 5 (Carlson J., 2006), and A. gambiae MOZ2a (Aultman et al., 2002) with RepeatMasker using the standard Repbase Drosophila library, RepeatRunner using the standard Repbase Drosophila library and also our ‘Dipteran + PILER’ library. Genomes were split into 100 kilobase (kb) segments, or smaller, and the number of masked bases was summed. The complete masked genome sequence and tab-delimited description of the masked genomes is provided in Supplemental Files 2a and 2b, respectively. We used the R software package (GNU-Project, 2006) to compute correlation coefficients and other statistical measures. We used Microsoft Excel to compute simple averages, deviations, and to generate graphs.
3. Results & Discussion
3.1 Overview of PILER-DF Predictions
PILER-DF detects intact interspersed elements that are found intact at least three times in a genome. We executed PILER-DF on the Release 3 D. melanogaster genome and preliminary assemblies of D. yakuba, D. simulans, D. sechellia, D. persimilis, D. virilis, D. pseudoobscura, D. ananassae, D. mojavensis, D. erecta, D. grimshawi, D. willistoni and A. gambiae genomes to identify known and novel interspersed repeats (Table 1). While D. melanogaster annotations already contained over 100 described TE families (Kaminker et al., 2002), there were very few transposable elements described in the other species.
PILER-DF generated 11,795 raw element predictions in the genomes studied (Figure 1). For each alignment produced by PILER-DF, we calculated the ‘centroid’ element (see Section 2.1). We chose to use centroid sequences instead of a simple consensus sequence because 100% identical copies of consensus sequences are often not found in the genome, whereas there is always at least one instance of a centroid; hence, a sufficiently degenerate consensus sequence might be less sensitive. There were 1595 distinct centroid predictions in all of the described genomes (Figure 1). In order to reduce the redundancy of similar predictions, we discarded PILER-DF predictions that were more than 90% similar to another prediction for that species over at least 90% of the length. After redundancy reduction there were 892 centroid predictions for all the Dipteran genomes (Table 2). Interestingly, most of the original centroid predictions for D. grimshawi were found to be redundant. Out of 1,027 raw elements and 288 centroids, only nine were determined to be non-redundant. Thus, it would appear that there was a substantial lineage-specific amplification of these repeats in D. grimshawi. The number of non-redundant centroid predictions varied widely between genomes, from only nine in D. simulans to 382 in A. gambiae (Table 2). The number of predictions found per genome was correlated with genome size (R=0.8 centroids (p<1.4×10-8), R=0.85 for raw elements (p<1.4×10-8)). Thus, as the genome size increases the number of PILER-DF predictions increases proportionally, at least for the Dipteran genomes studied.
PILER-DF predictions tended to be shorter than the canonical functional elements for a given class. It would appear that many PILER-DF TE predictions are non-functional or non-autonomous, since they are often too short to contain complete transposase or RT ORFs. For example, only 53/278 LTR and LINE predictions (19%) were greater than the 5 kb length typically found for functional elements in these classes (Kaminker et al., 2002). Likewise only 106/245 of TIR-type DNA transposon predictions (43%) fall within the 1-4.5 kb size range found for known functional TIR elements in D. melanogaster (Kaminker et al., 2002). The majority of the LTR/LINE/TIR predictions (364/523, 70%) fall outside of the size range that suggests they are autonomous and functional. Only 52/523 of these predictions (10%) meet the minimum length requirement for an autonomous TE and have LTR or TIR sequences that are 100% identical, which also suggests the possibility of a functional TE. PILER-DF predictions meeting minimal length criteria for known functional elements are provided in Supplemental File 1a.
These results underscore the difficulty of identifying novel, active TEs. In general it is expected that active TEs have perfectly matching terminal repeats and full-length ORFs encoding transposition machinery. While relatively few of the PILER-DF predictions meet these criteria, it is important to note that recent studies suggest a wide array of non-autonomous TEs that hijack the transposition machinery of a few ‘master’ TEs, such as MITE elements. Also, other repetitive sequences, such as Helitron elements, do not appear to have any of the hallmarks of canonically described TEs such as transposition ORFs or terminal repeats (Kapitonov and Jurka, 2001). The wide variety of short PILER-DF predictions we describe may represent similar classes of transposable elements with novel modes of replication.
3.2 Species-Specific TEs
We integrated results from several analyses in order to classify the PILER-DF predictions into convenient ‘Class’ and ‘Family’ designations similar to those defined in previous D. melanogaster studies (Kaminker et al., 2002; Quesneville et al., 2005). We used TBLASTX and BLASTN against elements described in Repbase and Drosophila TEs curated at the BDGP and BLASTX to identify predictions with similarity to repeat proteins described in Genbank. We conservatively chose only to determine the ‘Class’ for our predictions, for example LTR or TIR, instead of the individual repeat ‘Family’, unless 90% or more of the BLAST data indicated a single ‘Family’ for that prediction. In cases where a PILER-DF prediction was 99% subsumed by a known ‘Family’, we assigned the prediction to that ‘Family’. Overall, 41/892 (4.6%) of the non-redundant PILER-DF predictions had 99% or more identity to a known repeat (8 in A. gambiae, 27 in D. melanogaster, 1 in D. sechellia, 1 in D. simulans, 1 in D. virilis, 3 in D. willistoni). The high percentage of D. melanogaster predictions with at least 99% identity to other elements was expected, since TEs are more highly curated for this species.
The majority of predictions could be classified into a TE ‘Class’ based on homology to a previously described element. Overall, 519/892 (58%) of predictions had significant similarity to a known TE ‘Class’ and 195/892 (22%) of predictions could be directly categorized into a specific ‘Class’ by 90% or greater sequence identity. In order to improve identification, we calculated an ‘Integrated’ score by summing the number of ‘Class’ assignments for all significant BLAST HSPs used in our annotation. Using this approach, 348/892 (39%) had an ‘Integrated’ score of 90% or greater compared to 22% if we only summed raw BLAST results (Supplemental File 1a). Thus, the use of parallel database search strategies with a simple data integration step significantly improved our ability to predict the ‘Class’ for predictions. In total, 273/892 (31%) of the predictions can be classified as retrotransposable elements, including 149/892 (17%) LTR Class TEs and 124/892 (14%) LINE elements. In D. melanogaster 83% of the predictions were retrotransposons; D. simulans (78%), and D. sechellia (44%) also had relatively more retrotransposable elements predicted when compared to other Dipterans, with the exception of D. virilis (55%). The higher percentage of LTR/LINE class repeats in these three closely related species may indicate a recent expansion of retrotransposons in the melanogaster lineage relative to more distantly diverged Drosophilids. However, the ability of PILER-DF to detect a given class of repeats tends to vary from species to species and therefore may bias this observation.
Overall, 243/892 (27%) of predictions have similarity to DNA TEs, which include TIRs, Foldback (FB) elements, and Helitrons. Interestingly, DNA type predictions were overrepresented in several species including D. yakuba (33% of predictions), D. erecta (29%), D. ananassae (28%), D. persimilis (45%), D. pseudoobscura (40%). D. mojavensis (45%), and D. willistoni (29%) (Table 2). This is in contrast to other Drosophila species and A. gambiae where only 11-22% of PILER-DF predictions are DNA transposons. This may provide evidence for an expansion of non-DNA TEs in D. melanogaster and other closely related species. Interestingly, rolling-circle DNA transposons, typified by the Helitron repeat were only found in D. ananassae (8), A. gambiae (4), D. grimshawi (1), and D. willistoni (1) (Table 2).
3.3 Novel PILER-DF Predictions
Of the 892 non-redundant centroid predictions we annotated, 26% (229/892) do not have significant sequence similarity to any known repeat, TE protein, or annotated gene (Table 2). We specifically looked for terminal repeats (TIRs and LTRs), which were found in 154/892 (17%) of the non-redundant centroid predictions. Furthermore, 73/892 (8.2%) of all predictions had terminal repeats that were at least 99% identical, suggesting that these elements may be functional or recently inactivated. Some of the ‘Novel’ predictions had TRs detected: 5/229 (2.2%) of novel predictions had LTRs and 19/229 (8.3%) had TIRs. Thus, 205/229 (90%) of ‘Novel’ predictions had no detectable sequence similarity to a known TE or gene within our chosen e-value cut-off.
The fraction of ‘Novel’ elements varied somewhat between species. No novel elements were found in D. melanogaster, indicating that the high level of annotation of D. melanogaster has likely identified most TE sequences. The highest fractions of novel predictions were for D. willistoni (35%), D. ananassae (32%), A. gambiae (33%), D. pseudoobscura (28%), D. erecta (29%), and D. sechellia (33%) (Table 2). The majority of novel TE predictions for all species were found in A. gambiae (76/229, 33%), D. ananassae (64/229, 28%), D. willistoni (34/229, 15%), and D. pseudoobscura (24/229, 10%).
3.4 False Positives & Tandem Repeats
Automated repeat analysis followed by bioinformatic analyses against biological datasets provides a valuable quality check for new repeat and gene annotations. In many cases repeats that are misannotated as TE’s cannot be directly tested in laboratories. Flagging annotations as potential TE’s provides one way to prioritize experiments that will unambiguously determine if they represent coding genes or TEs. Refinement of annotation sets to clearly distinguish functional genes from other genome features is an ongoing and necessary step for keeping models of the genome current.
There were two types of PILER-DF predictions that we considered to be false positives: sequences with close similarity to annotated genes of known function, and sequences containing a significant fraction of short tandem repeats, as determined by TRF (Benson, 1999). PILER-DF is expected to have a low false positive rate, which was confirmed by our observation that only 24/892 (2.7%) of predictions had significant similarity to a known gene. False-positive gene homologies ranged from 0% for D. simulans, D. ananassae, D. willistoni, and D. erecta to 27% of D. persimilis predictions. In other words, predictions made by PILER-DF are very likely to represent bona fide TE sequences and not protein-coding genes. However, the requirement for there to be three intact copies of a TE, all of which are surrounded by unique sequence, is sufficiently stringent that many TEs are not identified.
We searched curated protein-coding gene datasets from D. melanogaster, D. pseudoobscura, and A. gambiae to screen out false positive predictions, however we only considered similarity to a D. melanogaster gene as criteria for false positives, since these annotations are the best curated of the annotation sets used. In general, PILER-DF predictions that were legitimate false positives had homology to a few types of genes. Several had significant similarity to trypsins, lysozymes, and actin, all known to be members of gene families (Misra et al., 2002). While a few false positives due to repeated exons was expected, most repeated exons did not result in false positives. While our false-positive rate was low, these results nonetheless emphasize the difficulty in discerning a ‘real’ protein-coding gene from a transposable element in some circumstances. Annotations from D. pseudoobscura and A. gambiae that had similarity to our PILER-DF predictions may represent bad gene annotations and are outlined in Supplemental File 1a.
The second type of false positives we attempted to identify was tandem repeats. We assumed that predictions that were composed of a large percentage of tandem repeats were not as likely to represent bona fide TEs, and instead represented cases of tandem repeats that happened to be of similar size or were flanked by similar TEs. We measured the tandem repeat content of our non-redundant centroid predictions using TRF and excluded predictions with greater than 25% tandem repeat content. This cut-off was determined empirically by choosing a tandem repeat content higher than observed in the gene annotation dataset for D. melanogaster (data not shown). Overall, 83/892 (9%) of our centroid PILER-DF predictions were classified as tandem repeats. Of course, further functional experiments are required to validate whether a given prediction is a legitimate gene, a tandem repeat, or a TE. The relatively high percentage of tandemly duplicated sequence in the PILER-DF predictions may indicate the sequence bias that these elements exhibit when they transpose.
3.5 Repeatmasking Genomes
We used both RepeatMasker and RepeatRunner with our enhanced, species-specific PILER-DF repeat libraries to mask thirteen Dipteran genomes. Recently, a web-based version of RepeatMasker has been made available that uses protein-based searches (Smit, 2005). However, a standalone application that is capable of processing full genomes is not yet available. RepeatRunner solves this problem by integrating protein searches with RepeatMasker results, thus greatly enhancing the ability to mask genomes, even without large libraries of known repeats.
When species-specific libraries are used with RepeatRunner there is dramatic improvement in repeat identification (Figure 2). In almost all cases, the use of RepeatRunner resulted in more repeat identification for the genome studies, and the use of PILER-DF libraries further enhances this effect. Repeatmasking improvement was dramatic for D. ananassae, which jumped from 20% repetitive sequence using RepeatMasker to 35% using RepeatRunner with the PILER-DF predictions (Figure 2). We observed more than a two-fold increase in the amount of sequences identified as repeats for A. gambiae when using RepeatRunner + PILER, versus using RepeatMasker alone (Figure 2). Although the total amount of repetitive sequence identified for species with few TE predictions, such as D. simulans, was modest (20% RepeatMasker, 21% RepeatRunner, 22% RepeatRunner + PILER), even a 1% increase in masking for D. simulans translates into an additional 1.4Mb of masked genomic sequence. Given that the average protein-coding gene density is ∼115/Mb in D. melanogaster euchromatin (Misra et al., 2002) and ∼20/Mb in heterochromatin (Hoskins et al., 2002), this means that each additional percent of masked sequence translates into 20-115 predicted genes that may, in fact, be TEs. Improvement of repeat identification is an essential step to refine the gene-finding process for these and other newly sequenced, unannotated genomes.
Figure 2.
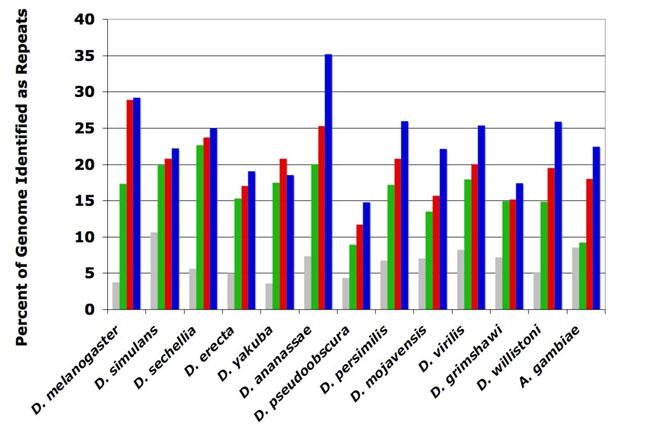
Total Genomic Repeat Content for Thirteen Dipteran Genomes
Frequency histogram showing the total ‘N’ content of Dipteran genomes before repeatmasking and after using RepeatMasker, RepeatRunner, and RepeatRunner + PILER-DF predictions. The percent frequency of uncalled base pairs and sized sequence gaps (grey) often result from genome assembly collapses due to repetitive regions and indicate a lower limit to the potential genomic repeat content. The total percent of masked sequence using RepeatMasker (green), RepeatRunner (red), and RepeatRunner + PILER-DF libraries (blue) are shown.
The results of our RepeatRunner masking of the D. melanogaster genome suggest that the euchromatin regions still contain some unidentified repeats. While previous studies suggested that 3.86% of the D. melanogaster euchromatin genome regions are composed of TE-like sequence (Kaminker et al., 2002), RepeatRunner reported an additional 2.5Mb (6.3% total, Figure 3). Data from D. melanogaster also suggests that RepeatRunner has its greatest effect on heterochromatin sequence. While previous studies using RepeatMasker identified only 52% of heterochromatin as repetitive (Hoskins et al., 2002), the ability to use protein alignments with RepeatRunner increased the amount of repetitive sequence identified to 79% (Figure 3). However, the addition of PILER-DF libraries did not significantly increase the amount of identified repeat sequence for either the euchromatin (6.4%) or the heterochromatin (79%). We conclude that the highly homogenized, fragmented, nested TEs within heterochromatin mask poorly using the nucleotide alignments provided by RepeatMasker most likely because the complex repeats are too divergent.
Figure 3.
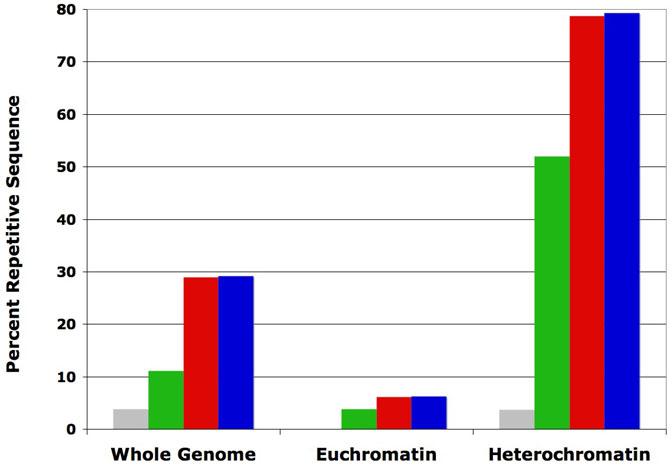
Comparison of D. melanogaster euchromatin and heterochromatin
Frequency histogram showing the total ‘N’ content of D. melanogaster euchromatin and heterochromatin before masking and after using RepeatMasker, RepeatRunner, and RepeatRunner + PILER-DF predictions. The percent frequency of uncalled base pairs and sized sequence gaps (grey) often result from genome assembly collapses due to repetitive regions and indicate a lower limit to the potential genomic repeat content. The total percent of masked sequence measured for Release 3 (green), RepeatRunner (red), and RepeatRunner + PILER-DF libraries (blue) are shown.
4. Conclusions
We have described a TE prediction pipeline based on PILER-DF to generate predictions for thirteen Dipteran genomes.
We used these predictions to aggressively mask repeats in thirteen genomes using our program, RepeatRunner, which we show especially effective at identifying fragmented repeats in D. melanogaster heterochromatin.
We have shown that the majority of our TE predictions are species-specific examples of repeats described in other Dipterans, but that PILER-DF also predicts novel repeat families with no homology to known TEs.
Supplementary Material
Acknowledgements
This work was supported by National Human Genomes Research Institute grant HG000747 (to C.D. Smith, S.E. Celniker and G.H. Karpen, PI). Mark Yandell funding was supported by the Howard Hughes Medical Institute. We’d also like to acknowledge Erwin Frise who maintains the hardware used for these analyses.
Abbreviations
- TE
transposable element
- TIR
terminal inverted repeat
- LTR
long terminal repeat
- LINE
long interspersed nuclear element
- FB
foldback element
- TRF
Tandem Repeat Finder
- CAF1
Comparative Analysis Freeze 1
- MITE
Miniature Transposable Element
- Mb
Megabase
- kb
kilobase
- RNAi
RNA interference
- HSP
High-scoring-pair
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AAA Assembly, Alignment, and Annotation of Drosophilid Genomes. 2006 http://rana.lbl.gov/drosophila/caf1.html.
- Agencourt B. The genomes of D. mojavensis, D. ananassae. 2005 http://www.agencourt.com/.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ashburner M. The Natural Transposable Element Project. 2005 http://www.bdgp.org/p_disrupt/TE.html.
- Aultman KS, Gottlieb M, Giovanni MY, Fauci AS. Anopheles gambiae genome: completing the malaria triad. Science. 2002;298:13. doi: 10.1126/science.298.5591.13. [DOI] [PubMed] [Google Scholar]
- BDGP Berkeley Drosophila Genome Project. 2006 http://www.bdgp.org.
- Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441:87–90. doi: 10.1038/nature04696. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 2005;19:1635–55. doi: 10.1101/gad.1324305. [DOI] [PubMed] [Google Scholar]
- Britten R. Transposable elements have contributed to thousands of human proteins. Proc Natl Acad Sci U S A. 2006;103:1798–803. doi: 10.1073/pnas.0510007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Carlson J,HR, Wan K, George R, Svirskas R, Kennedy C, Karpen GH, Rubin GM, Celniker SE. 2006. The Release 5 genome sequence of D. melanogaster.
- Casacuberta E, Casacuberta JM, Puigdomenech P, Monfort A. Presence of miniature inverted-repeat transposable elements (MITEs) in the genome of Arabidopsis thaliana: characterisation of the Emigrant family of elements. Plant J. 1998;16:79–85. doi: 10.1046/j.1365-313x.1998.00267.x. [DOI] [PubMed] [Google Scholar]
- Casacuberta E, Pardue ML. RNA interference has a role in regulating Drosophila telomeres. Genome Biol. 2006;7:220. doi: 10.1186/gb-2006-7-5-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Pachter L. Identification of transposable elements using multiple alignments of related genomes. Genome Res. 2006;16:260–70. doi: 10.1101/gr.4361206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, Rubin GM. The Drosophila melanogaster genome. Annu Rev Genomics Hum Genet. 2003;4:89–117. doi: 10.1146/annurev.genom.4.070802.110323. [DOI] [PubMed] [Google Scholar]
- Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003;13:651–8. doi: 10.1016/j.gde.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Myers EW. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21(Suppl 1):i152–8. doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- Engels WR. The P family of transposable elements in Drosophila. Annu Rev Genet. 1983;17:315–44. doi: 10.1146/annurev.ge.17.120183.001531. [DOI] [PubMed] [Google Scholar]
- GNU-Project R - An open source environment for graphing and statistics. 2006 http://www.r-project.org/.
- Grady DL, Ratliff RL, Robinson DL, McCanlies EC, Meyne J, Moyzis RK. Highly conserved repetitive DNA sequences are present at human centromeres. Proc Natl Acad Sci U S A. 1992;89:1695–9. doi: 10.1073/pnas.89.5.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS. X inactivation-specific methylation of LINE-1 elements by DNMT3B: implications for the Lyon repeat hypothesis. Hum Mol Genet. 2003;12:2559–67. doi: 10.1093/hmg/ddg268. [DOI] [PubMed] [Google Scholar]
- Hikosaka A, Yokouchi E, Kawahara A. Extensive amplification and transposition of a novel repetitive element, xstir, together with its terminal inverted repeat in the evolution of Xenopus. J Mol Evol. 2000;51:554–64. doi: 10.1007/s002390010119. [DOI] [PubMed] [Google Scholar]
- Hoskins RA, et al. Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0085. RESEARCH0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, Ashburner M, Celniker SE. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0084. RESEARCH0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A. 2001;98:8714–9. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–32. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Misra S, et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0083. RESEARCH0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Eskin E, Pevzner PA. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res. 2004;14:2245–52. doi: 10.1101/gr.2693004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol. 2005;1:166–75. doi: 10.1371/journal.pcbi.0010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Richards S, et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005;15:1–18. doi: 10.1101/gr.3059305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JD, Preston BD, Johnston LA, Soni A, Loeb LA, Kunkel TA. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol Cell Biol. 1989;9:469–76. doi: 10.1128/mcb.9.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 19962004 http://www.repeatmasker.org.
- Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 2005 http://www.repeatmasker.org/cgi-bin/RepeatProteinMaskRequest.
- Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–7. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Lev-Maor G, Reznik M, Dagan T, Belinky F, Graur D, Ast G. Minimal conditions for exonization of intronic sequences: 5′ splice site formation in alu exons. Mol Cell. 2004;14:221–31. doi: 10.1016/s1097-2765(04)00181-9. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. The Berkeley Drosophila Genome Project gene disruption project: Single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999;153:135–77. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg BG, Gotea V, Makalowski W. Transposable elements as a significant source of transcription regulating signals. Gene. 2006;365:104–10. doi: 10.1016/j.gene.2005.09.036. [DOI] [PubMed] [Google Scholar]
- TIGR. Genome sequence of D. willistoni. 2005 http://www.tigr.org.
- Tu Z. Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedes aegypti. Proc Natl Acad Sci U S A. 1997;94:7475–80. doi: 10.1073/pnas.94.14.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z. Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito, Anopheles gambiae. Proc Natl Acad Sci U S A. 2001;98:1699–704. doi: 10.1073/pnas.041593198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- WUSTL. Genome sequence of D. yakuba and D. simulans. 2005 http://genome.wustl.edu/.
- Yandell M. Comparative Genomics Library-RepeatRunner. 2006 http://www.yandell-lab.org/repeat_runner/index.html.
- Yandell M, Mungall CJ, Smith C, Prochnik S, Kaminker J, Hartzell G, Lewis S, Rubin GM. Large-scale trends in the evolution of gene structures within 11 animal genomes. PLoS Comput Biol. 2006;2:e15. doi: 10.1371/journal.pcbi.0020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.