

Abstract
Protein kinase B (Akt) activation is well known for its protective effects against apoptosis. However, the role of Akt in regulation of necrosis is unknown. This study was designed to test whether Akt activation protects against nephrotoxicant-induced injury and death in renal proximal tubular cells (RPTC). Exposure of primary cultures of RPTC to the nephrotoxic cysteine conjugate, S-(1,2-dichlorovinyl)-l-cysteine (DCVC), resulted in 9% apoptosis and 30% necrosis at 24 h following the exposure. Akt was activated during 8 h but not at 24 h following toxicant exposure. No RPTC necrosis was observed during Akt activation. Blocking Akt activation using a phosphatidylinositol 3-kinase inhibitor, LY294002 (20 μM), or expressing dominant negative (inactive) Akt increased DCVC-induced RPTC necrosis to 42%. In contrast, Akt activation by expression of constitutively active Akt diminished necrosis to 15%. Modulation of Akt activity had no effect on DCVC-induced apoptosis. DCVC-induced RPTC injury was accompanied by decreases in respiration (51% of controls) and ATP levels (57% of controls). Akt inhibition exacerbated decreases in RPTC respiration and intracellular ATP content (both to 30% of controls). In contrast, Akt activation reduced DCVC-induced decreases in respiration (80% of controls) and prevented decline in ATP content. These data show that in RPTC, Akt activation reduces 1) toxicant-induced mitochondrial dysfunction, 2) decreases in ATP levels, and 3) necrosis. We conclude that Akt activation plays a protective role against necrosis caused by nephrotoxic insult in RPTC. Furthermore, we identified mitochondria as a subcellular target of protective actions of Akt against necrosis.
Keywords: renal proximal tubular cells; S-(1,2-dichlorovinyl)-l-cysteine; apoptosis; necrosis; ATP; mitochondria
kidney exposure to ischemia-reperfusion and toxic insults is a major cause of acute renal injury that results in necrosis and/or apoptosis (23, 46). Ischemic and toxic injury in the kidney results in rapid decrease in adenine nucleotide (ATP, ADP, and AMP) levels and often leads to acute renal failure (46). Within the kidney, the renal proximal tubule cells (RPTC) are the most susceptible to inhibition of cellular energy metabolism due to their dependence on oxidative metabolism for ATP production and their low capacity for glycolysis (1, 3). Exposure of renal proximal tubules to various nephrotoxicants including S-(1,2-dichlorovinyl)-l-cysteine (DCVC), a nephrotoxic metabolite of common environmental contaminants trichloroethylene and dichloroacetylene, results in selective RPTC injury and death due to both apoptosis and necrosis (4, 10, 17, 19, 20). The mechanisms of DCVC-induced injury involve mitochondrial dysfunction, decrease in ATP levels, thiol depletion, loss of calcium homeostasis, decreases in Na+-K+-ATPase activity and active Na+ transport, lipid peroxidation, and DNA damage (4, 19, 21, 24, 32, 33, 43). However, signaling pathways mediating these pathological changes and mechanisms that may render protection against nephrotoxicant-induced injury and death in renal proximal tubules remain poorly understood.
Protein kinase B (PKB, Akt) is a serine/threonine kinase that is activated in the phosphoinositide-3 kinase (PI3K)-dependent pathway (22, 41). Activation of Akt commences when PI3K catalyzes production of phosphatidylinositol bisphosphates and trisphosphates. This recruits phosphoinositide-dependent kinase (PDK) and Akt to migrate to the plasma membrane, which facilitates PDK-mediated phosphorylation of Akt at serine 473 and threonine 308 (22, 41). Activation of Akt has been implicated in cell proliferation, growth, differentiation, and survival (22, 41). Akt activation by hypoxia/reoxygenation, ischemia-reperfusion, and oxidative stress has been shown in cardiac myocytes, neuronal cells, pulmonary epithelial cells, and human renal tubular epithelial cells (30, 39, 42). Akt is also activated by drugs, toxicants, and free radicals including lithium, valproate, Clostridium difficile toxin, tert-butylhydroquinone, singlet oxygen, and nitric oxide in various cell types (5, 14, 15, 49).
The protective role of Akt against apoptosis initiated by mitochondrial damage is well established. For example, Akt activation reduces ischemia-reperfusion and hypoxia/reoxygenation-induced apoptosis in cardiomyocytes and neuronal cells by inhibiting cytochrome c release and activation of caspase-9 and caspase-3 (12, 30, 42). Several proapoptotic (Bad, Bax, and caspase-9) and anti-apoptotic (Bcl-xL and Bcl-2) proteins serve as the downstream targets of Akt (9, 12, 47). Activation of Akt is critical for survival of RPTC and progressive inhibition of Akt is the major factor responsible for apoptosis due to withdrawal of survival factors (40). In cell lines of renal proximal tubular origin, such as LLC-PK1 and HK-2, Akt inactivation activates Bad, accelerates caspase-9 and caspase-3 activation and apoptosis through the mitochondrial pathway and Akt activation, and attenuates both apoptosis and necrosis induced by oxidative stress and serum withdrawal (16, 39). Growth factors including hepatocyte growth factor (HGF) execute their anti-apoptotic effects in RPTC by enhancing activation of Akt and increasing expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL as well as inhibiting expression of proapoptotic protein Bad (8, 25, 27). HGF-induced Akt activation protects against folic acid-induced renal tubular epithelial cell injury and death and promotes regeneration of these cells following the injury (8).
In contrast, not much is known about the involvement of Akt in the mechanisms of necrosis. It has been shown that expression of active Akt protects against ischemia-reperfusion-induced necrosis in rat liver and in cardiomyocytes (11, 36). Protective effects of lipoic acid against ischemia-reperfusion injury and necrosis in the liver are mediated through Akt activation (29). On the contrary, Akt inhibition by an anti-tumor agent kahalalide F induces necrosis in breast cancer cells (13). Our previous studies have shown that epidermal growth factor (EGF) promotes recovery of RPTC mitochondrial function and active Na+ transport following DCVC injury (33). Activation of EGF receptor in RPTC results in Akt phosphorylation, which mediates RPTC proliferation (48). However, the role of Akt in necrosis of RPTC is not known. We hypothesized that Akt activation protects against RPTC injury and death and/or promotes the recovery of RPTC following DCVC injury. Hence, the aim of this study was to determine whether Akt plays a role in nephrotoxicant-induced RPTC injury and death and whether activation of Akt protects against RPTC death following nephrotoxicant exposure.
MATERIALS AND METHODS
Materials
Female New Zealand White rabbits (2.0–2.5 kg) were purchased from Myrtle's Rabbitry (Thompson Station, TN). The cell culture media, DMEM, MEM, a 50:50 mixture of DMEM and Ham's F-12 nutrient mix without phenol red, pyruvate, and glucose, and FBS were purchased from MediaTech Cellgro (Herndon, VA). PI3 kinase inhibitor (LY294002), Akt activity assay kit, polyclonal anti-Akt and anti-phospho-Akt(Ser473) antibodies, anti-phospho-Akt(Ser473) antibody conjugated to Alexa Fluor 488, and cleaved caspase 3 antibody were supplied by Cell Signaling Technologies (Beverly, MA). Antibody that recognizes both caspase 9 and cleaved caspase 9 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Annexin V-fluorescein isothiocyanate (FITC) was purchased from BioVision (Mountain View, CA). ATP Bioluminescence Assay Kit HS II and protease inhibitors cocktail were obtained from Roche (Mannheim, Germany). Phosphatase inhibitors cocktail and calf thymus DNA were supplied by Sigma (St. Louis, MO). The supersignal chemiluminescent system was obtained from Pierce (Rockford, IL). PicoGreen dsDNA quantitation reagent was purchased from Invitrogen (Carlsbad, CA). Adenoviral vectors encoding dominant negative (inactive) Akt [HA-Akt (K179M)] and constitutively active Akt (HA-myr Akt) were constructed and provided by Dr. J. Sadoshima (University of Medicine and Dentistry of New Jersey, Newark, NJ). Adenovirus carrying an empty pShuttle vector was obtained from BD Biosciences Clontech (Palo Alto, CA). AD293 and HEK293 cells were purchased from ATCC (Manassas, VA). Broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone, zVAD-FMK, was purchased from Biomol International (Plymouth Meeting, PA). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from Molecular Probes (Eugene, OR). The source of other reagents has been previously described (31, 35).
DCVC
DCVC was synthesized by the method of McKinney et al. (26). Briefly, anhydrous ammonia (250 ml) was stirred under nitrogen while cooling in a dry ice/acetone bath. Sodium metal (0.2 mol) was slowly added, and the stirring was continued until the sodium had dissolved, giving a blue color. l-Cysteine (0.1 mol) was added portion wise and stirred until the mixture was colorless. Then trichloroethylene (0.1 mol) was added, and stirring was continued for 90 min. The dry ice/acetone bath was removed, and the ammonia was evaporated under a continuous stream of air. The remaining solid was dissolved in 300 ml of water and the remaining ammonia was removed in vacuo. The pH was adjusted to 5.0 using glacial acetic acid to afford a precipitate. Ethanol (300 ml) was added, and the mixture was refrigerated overnight. The precipitate was filtered, dissolved in 700 ml of hot water (70°C), treated with 1 g of activated charcoal (10 min), and filtered while hot. Ethanol (700 ml) was added, and the mixture was refrigerated overnight. The resulting crystals were filtered and dried in vacuo to give 16.8 g (78%), mp 156° dec (lit 156–160° dec) (26).
Isolation and culture of RPTC
Renal proximal tubules were isolated from female New Zealand White rabbit kidneys by the iron-oxide perfusion method as previously described (37). Isolated proximal tubules were grown in 35-mm dishes under improved culture conditions as previously described (35). The culture medium used was a 50:50 mixture of DMEM and Ham's F-12 nutrient mix without phenol red, glucose, and pyruvate and supplemented with 15 mM HEPES, 15 mM NaHCO3, 2.5 mM glutamine, 6 mM lactate (pH 7.4, 295 mosmol/kgH2O). Human transferrin (5 μg/ml), selenium (5 ng/ml), hydrocortisone (50 nM), bovine insulin (10 nM), and l-ascorbic acid-2-phosphate (50 μM) were added before daily media change. Culture plates were constantly shaken (80 rpm) to improve oxygenation of the culture media.
Toxicant treatment of RPTC monolayer
Insulin was removed from the culture media 24 h before the DCVC treatment. Confluent quiescent RPTC monolayers were exposed to 240 μM DCVC for 90 min. DCVC exposure was terminated by aspirating the culture media and adding fresh warm culture media containing all hormones except for insulin. Samples were taken at different time points after DCVC exposure for assessment of cell death, mitochondrial functions, ATP and DNA content, immunoblot analysis, and immunoprecipitation. Akt activation was inhibited by adding 20 μM LY294002 (PI3 kinase inhibitor) or expressing dominant inactive Akt using adenoviral vector encoding dominant negative Akt (a mutant form of Akt rendered inactive by replacing the phosphorylatable serine and threonine residues with nonphosphorylatable alanine residue) before and after DCVC exposure (MOI = 21). Akt activation in RPTC was increased by expressing constitutively active Akt using adenoviral vector encoding a myristylated (at the NH2 terminus) form of Akt with enhanced basal kinase activity before and after DCVC exposure (MOI = 27). Infection with adenovirus encoding an empty pShuttle vector (MOI = 21) was used as a negative control. Adenoviral infection of confluent RPTC was carried out 48 h before DCVC exposure followed by the second infection immediately after removal of DCVC. At these concentrations, adenoviral particles did not produce any cytotoxic effect in RPTC. Pan caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-FMK; 50 μM), was added 1 h before DCVC exposure and immediately after DCVC exposure.
Adenoviral amplification
Adenoviruses carrying an empty pShuttle vector and cDNAs coding for dominant negative and constitutively active Akt were amplified in AD293 cells followed by another amplification in HEK293 cells. AD293 cells were grown at 37°C and 95% air-5% CO2 in DMEM supplemented with 10% FBS until 90% confluent, infected with adenoviral stock solution, and cultured until the cytopathic effect (cells rounding up and detaching from the monolayer) was observed. The AD293 cells were then collected, centrifuged at 150 g for 5 min, and lysed in PBS, pH 7.4, by three consecutive freeze-thaw cycles. The cell lysate was centrifuged at 200 g for 5 min, and the supernatant was used for amplification in HEK293 cells. HEK293 cells were grown at 37°C and 95% air-5% CO2 in Eagle's MEM supplemented with 10% horse serum until 90% confluent, infected with cell lysates carrying adenoviral particles obtained from first amplification in AD293 cells, and cultured until the cytopathic effect was observed. HEK293 cells were then collected and processed as described above for AD293 cells. Adenoviral particles were isolated and purified from HEK293 lysates by centrifugation in CsCl density gradient (7.5 and 8.3 M) at 175,500 g for 1 h. The viral titer in the purified fraction was determined by the end point dilution assay.
Assessment of RPTC death
RPTC apoptosis was evaluated by measuring phosphatidylserine externalization on the plasma membrane using annexin V/propidium iodide binding assay as previously described (34). Briefly, RPTC monolayers were washed with ice-cold binding buffer (10 mM HEPES, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2, pH 7.4) and incubated for 15 min on ice in the presence of propidium iodide diluted in the binding buffer (2 μg/ml). The monolayers were washed with the binding buffer and incubated for 10 min at room temperature in the presence of annexin V-FITC (125 ng/ml). RPTC were then washed twice with the ice-cold binding buffer, scraped off the dishes, suspended in ice-cold binding buffer, and processed for flow cytometry. Propidium iodide and annexin V-FITC fluorescence were quantified by flow cytometry (Beckton Dickinson's FACS Calibur) using excitation at 488 nm and emission at 590 and 530 nm, respectively. For each sample, 10,000 events were counted. Cells positive for annexin V and negative for propidium iodide were considered apoptotic. Cells positive for propidium iodide and negative for annexin V were considered necrotic.
Assessment of LDH release
Release of lactate dehydrogenase (LDH) into the culture medium was used as another marker of RPTC necrosis. LDH activity in culture media and RPTC lysates was determined spectrophotometrically at 340 nm by measuring NADH (0.3 mM) oxidation in the presence of 1.8 mM pyruvate as a substrate as described by Moran and Schnellmann (28).
DAPI staining
Nuclear morphology was visualized by DAPI staining. RPTC monolayers were fixed in 3.7% formaldehyde for 15 min, rinsed with PBS, and incubated in the presence of 8 μM DAPI for 2 h at room temperature. Following staining, RPTC monolayers were rinsed with PBS, coverslips were mounted, and the nuclei were evaluated under a Zeiss Fluorescent Microscope.
Immunoblot analysis
Phosphorylation and protein levels of Akt in RPTC lysates were assessed by immunoblot analysis. RPTC samples were collected at different time points during and following DCVC exposure. The monolayers were washed with ice-cold PBS and lysed on ice in the modified radioimmune precipitation assay (RIPA) buffer (50 mM Tris·HCl, 150 mM NaCl, 1 mM EGTA, 2% Triton X-100, 1 mM Na3VO4, 1 mM NaF, and the protease and phosphatase inhibitor cocktails; pH 7.4) for 10 min. The cell lysates were centrifuged at 18,400 g for 30 min at 4°C to pellet the detergent-insoluble fraction, and the supernatants were boiled for 10 min at 95°C in Laemmli sample buffer (60 mM Tris·HCl, pH 6.8, 2% SDS, 10% glycerol, 100 mM β-mercaptoethanol, and 0.01% bromphenol blue) (18). Aliquots of each sample containing equal amounts of protein (20 μg) were subjected to SDS-PAGE followed by electroblotting of proteins to nitrocellulose membrane. Blots were blocked in Tris-buffered saline (50 mM Tris·HCl, 150 mM NaCl, pH 7.5) containing 0.05% Tween 20 (TBS-T) supplemented with 0.5% casein (blocking buffer) and incubated overnight at 4°C with primary antibodies diluted in the blocking buffer. Following incubation, blots were washed in TBS-T, incubated with secondary IgG coupled to horseradish peroxidase diluted in the blocking buffer, and washed again in TBS-T. The supersignal chemiluminiscent system was used for detection of immunoblots and the results were quantified using scanning densitometry.
Immunocytochemistry
RPTC monolayers were washed three times with ice-cold PBS and fixed with 3.7% formaldehyde for 10 min on ice. Fixed monolayers were permeabilized with ice-cold 100% methanol for 30 min on ice, blocked in PBS containing 0.5% BSA for 1 h, and incubated for 1 h at room temperature in the presence of the anti-phospho-Akt (Ser473) antibody conjugated to Alexa Fluor 488 diluted in PBS containing 0.5% BSA. Monolayers were washed with PBS containing 0.5% BSA, mounting media added, and coverslips mounted. RPTC monolayers were examined and images taken using a Zeiss LSM410 confocal laser-scanning microscope at a magnification of ×400.
Immunoprecipitation and measurement of Akt activity
Akt activity in RPTC lysates was determined using nonradioactive Akt kinase assay kit following immunoprecipitation of Akt from RPTC lysates using Akt antibody cross linked to agarose hydrazide beads. RPTC monolayers were washed with ice-cold PBS, lysed in ice-cold cell lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride, pH 7.5) for 5 min, and centrifuged for 5 min at 18,400 g at 4°C. The supernatants containing equal amounts of cellular protein (500 μg) were incubated with the immobilized Akt antibody for 2 h at 4°C with constant mixing on a rotating shaker at 60 rpm. The immunocomplexes were pelleted by centrifugation at 18,500 g for 30 s at 4°C, washed twice with the lysis buffer and twice with the kinase assay buffer (25 mM Tris, 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, 10 mM MgCl2, pH 7.5). The kinase assay used glycogen synthase kinase-3 (GSK-3) fusion protein (GSK-3α/β crosstide corresponding to residues surrounding serines 21/9: CGPKGPGRRGRRRTSSFAEG) as the Akt substrate. The Akt immunocomplexes were incubated with GSK-3 fusion protein (1 μg/50 μl) and 200 μM ATP for 30 min at 30°C. The reaction was terminated by adding Laemmli sample buffer and boiling for 5 min at 95°C. The samples were spun down at 14,000 g for 2 min and subjected to SDS-PAGE followed by immunoblotting. Phosphorylation of GSK-3 fusion protein was assessed using phospho-GSK-3α/β(Ser21/9) antibody.
Measurement of oxygen consumption
Mitochondrial function was assessed by measuring basal QO2 as a marker (38). At 24 h after DCVC exposure, RPTC monolayers were gently detached from culture dishes with the use of rubber policeman, suspended in the culture media, and transferred to the QO2 measurement chamber. QO2 was measured polarographically using a Clark-type electrode as described previously (31, 35).
Measurement of intracellular ATP content
ATP content in RPTC was measured by the luciferase method using the ATP bioluminescence assay kit supplied by Roche (Indianapolis, IN) and instructions provided by the manufacturer as described previously (24).
Monolayer DNA content
Monolayer DNA content was used as a marker of cell number. DNA concentration in RPTC lysates was determined by PicoGreen dsDNA quantitation reagent using the manufacturer's protocol and calf thymus dsDNA as the standard. PicoGreen dsDNA reagent exhibits a large fluorescent enhancement upon binding to double-strand DNA and provides the most sensitive method available for measuring dsDNA concentrations. RPTC monolayers were washed with ice-cold PBS, solubilized in 1 ml solubilization buffer (100 mM Tris, 150 mM NaCl, 0.05% Triton X-100; pH 7.5), sonicated, and centrifuged at 11,300 g for 2 min at 4°C. RPTC lysates and DNA standards were diluted in Tris/EDTA buffer (50 mM Tris, 2 M NaCl, 2 mM EDTA, pH 7.4) and incubated in the presence of PicoGreen solution for 5 min at room temperature. Fluorescence was evaluated using excitation at 485 nm and emission at 535 nm.
All results were normalized to cellular protein, which was measured by bicinchoninic acid (BCA) assay using BSA as the standard.
Statistical analysis
Data are presented as means ± SE and were analyzed for significance by one-way ANOVA. Multiple means were compared using Student-Newman-Keuls test. The level of significance was set at P < 0.05. RPTC isolated from an individual rabbit represented one experiment (n = 1).
RESULTS
Effect of DCVC exposure on RPTC death
Exposure of RPTC to different concentrations of DCVC (5–500 μM for 90 min) resulted in a concentration-dependent increase in both apoptosis and necrosis. Exposure of RPTC to 100, 240, and 500 μM DCVC induced 19 ± 4, 22 ± 2, and 14 ± 3% of apoptosis, respectively, as determined by annexin V binding to the plasma membrane. DCVC also produced necrosis in a concentration-dependent manner (10 ± 3, 30 ± 2, and 47 ± 6% at 24 h after exposure to 100, 240, and 500 μM DCVC, respectively). Figure 1 shows that DCVC (240 μM)-induced RPTC necrosis (measured using propidium iodide binding and LDH release as markers) was time dependent and was preceded by initiation of apoptosis. Exposure of RPTC to DCVC initiates apoptosis at 4 h (10.3 ± 3% of annexin V-positive cells; Fig. 1A), which is followed by necrosis starting at 8 h (8 ± 2% LDH release) and increasing through 12 and 24 h (20 ± 3 and 30 ± 4% of LDH release) following the exposure (Fig. 1B). Similarly, flow cytometry analysis of propidium iodide binding show that DCVC-induced necrosis increases at 8, 12, and 24 h following the exposure (Fig. 1B).
Fig. 1.
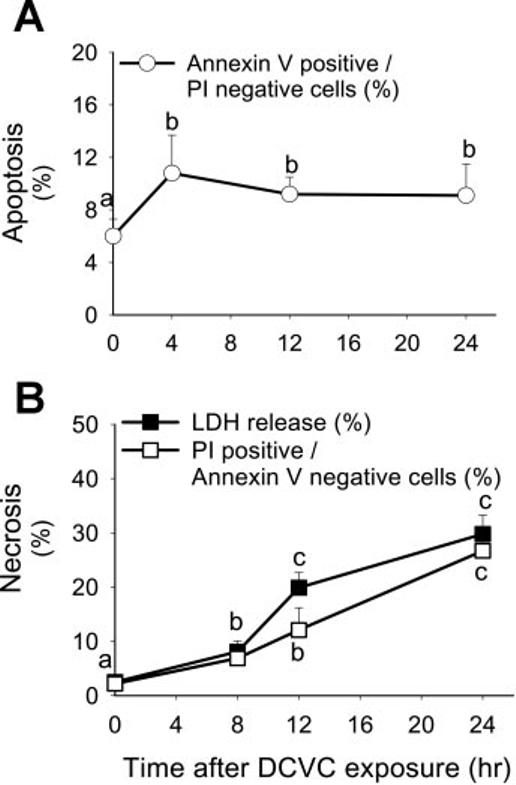
Effect of DCVC (240 μM) on renal proximal tubular cells (RPTC) apoptosis (A; annexin V-FITC positive/propidium iodide-negative cells) and necrosis (B; measured by evaluating LDH release and quantifying propidium iodide positive/annexin V-FITC-negative RPTC) at different time points following the exposure. The results are means ± SE (n = 6). Values with dissimilar superscripts (a, b, c) are significantly different (P < 0.05) from each other.
Activation of Akt by DCVC
The phosphorylation status of Akt was used as an initial indicator of Akt activation in DCVC-injured RPTC. Exposure of RPTC to DCVC resulted in a time-dependent increase in phosphorylation of Akt at Ser473 (phosphorylation of this residue stabilizes the active conformation of Akt leading to its full activation). Figure 2 shows that protein levels of phosphorylated Akt increased three-, four-, and sixfold at 15, 45, and 90 min of DCVC exposure in RPTC, respectively, and remained at increased levels for at least 8 h following DCVC exposure (Fig. 2, A and B). The ratio of phosphorylated Akt to total Akt following DCVC exposure was increased 1.5-, 2.5-, and 2-fold at 1, 4, and 6 h, respectively, compared with controls (Fig. 2C). The treatment of RPTC with PI3K inhibitor, 20 μM LY294002, before DCVC exposure blocked the phosphorylation of Akt at all time points during and following DCVC exposure demonstrating that LY294002 inhibits Akt activation (Fig. 2D and see Fig. 4B).
Fig. 2.
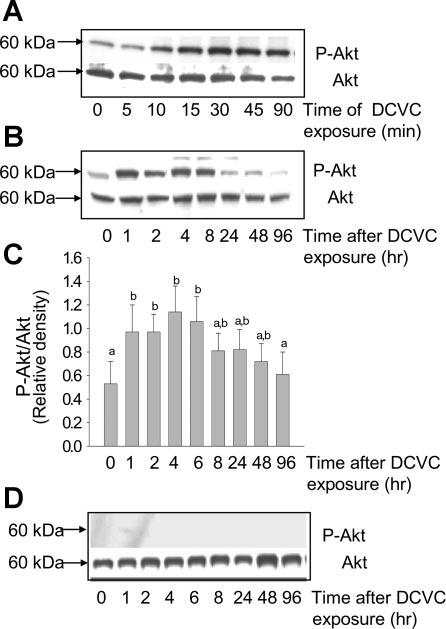
Phosphorylation of Akt(Ser473) in DCVC injured RPTC. A: protein levels of phosphorylated and total Akt in RPTC at different time points of DCVC (240 μM) exposure (90 min). B: protein levels of phosphorylated and total Akt in RPTC at different time points after DCVC exposure. C: ratio of phospho-Akt(Ser473) to total Akt at different time points following DCVC injury in RPTC. The results (quantified by densitometry) are means ± SE (n = 3). Values with dissimilar superscripts (a, b) are significantly (P < 0.05) different from each other. D: effect of PI3 kinase inhibitor (LY294002, 20 μM) on Akt phosphorylation following DCVC injury in RPTC. Blots are representative of 4 independent experiments.
Fig. 4.
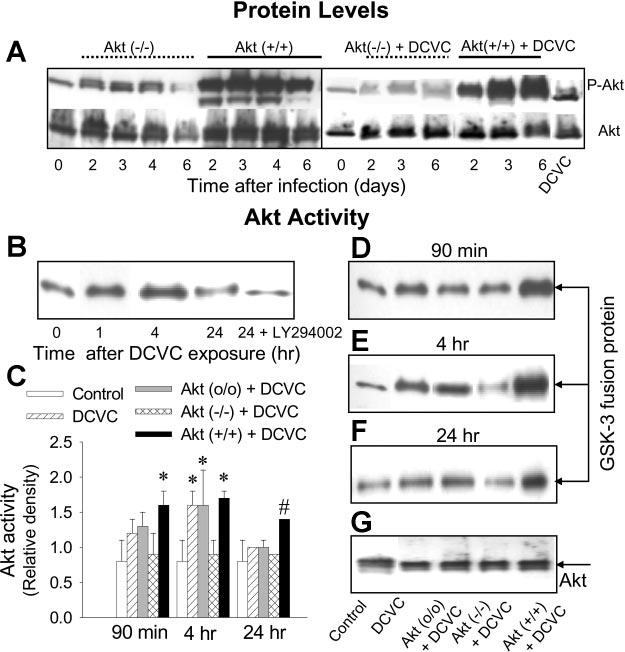
Protein levels of phosphorylated and total Akt in RPTC expressing dominant negative Akt [MOI = 21; Akt (−/−)] and constitutively active Akt [MOI = 27; Akt (+/+)] in the absence of and presence of DCVC (A) at various time points following infection with adenovirus carrying Akt cDNA. Akt activity phosphorylation of GSK-3α/β in RPTC at various time points following DCVC (240 μM) exposure in the absence and presence of 20 μM LY294002 (B), at 90 min (D), 4 h (E), and 24 h (F) following DCVC exposure. G: protein levels of Akt immunoprecipitated from RPTC homogenates. Akt (o/o)-RPTC infected with adenovirus carrying an empty plasmid vector (MOI = 21). The blots are representative of 3 independent experiments. C: graph showing Akt activity [measured as ratio of phospho-GSK-3α/β to total Akt (quantified by densitometry) in immunoprecipitates from RPTC] in RPTC expressing Akt (−/−) and Akt (+/+).Values with dissimilar superscripts are significantly (P < 0.05) different from each other.
Immunocytochemical analysis of Akt phosphorylation using antibody recognizing phospho-Akt (Ser473) confirmed that DCVC exposure increased the levels of phosphorylated Akt in RPTC. In addition, the number of cells that tested positive for phosphorylated Akt increased in DCVC-exposed RPTC monolayers compared with controls (Fig. 3, A and B). Akt phosphorylation was still increased at 4 h (Fig. 3C) but decreased at 24 h following DCVC exposure to the levels observed in controls (Fig. 3D). Collectively, these data suggest that Akt is activated by DCVC exposure, but this activation is not sustained at later time points of the injury.
Fig. 3.
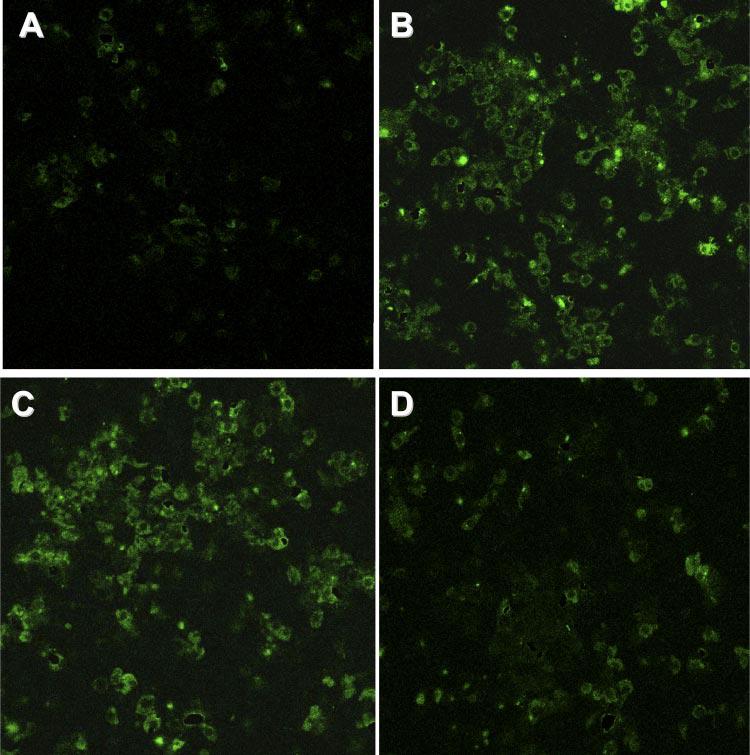
Immunocytochemical assessment of Akt phosphorylation in DCVC (240 μM)-injured RPTC. A: control RPTC. B: RPTC exposed to DCVC for 90 min. C: RPTC at 4 h following DCVC withdrawal. D: RPTC at 24 h following DCVC withdrawal. Magnification ×400. Confocal laser-scanning images are representative of 3 independent experiments.
Measurements of Akt activity showed that DCVC-induced Akt phosphorylation results in the activation of Akt as indicated by the phosphorylation of a specific Akt substrate, GSK-3α/β(Ser21/9), by Akt immunoprecipitated from RPTC. Akt was activated during (Fig. 4B) and following (4 h) DCVC exposure, but this activation subsided 24 h after exposure (Fig. 4, B and F).
Expressing constitutively active and inactive Akt in RPTC
To determine the role of Akt in DCVC-induced RPTC injury and death, protein levels of constitutively active and inactive Akt were increased by infecting RPTC with adenovirus carrying cDNA coding for constitutively active or dominant negative (inactive) Akt. RPTC were infected 48 h before the toxicant exposure and immediately after the exposure to maintain Akt activity throughout the whole experiment. Expressing constitutively active Akt, Akt(+/+), increased Akt phosphorylation in RPTC for at least 6 days postinfection (Fig. 4A) and resulted in increased Akt activity in DCVC-treated cells (Fig. 4, D-F). In contrast, expressing dominant negative Akt, Akt(−/−), blocked DCVC-stimulated Akt phosphorylation (Fig. 4A) and decreased Akt activity in DCVC-treated RPTC (Fig. 4, D-F). Adenovirus containing an empty vector, Akt(o/o), had no effect on Akt activity demonstrating that effects of Akt(+/+) and Akt(−/−) were specific (Fig. 4, D-F).
These results show that Akt is activated in RPTC during DCVC exposure and early time points following the treatment, but Akt activation subsides 24 h after exposure. Treatment of RPTC with PI3 kinase inhibitor (20 μM LY294002) or expressing inactive Akt prevents the activation of Akt by DCVC. In contrast, expressing constitutively active Akt increases DCVC-induced Akt activation in RPTC.
Lack of Akt involvement in DCVC-induced RPTC apoptosis
To determine whether Akt plays a role in DCVC-induced RPTC apoptosis, Akt activation was blocked and RPTC apoptosis was examined following DCVC injury by 1) quantifying phosphatidylserine externalization on the plasma membrane by flow cytometric analysis of annexin V-FITC binding, 2) assessing changes in nuclear morphology using DAPI staining, and 3) immunoblot analysis of caspase 9 and caspase 3 cleavage which is indicative of caspase activation. Blocking Akt activation using LY294002 showed no effect on DCVC-induced phosphatidylserine externalization (marker of apoptosis) at 24 h after DCVC exposure (19 ± 3 vs. 22 ± 2% in the presence and absence of LY294002, respectively; see Fig. 6A). Likewise, decreasing Akt activation (by expressing dominant negative Akt) or increasing Akt activation (by expressing constitutively active Akt) had no effect on DCVC-induced apoptosis at any time point following DCVC exposure (see Fig. 7C).
Fig. 6.
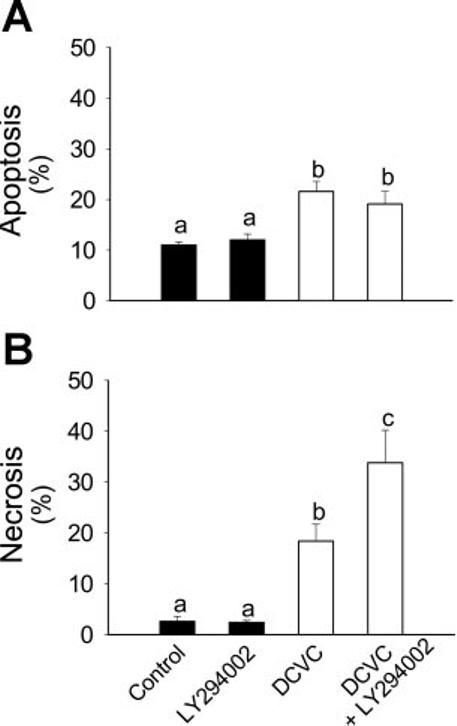
Effect of inhibition of Akt activation (20 μM LY294002) on DCVC-induced RPTC apoptosis (A) and necrosis (B) at 24 h following DCVC (240 μM) exposure. The results are means ± SE (n = 5). Values with dissimilar superscripts (a, b, c) are significantly (P < 0.05) different from each other.
Fig. 7.
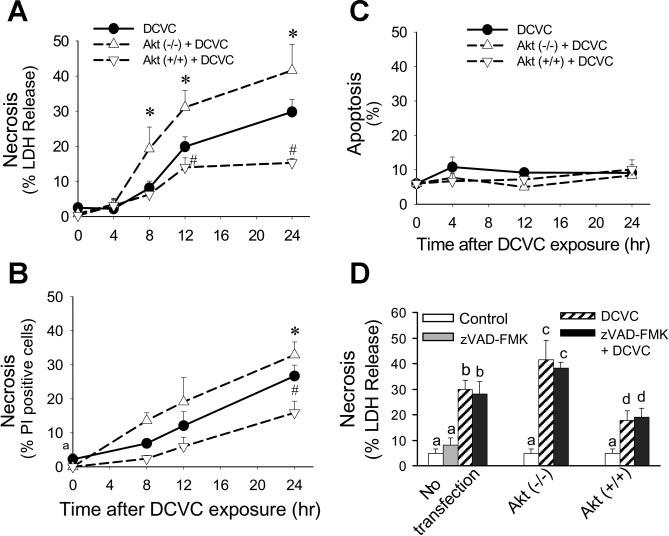
Effect of modulation of Akt activation on RPTC necrosis (A: measured using LDH release as marker, B: measured by flow cytometric analysis of propidium iodide-positive and annexin V-FITC-negative cells) and apoptosis (C) at different time points following DCVC (240 μM) exposure. D: effect of caspase inhibition (50 μM, zVAD-FMK) on DCVC-induced necrosis in RPTC at 24 h following DCVC exposure. Akt (−/−) RPTC infected with adenovirus carrying dominant negative (inactive) Akt (MOI = 21). Akt (+/+) RPTC infected with adenovirus carrying constitutively Akt (MOI = 27). The results are means ± SE (n = 5). Values with dissimilar superscripts (a, b, c, d) are significantly (P < 0.05) different from each other.
DCVC-induced apoptosis was also determined by assessing changes in nuclear morphology (using DAPI staining followed by fluorescence microscopy). Figure 5 shows that DCVC injury results in increased nuclear fragmentation in RPTC as determined at 24 h following the injury. Inhibition of Akt (by LY294002 or inactive Akt) had no effect on chromatin condensation and nuclear fragmentation in DCVC-injured RPTC (Fig. 5, C and D). Likewise, Akt activation had no effect on DCVC-induced changes in nuclear morphology in RPTC (Fig. 5E).
Fig. 5.
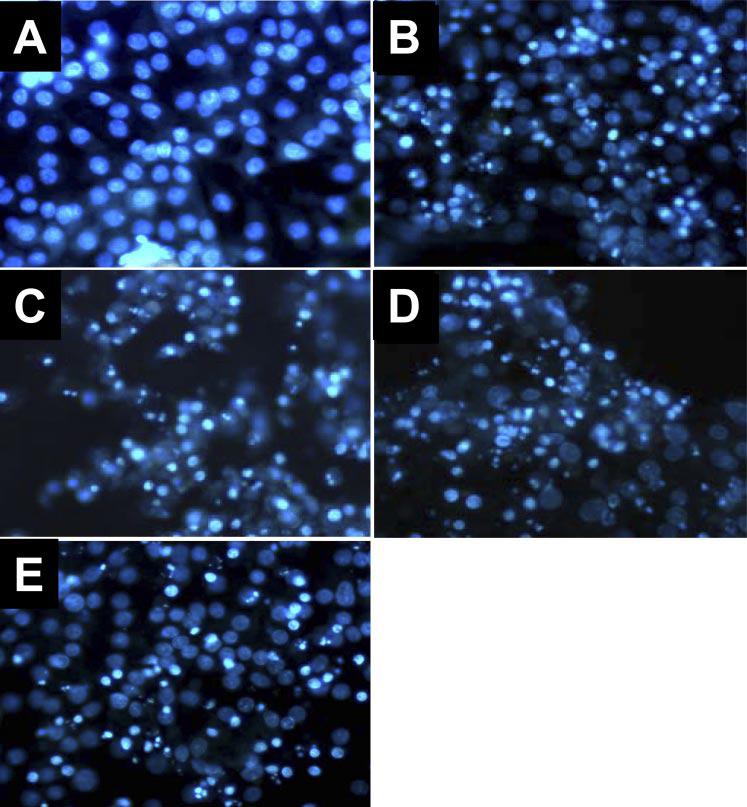
Effect of modulation of Akt activation on DCVC-induced changes in nuclear morphology at 24 h following DCVC exposure in RPTC. Control RPTC (A), DCVC-injured RPTC (B), DCVC-injured RPTC treated with LY294002 (C), DCVC-injured RPTC expressing inactive Akt (D), and DCVC-injured RPTC expressing constitutively active Akt (E). Magnification ×400. Microphotographs are representative of results obtained from 3 different experiments.
Taken together, these results demonstrate that Akt does not regulate DCVC-induced apoptosis in RPTC. Caspase 9 and caspase 3 cleavages (indicative of caspase activation) were evaluated by immunoblot analysis and were used as another marker of apoptosis in DCVC-injured RPTC. Compared with controls, no increase in protein levels of cleaved (active) caspase 9 was observed at any time point following DCVC exposure in injured RPTC (see Fig. 10C). Similarly, no cleaved caspase 3 was detected in DCVC-injured RPTC at any time point following toxicant exposure (see Fig. 10C). These results demonstrate that DCVC-induced apoptosis in RPTC is not associated with activation of caspases 9 and 3.
Fig. 10.
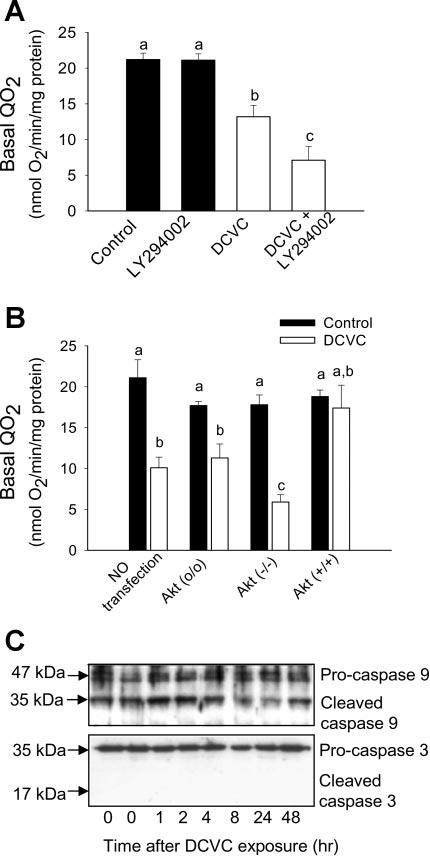
Effect of blocking and increasing Akt activation on cellular respiration in RPTC at 24 h following DCVC (240 μM) exposure. A: basal oxygen consumption (QO2) in DCVC-injured RPTC in the presence and absence of 20 μM LY294002. B: basal QO2 in DCVC-injured RPTC expressing dominant negative Akt [Akt (−/−)] and constitutively active Akt [Akt (+/+)] at 24 h following the exposure. Akt (o/o) RPTC infected with adenovirus carrying an empty plasmid vector. The results are means ± SE (n = 5). Values with dissimilar superscripts are significantly (P < 0.05) different from each other. C: immunoblot analysis of caspase 9 and caspase 3 cleavage in RPTC at different time points following DCVC exposure. The blots shown are representative of 4 individual experiments.
Role of Akt in DCVC-induced RPTC necrosis
In contrast to apoptosis, DCVC-induced necrosis (measured using both propidium iodide binding and LDH release as markers) was dependent on Akt activation status. DCVC-induced necrosis increased from 19 ± 3 to 34 ± 6% when Akt activation was blocked by LY294002 (Fig. 6B). LY294002 alone had no effect on RPTC death, which indicates that the increase in RPTC necrosis following DCVC injury was due to Akt inhibition and not due to the nonspecific effects of LY294002 (Fig. 6, A and B). Likewise, DCVC-induced necrosis at 24 h following the exposure was increased from 30 ± 3 to 42 ± 7% in RPTC expressing inactive Akt (Fig. 7A). In contrast, increasing Akt activation by expressing constitutively active Akt decreased DCVC-induced necrosis from 30 ± 3 to 15 ± 1% at 24 h following DCVC injury (Fig. 7A). Similar results were obtained when the role of Akt in DCVC-induced necrosis was evaluated by the quantification of propidium iodide-positive cells (Fig. 7B). Infecting RPTC with adenovirus carrying an empty vector [Akt(o/o)] had no effect on DCVC-induced RPTC death indicating that the effects of Akt(+/+) and Akt(−/−) were specific.
To determine whether late apoptosis contributed to the assessment of necrosis in DCVC-injured RPTC, caspases were inhibited using pan caspase inhibitor zVAD-FMK (a broadspectrum caspase inhibitor) and necrosis (LDH release) was determined following DCVC injury. Figure 7D shows that inhibition of caspases had no effect on DCVC-induced necrosis in RPTC (30 ± 3 vs. 28 ± 3% in the absence and presence of zVAD-FMK, respectively) regardless of Akt activation status. Furthermore, caspase inhibition had no effect on DCVC-induced apoptosis (8.8 ± 1.8 vs. 9.4 ± 2.5% apoptosis in the presence and absence of 50 μM zVAD-FMK, respectively). These results demonstrate that DCVC-induced apoptosis is not dependent on caspase activation and that the protective effects of Akt activation against RPTC necrosis following DCVC injury are not due to inhibition of apoptosis.
The effect of Akt on DCVC-induced RPTC death and loss was also monitored by measurements of monolayer DNA content. DNA content in RPTC was decreased by 37% at 24 h following DCVC exposure (23 ± 2 vs. 36 ± 1 μg/plate in DCVC-injured and control RPTC). Increasing Akt activation reduced the decreases in monolayer DNA content in RPTC following DCVC exposure (Fig. 8).
Fig. 8.
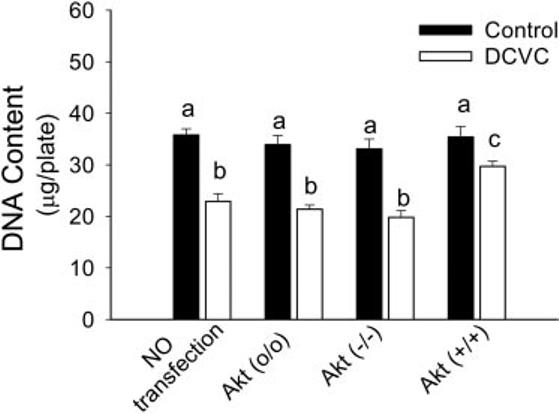
Effect of modulation of Akt activation on monolayer DNA content in RPTC at 24 h following the DCVC injury. Akt (o/o) RPTC infected with adenovirus carrying an empty plasmid vector (MOI = 21). Akt (−/−) RPTC infected with adenovirus carrying dominant negative (inactive) Akt (MOI = 21). Akt (+/+) RPTC infected with adenovirus carrying constitutively Akt (MOI = 27). Values with dissimilar superscripts are significantly (P < 0.05) different from each other.
These results show that Akt activation decreases RPTC necrosis and cell loss following DCVC injury and that Akt plays a protective role against nephrotoxicant-induced necrosis in RPTC. In contrast, Akt does not play a role in nephrotoxicant-induced apoptosis in RPTC.
Role of Akt in maintaining ATP levels following DCVC injury
The decrease in ATP levels is a critical factor initiating cellular events that eventually culminate in RPTC necrosis. To determine whether the protective role of Akt activation against RPTC necrosis is mediated through regulation of intracellular ATP levels, we determined intracellular ATP content in RPTC expressing active and inactive Akt following DCVC injury. As shown in Fig. 9, the exposure of RPTC to DCVC resulted in a decrease in intracellular ATP content to 57% of control levels (8 ± 3 vs. 15 ± 3 nmol/mg protein in DCVC-treated and control RPTC, respectively). Blocking Akt activation in DCVC-injured RPTC (using LY294002) resulted in further decrease in intracellular ATP content to 30% of controls (4 ± 2 nmol/mg protein; Fig. 9A). Similarly, decreasing Akt activation by expressing dominant negative Akt decreased intracellular ATP levels to 42% of controls (5.2 ± 1 nmol/mg protein). In contrast, increasing Akt activation prevented DCVC-induced decreases in intracellular ATP content (Fig. 9B). Expressing Akt(o/o) had no effect on the decreases in intracellular ATP levels in injured RPTC (Fig. 9B). These results show that Akt activation maintains intracellular ATP levels in DCVC-injured RPTC.
Fig. 9.
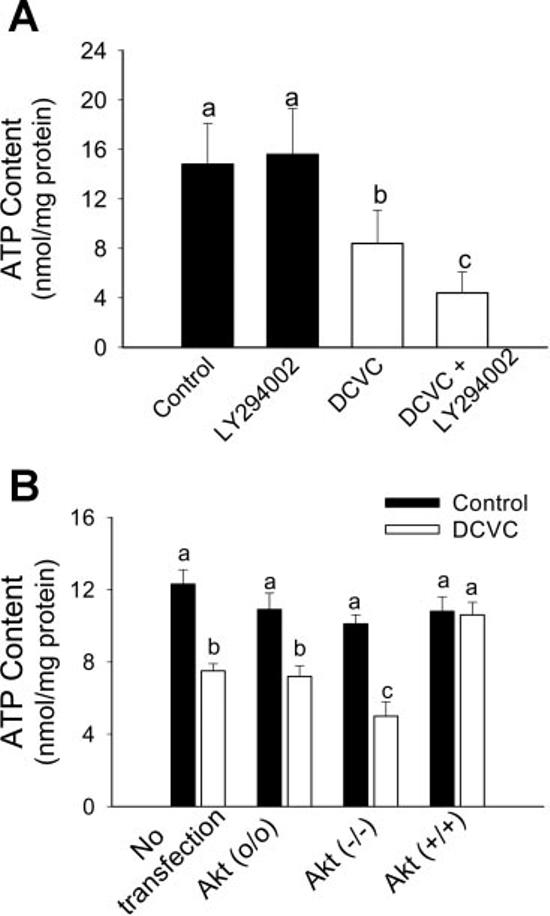
Effect of decreased and increased Akt activation on intracellular ATP content in RPTC at 24 h following DCVC (240 μM) exposure. A: ATP content in DCVC-injured RPTC in the presence and absence of 20 μM LY294002. B: ATP content in DCVC-injured RPTC expressing dominant negative Akt [Akt (−/−)] and constitutively active Akt [Akt (+/+)] at 24 h following the exposure. Akt (o/o) RPTC infected with adenovirus carrying an empty plasmid vector. The results are means ± SE (n = 8). Values with dissimilar superscripts (a, b, c) are significantly (P < 0.05) different from each other.
Role of Akt in maintaining mitochondrial function following DCVC injury
Previously, we showed that DCVC exposure decreases mitochondrial function in primary cultures of RPTC (24, 33). At 24 h following DCVC exposure, basal QO2 (used as a marker of overall mitochondrial function) decreased to 51% of control levels (Fig. 10, A and B). DCVC-induced decreases in RPTC respiration (Fig. 10) and intracellular ATP (Fig. 9) and increases in RPTC necrosis (Fig. 1B) at 24 h after the exposure were associated with the decrease in Akt activation (Figs. 2-4). To determine whether Akt activation plays a role in maintaining ATP levels in DCVC-injured RPTC through promotion of mitochondrial function, Akt activation in RPTC was blocked or increased and mitochondrial function was examined at 24 h following DCVC exposure. Blocking Akt activation (using LY294002 or expressing inactive Akt) in DCVC-injured RPTC further decreased basal QO2 (30 vs. 51% of controls in the presence and absence of inactive Akt, respectively; Fig. 10, A and B). In contrast, increased activation of Akt reduced DCVC-induced decreases in basal QO2 (80 vs. 51% of controls in the presence and absence of active Akt, respectively; Fig. 10B). These results demonstrate that inhibition of Akt activation exacerbates mitochondrial dysfunction following DCVC injury. In contrast, Akt activation promotes mitochondrial respiration in DCVC-injured RPTC, which suggests that Akt is involved in the regulation of mitochondrial function in nephrotoxicant-injured RPTC.
DISCUSSION
Protein kinase B/Akt has been implicated in protection against apoptosis in many cell types including cardiomyocytes, pulmonary and renal epithelial cells, and various cell lines (12, 16, 30, 42, 47). However, not much is known about the role of Akt in necrosis. The present study demonstrates the protective effects of Akt against necrosis induced by the nephrotoxicant, DCVC, in RPTC. Previous studies have shown that DCVC exposure in vivo results in renal proximal tubular necrosis and induces both apoptosis and necrosis in vitro in human RPTC (4, 10, 17, 20). Our previous and present data show that DCVC-induced apoptosis and necrosis in RPTC are associated with mitochondrial dysfunction and decreases in intracellular ATP levels (24, 33). Also, our previous studies have shown that EGF promotes the recovery of mitochondrial function and active Na+ transport in DCVC-injured RPTC (33). Because EGF activates a variety of signaling molecules in RPTC including Akt (48, 50), we tested whether Akt activation is protective against nephrotoxicant-induced RPTC injury.
This study is the first to demonstrate that activation of Akt decreases RPTC injury and necrosis induced by DCVC. Our data show that the exposure of RPTC to DCVC results in phosphorylation and activation of Akt. Phosphorylation of Akt is mediated through activation of PI3K, subsequent steps that involve production of phosphatidylinositol bisphosphates and trisphosphates, and activation of PDK1 and PDK2. Our immunoblot studies showed that inhibition of PI3 kinase using LY294002 blocks phosphorylation of Akt in RPTC during and following DCVC exposure. This indicates that DCVC-induced phosphorylation and hence activation of Akt in RPTC are mediated through PI3K pathway. Since PI3K activation is mediated through stimulation of cell membrane receptors, we speculate that DCVC binds to a membrane receptor that activates PI3K/Akt pathway. In addition, DCVC exposure increases intracellular calcium levels, which may activate intracellular signaling cascades leading to activation of PI3K and increased production of phosphatidylinositols activating Akt. However, the exact mechanism of Akt phosphorylation in RPTC after DCVC exposure is yet to be determined.
DCVC-induced phosphorylation and hence activation of Akt last 8 h with maximum Akt activation at 4 h following DCVC exposure. Surprisingly, at 4 h following DCVC exposure, we observed an induction of apoptosis, which preceded mitochondrial dysfunction and RPTC necrosis. No decreases in RPTC respiration or ATP levels were observed during Akt activation (data not shown). Furthermore, the mitochondrial membrane potential was still maintained at 4 h following DCVC exposure (24). Consistently, no significant amount of necrosis was found within 8 h following DCVC-induced injury. Interestingly, induction of apoptosis preceded DCVC-induced mitochondrial dysfunction and necrosis. As shown in Fig. 10C, DCVC-induced apoptosis was not accompanied by an increase in caspase 9 cleavage and no detectable levels of cleaved caspase 3 (an indicator of caspase 3 activation) were observed following the DCVC injury. In addition, no decreases in DCVC-induced apoptosis were observed in RPTC treated with pan caspase inhibitor, zVAD. Our unpublished observations also show that the activation of Akt in DCVC-injured RPTC has no effect on proapoptotic protein Bad known for its role in the mitochondrial apoptotic cascade. Therefore, our data suggest that DCVC does not induce apoptosis through the mitochondrial pathway and that DCVC-induced apoptosis proceeds through caspase-independent mechanisms.
The loss of Akt activation at later time points (24 h) following DCVC exposure was accompanied by decreases in RPTC respiration and intracellular ATP content as well as necrosis. To determine whether Akt activation plays a role in nephrotoxicant-induced RPTC injury and/or death, Akt activation was blocked by LY294002, a specific inhibitor of PI3 kinase, and RPTC injury and death were examined. LY294002 has been extensively utilized in many studies to block the phosphorylation of Akt in a variety of cell types. Our studies show that treatment with LY294002 blocks phosphorylation and activation of Akt in RPTC. Furthermore, expressing inactive Akt results in the inhibition of Akt activation, whereas expression of constitutively active Akt increases the levels of active Akt in DCVC-injured RPTC. This is the first report to demonstrate that Akt activation plays a protective role in toxicant-induced necrosis in RPTC. In contrast, inhibition of Akt activation increases DCVC-induced necrosis. This is in agreement with the previous report demonstrating that inhibition of Akt increases necrosis and abrogates the protective effect of erythropoietin against H2O2-induced necrosis in human renal tubular epithelial cells (HK-2) in vitro (39). The treatment of control RPTC with LY294002 or infection with adenoviral vector alone did not affect RPTC viability suggesting that the changes in DCVC-induced necrosis were the result of changes in Akt activity and not due to nonspecific effects of LY294002 and the adenoviral or plasmid DNA.
To determine whether Akt-mediated protection against DCVC-induced necrosis was due to the regulation of ATP levels, we examined the effects of Akt activation and inhibition on intracellular ATP content in DCVC-injured RPTC. Expressing active Akt in RPTC prevented decreases in intracellular ATP levels. In contrast, inhibition of Akt activation exacerbated the decreases in intracellular ATP content. Thus, these results demonstrate that Akt activation maintains ATP content in DCVC-injured RPTC and suggest that Akt activation protects against necrosis, in part, by maintaining intracellular ATP levels. Similarly, it was previously suggested that the protective effects of Akt activation against ischemia-reperfusion-induced necrosis in hepatocytes are mediated through maintaining intracellular ATP levels (29). Intracellular ATP content is dependent on mitochondrial function and oxidative phosphorylation. Our previous studies have shown that DCVC caused mitochondrial dysfunction and decreased ATP production and ATP content in RPTC (24, 33). Therefore, we hypothesized that preservation of intracellular ATP levels in DCVC-injured RPTC by active Akt is due to protective effects on mitochondrial function in toxicant-injured RPTC. Measurements of mitochondrial function showed that inhibition of Akt activation exacerbates DCVC-induced decreases in basal QO2, whereas increasing Akt activation prevents the decreases in RPTC respiration following DCVC injury. These results suggest that Akt activation maintains mitochondrial function and mitochondrial ATP production, thus preserving intracellular ATP in RPTC during nephrotoxicant injury. Active Akt has been shown to accumulate in the mitochondria and form a complex with the catalytic β-subunit of F1F0-ATPase in human neuroblastoma and embryonic kidney cells (2). Thus, it is likely that Akt activation promotes mitochondrial respiration and ATP production by stimulating the function of F1F0-ATPase and intracellular ATP levels. Also, it is possible that Akt promotes mitochondrial function through phosphorylating and regulating proteins involved in the assembly and/or opening of the mitochondrial permeability transition pore and maintaining ΔΨm. It needs to be emphasized that increased ATP levels in injured RPTC were not due to induction of glycolysis as the culture media used in our study did not contain glucose.
Numerous reports have shown that Akt activation protects against apoptosis and that inhibition of Akt increases apoptosis induced by hypoxia/reoxygenation, ischemia-reperfusion, oxidative stress, chemotherapeutic drugs, toxicants, and growth or survival factor withdrawal in a variety of cell types including cardiomyocytes, neurons, pulmonary epithelial cells, tumor and malignant cells, and RPTC (5, 9, 12, 16, 30, 39, 40, 42). However, our data show that neither inhibition nor activation of Akt has any effect on RPTC apoptosis induced by DCVC and that DCVC-induced apoptosis occurs in the absence of caspase activation. It has been established that anti-apoptotic actions of Akt are mediated mostly through phosphorylation and inhibition of proapoptotic proteins such as Bad, Bax, and caspase 9. Caspase-independent apoptosis (induced by cisplatin) in RPTC has been demonstrated previously (6, 7, 34). Caspase-independent mechanism of DCVC-induced apoptosis in RPTC may explain the failure of Akt activation to decrease apoptosis. Previous studies have shown that DCVC induces DNA damage in RPTC (4, 43). A recent report showed that ERK1/2, but not the Akt pathway, is involved in the protection against DNA damage-mediated apoptosis in neuronal cells (44). Our unpublished data show that DCVC exposure induces phosphorylation of ERK1/2 in RPTC. It is likely that DCVC-induced apoptosis is regulated by MEK1/2-ERK1/2 pathway in the PI3 kinase/Akt-independent manner.
In conclusion, this is the first report showing that the activation of Akt serves as a protective mechanism against nephrotoxicant-induced energy deficits and necrosis in RPTC. Our study identified mitochondria as a target of protective actions of Akt during nephrotoxicant-induced injury. Akt activation diminishes mitochondrial dysfunction, prevents decreases in intracellular ATP content, and reduces necrosis in injured RPTC. Our study suggests that Akt activation may represent means to diminish RPTC injury and necrosis due to exposure to nephrotoxic compounds.
ACKNOWLEDGMENTS
We thank Dr. J. Sadoshima (University of Medicine and Dentistry of New Jersey, Newark, NJ) for the generous gift of adenoviruses carrying dominant negative and constitutively active Akt cDNAs. We also thank Dr. P. M. Price (University of Arkansas for Medical Sciences, Little Rock, AR) for assistance in adenoviral amplifications and purifications and A. Mangu for assisting in S-(1,2-dichlorovinyl)-l-cysteine synthesis.
GRANTS
This work was supported by National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-59558 (to G. Nowak) and a Predoctoral Fellowship from the American Heart Association, Heartland Affiliate (to Z. P. Shaik) and a Committee for Allocation of Graduate Student Research Funds grant at the University of Arkansas for Medical Sciences in Little Rock, AR (to Z. P. Shaik). The use of the facilities in the University of Arkansas for Medical Sciences, Digital and Confocal Microscopy Laboratory supported by NIH Grant 2 P20-RR-16460 (INBRE) is also acknowledged.
REFERENCES
- 1.Bagnasco S, Good D, Balaban R, Burg M. Lactate production in isolated segments of the rat nephron. Am J Physiol Renal Fluid Electrolyte Physiol. 1985;248:F522–F526. doi: 10.1152/ajprenal.1985.248.4.F522. [DOI] [PubMed] [Google Scholar]
- 2.Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonventre JV. Pathogenetic and regenerative mechanisms in acute tubular necrosis. Kidney Blood Press Res. 1998;21:226–229. doi: 10.1159/000025861. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Cai J, Anders MW, Stevens JL, Jones DP. Role of mitochondrial dysfunction in S-(1,2-dichlorovinyl)-l-cysteine-induced apoptosis. Toxicol Appl Pharmacol. 2001;170:172–180. doi: 10.1006/taap.2000.9107. [DOI] [PubMed] [Google Scholar]
- 5.Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann NY Acad Sci. 2005;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- 6.Cummings BS, Kinsey GR, Bolchoz LJC, Schnellmann RG. Identification of caspase independent apoptosis in epithelial and cancer cells. J Pharmacol Exp Ther. 2004;310:126–134. doi: 10.1124/jpet.104.065862. [DOI] [PubMed] [Google Scholar]
- 7.Cummings BS, Schnellmann RG. Cisplatin induced renal cell apoptosis: caspase 3-dependent pathways. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- 8.Dai C, Yang J, Liu Y. Single injection of naked plasmid encoding hepatocyte growth factor prevents cell death and ameliorates acute renal failure in mice. J Am Soc Nephrol. 2002;13:411–422. doi: 10.1681/ASN.V132411. [DOI] [PubMed] [Google Scholar]
- 9.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 10.Elfarra AA, Jakobson I, Anders MW. Mechanism of S-(1,2-dichlorovinyl)glutathione-induced nephrotoxicity. Biochem Pharmacol. 1986;35:283–288. doi: 10.1016/0006-2952(86)90527-7. [DOI] [PubMed] [Google Scholar]
- 11.Harada N, Hatano E, Koizumi N, Nitta T, Yoshida M, Yamamoto N, Brenner DA, Yamaoka Y. Akt activation protects rat liver from ischemia-reperfusion injury. J Surg Res. 2004;121:159–170. doi: 10.1016/j.jss.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Hirai K, Hayashi T, Chan PH, Zeng J, Yang GY, Basus VJ, James TL, Litt L. PI3K inhibition in neonatal rat brain slices during and after hypoxia reduces phospho-Akt and increases cytosolic cytochrome c and apoptosis. Brain Res Mol Brain Res. 2004;124:51–61. doi: 10.1016/j.molbrainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Janmaat ML, Rodriguez JA, Jimeno J, Kruyt FA, Giaccone G, Kahalalide F. induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol Pharmacol. 2005;68:502–510. doi: 10.1124/mol.105.011361. [DOI] [PubMed] [Google Scholar]
- 14.Kang KW, Cho MK, Lee CH, Kim SG. Activation of phosphatidylinositol 3-kinase and Akt by tert-butylhydroquinone is responsible for anti-oxidant response element-mediated rGSTA2 induction in H4IIE cells. Mol Pharmacol. 2001;59:1147–1156. doi: 10.1124/mol.59.5.1147. [DOI] [PubMed] [Google Scholar]
- 15.Kang KW, Choi SH, Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide. 2002;7:244–253. doi: 10.1016/s1089-8603(02)00117-9. [DOI] [PubMed] [Google Scholar]
- 16.Kaushal GP, Kaushal V, Hong X, Shah SV. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 2001;60:1726–1736. doi: 10.1046/j.1523-1755.2001.00026.x. [DOI] [PubMed] [Google Scholar]
- 17.Kruidering M, Maasdam DH, Prins FA, de Heer E, Mulder GJ, Nagelkerke JF. Evaluation of nephrotoxicity in vitro using a suspension of highly purified porcine proximal tubular cells and characterization of the cells in primary culture. Exp Nephrol. 1994;2:324–344. [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Lash LH, Anders MW. Mechanism of S-(1,2-dichlorovinyl)-l-cysteineand S-(1,2-dichlorovinyl)-l-homocysteine-induced renal mitochondrial toxicity. Mol Pharmacol. 1987;32:549–556. [PubMed] [Google Scholar]
- 20.Lash LH, Hueni SE, Putt DA. Apoptosis, necrosis, and cell proliferation induced by S-(1,2-dichlorovinyl)-l-cysteine in primary cultures of human proximal tubular cells. Toxicol Appl Pharmacol. 2001;177:1–16. doi: 10.1006/taap.2001.9295. [DOI] [PubMed] [Google Scholar]
- 21.Lash LH, Xu Y, Elfarra AA, Duescher RJ, Parker JC. Glutathione-dependent metabolism of trichloroethylene in isolated liver and kidney cells of rats and its role in mitochondrial and cellular toxicity. Drug Metab Dispos. 1995;23:846–853. [PubMed] [Google Scholar]
- 22.Li Q, Zhu GD. Targeting serine/threonine protein kinase B/Akt and cell-cycle checkpoint kinases for treating cancer. Curr Top Med Chem. 2002;2:939–971. doi: 10.2174/1568026023393318. [DOI] [PubMed] [Google Scholar]
- 23.Lieberthal W, Koh JS, Levine JS. Necrosis and apoptosis in acute renal failure. Semin Nephrol. 1998;18:505–518. [PubMed] [Google Scholar]
- 24.Liu X, Godwin ML, Nowak G. Protein kinase C-α inhibits the repair of oxidative phosphorylation after S-(1,2-dichlorovinyl)-l-cysteine injury in renal cells. Am J Physiol Renal Physiol. 2004;287:F64–F73. doi: 10.1152/ajprenal.00216.2003. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y. Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol Renal Physiol. 1999;277:F624–F633. doi: 10.1152/ajprenal.1999.277.4.F624. [DOI] [PubMed] [Google Scholar]
- 26.McKinney LL, Picken JC, Jr, Weakley FB, Eldridge AC, Campbell RE, Cowan JC, Biester HE. Toxic factor of trichloroethylene-extracted soybean oil meal. J Am Chem Soc. 1959;81:909–915. [Google Scholar]
- 27.Mizui M, Isaka Y, Takabatake Y, Mizuno S, Nakamura T, Ito T, Imai E, Hori M. Electroporation-mediated HGF gene transfer ameliorated cyclosporine nephrotoxicity. Kidney Int. 2004;65:2041–2053. doi: 10.1111/j.1523-1755.2004.00625.x. [DOI] [PubMed] [Google Scholar]
- 28.Moran JH, Schnellmann RG. A rapid beta-NADH-linked fluorescence assay for lactate dehydrogenase in cellular death. J Pharmacol Toxicol Methods. 1996;36:41–44. doi: 10.1016/1056-8719(96)00071-8. [DOI] [PubMed] [Google Scholar]
- 29.Muller C, Dunschede F, Koch E, Vollmar AM, Kiemer AK. Alpha-lipoic acid preconditioning reduces ischemia-reperfusion injury of the rat liver via the PI3-kinase/Akt pathway. Am J Physiol Gastrointest Liver Physiol. 2003;285:G769–G778. doi: 10.1152/ajpgi.00009.2003. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima T, Iwabuchi S, Miyazaki H, Okuma Y, Kuwabara M, Nomura Y, Kawahara K. Preconditioning prevents ischemia-induced neuronal death through persistent Akt activation in the penumbra region of the rat brain. J Vet Med Sci. 2004;66:521–527. doi: 10.1292/jvms.66.521. [DOI] [PubMed] [Google Scholar]
- 31.Nowak G, Schnellmann RG. Improved culture conditions stimulate gluconeogenesis in primary cultures of renal proximal tubule cells. Am J Physiol Cell Physiol. 1995;268:C1053–C1061. doi: 10.1152/ajpcell.1995.268.4.C1053. [DOI] [PubMed] [Google Scholar]
- 32.Nowak G, Carter CA, Schnellmann RG. Ascorbic acid promotes recovery of cellular functions following toxicant-induced injury. Toxicol Appl Pharmacol. 2000;167:37–45. doi: 10.1006/taap.2000.8986. [DOI] [PubMed] [Google Scholar]
- 33.Nowak G, Keasler KB, McKeller DE, Schnellmann RG. Differential effects of EGF on repair of cellular functions after dichlorovinyl-l-cysteine-induced injury. Am J Physiol Renal Physiol. 1999;276:F228–F236. doi: 10.1152/ajprenal.1999.276.2.F228. [DOI] [PubMed] [Google Scholar]
- 34.Nowak G, Price PM, Schnellmann RG. Lack of a functional p21WAF1/CIP1 gene accelerates caspase-independent apoptosis induced by cisplatin in renal cells. Am J Physiol Renal Physiol. 2003;285:F440–F450. doi: 10.1152/ajprenal.00233.2002. [DOI] [PubMed] [Google Scholar]
- 35.Nowak G, Schnellmann RG. l-Ascorbic acid regulates growth and metabolism of renal cells: improvements in cell culture. Am J Physiol Cell Physiol. 1996;271:C2072–C2080. doi: 10.1152/ajpcell.1996.271.6.C2072. [DOI] [PubMed] [Google Scholar]
- 36.Rajesh KG, Suzuki R, Maeda H, Yamamoto M, Yutong X, Sasaguri S. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a PI3K/Akt dependent pathway. J Mol Cell Cardiol. 2005;39:766–776. doi: 10.1016/j.yjmcc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Rodeheaver DP, Aleo MD, Schnellmann RG. Differences in enzymatic and mechanical isolated rabbit renal proximal tubules: comparison in long-term incubation. In Vitro Cell Dev Biol. 1990;26:898–904. doi: 10.1007/BF02624615. [DOI] [PubMed] [Google Scholar]
- 38.Schnellmann RG. Measurement of oxygen consumption. Methods Toxicol. 1994;12:128–139. [Google Scholar]
- 39.Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol. 2004;15:2115–2124. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- 40.Sinha D, Bannergee S, Schwartz JH, Lieberthal W, Levine JS. Inhibition of ligand-independent ERK1/2 activity in kidney proximal tubular cells deprived of soluble survival factors upregulates Akt and prevents apoptosis. J Biol Chem. 2004;279:10962–10972. doi: 10.1074/jbc.M312048200. [DOI] [PubMed] [Google Scholar]
- 41.Storz P, Toker A. 3′-Phosphoinositide-dependent kinase-1 (PDK-1) in PI 3-kinase signaling. Front Biosci. 2002;7:D886–D902. doi: 10.2741/storz. [DOI] [PubMed] [Google Scholar]
- 42.Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–3049. doi: 10.1161/01.CIR.0000130647.29030.90. [DOI] [PubMed] [Google Scholar]
- 43.Vamvakas S, Bittner D, Dekant W, Anders MW. Events that precede and that follow S-(1,2-dichlorovinyl)-l-cysteine-induced release of mitochondrial Ca2+ and their association with cytotoxicity to renal cells. Biochem Pharmacol. 1992;44:1131–1138. doi: 10.1016/0006-2952(92)90377-u. [DOI] [PubMed] [Google Scholar]
- 44.Wang CX, Song JH, Song DK, Yong VW, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differentiation. doi: 10.1038/sj.cdd.4401804. In press. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 46.Weinberg JM. The cell biology of ischemic renal injury. Kidney Int. 1991;39:476–500. doi: 10.1038/ki.1991.58. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20:7779–7786. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- 48.Zhuang S, Dang Y, Schnellmann RG. Requirement of the epidermal growth factor receptor in renal epithelial cell proliferation and migration. Am J Physiol Renal Physiol. 2004;287:F365–F372. doi: 10.1152/ajprenal.00035.2004. [DOI] [PubMed] [Google Scholar]
- 49.Zhuang S, Kochevar IE. Singlet oxygen-induced activation of Akt/protein kinase B is independent of growth factor receptors. Photochem Photobiol. 2003;78:361–371. doi: 10.1562/0031-8655(2003)078<0361:soaopk>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 50.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am J Physiol Renal Physiol. 2004;286:F858–F865. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]