

Abstract
Pro-inflammatory cytokines such as TNF-α play an important role in the pathophysiology of diseases such as Crohn's and ulcerative colitis which cause increased risk of colorectal cancer. However, the mechanisms underlying colon carcinogenesis are not well understood. Herein we report that inhibition/antisense abolition of polyol pathway enzyme, aldose reductase (AR) inhibited the TNF-α–induced synthesis of prostaglandin E2 and the activity of cyclooxygenase (Cox) in human colon cancer cells, Caco-2. Inhibition of AR prevented TNF-α -induced activation of PKC and NF-κB which resulted in the abrogation of Cox-2 mRNA and protein expression. These results suggest that inhibition of AR could be a novel chemopreventive approach to colon cancer.
Keywords: Aldose reductase, Inflammation, Colon cancer, TNF-α, PGE2, PKC
1. Introduction
Prostaglandin E2 (PGE2) is one of the main inflammatory markers that plays an important role as a local chemical messenger to cause inflammation in colon cancer [1]. In the colon, intestinal epithelial cells (IECs) are the first host defense against various pathogens, toxins and chemical stimuli [2,3]. For example bacterial infections in IECs up-regulate expression of a battery of NF-κB-dependent proinflammatory cytokines such as TNF-α, IL-1, IL-6 and granulocyte macrophage-colony stimulating factor (GM-CSF) including PGE2 and nitric oxide [2-6]. The cytokines and chemokines by autocrine and paracrine fashion elevate the host immune response. Among the proinflammatory cytokines, TNF-α is a central mediator in the pathophysiology of chronic inflammatory bowel diseases (IBD) such as Crohn's and ulcerative colitis [4,7]. The enzyme Cox-2 has been shown to be over-expressed in IBD and most colorectal adenocarcinomas [5,8]. The cyclooxygenase isoforms 1 & 2 catalyze the first two steps in the biosynthesis of prostaglandins from arachidonic acid which are known to be the major cause of inflammation [8]. The de novo synthesis of Cox-2 is triggered by the exposure of cells to cytokines such as TNF-α and IL-6 which independently activate the nuclear factor–κB (NF-κB), a redox sensitive transcription factor, that activates a number of genes such as IL-6, inducible nitric oxide synthase (iNOS) and aldose reductase (AR) [3,5,9]. In colonic epithelial cell line, HT-29, TNF-α-induced over-expression of Cox-2 is completely dependent on NF-κB activation [10].
We have earlier shown that AR, a member of the aldo-keto reductase (AKR) superfamily, reduces one of the most abundant and toxic lipid aldehyde, 4-hydroxy-trans-2-nonenal (HNE), and its conjugate with glutathione (GS-HNE) to 1, 4-dihydroxynonane (DHN) and GS-DHN, respectively with a Km in the range of 10-30 μM [11]. This enzyme also catalyzes the first and rate-limiting step of the polyol pathway of glucose metabolism [11]. Inhibition of this enzyme by two structurally distinct pharmacological inhibitors, sorbinil and tolrestat or by AR SiRNA prevents growth factors- and TNF-α-induced phosphorylation and degradation of IκB-α and activation of NF-κB and PKC, proliferation of vascular smooth muscle cells (VSMC), and apoptosis of vascular endothelial cells (VEC) [12-14]. Our results indicate that GS-DHN formed by the reduction of GS-HNE catalyzed by AR, mediates cytokines-, chemokines- and growth factors-induced NF-κB activation that would result in increased formation of inflammatory markers. However, the mechanisms by which inhibition of AR prevents TNF-α-induced NF-κB activation and cytotoxicity in colon cancer remain unclear. Similarly, various antioxidants and compounds that inhibit NF-κB activation have been shown to be beneficial in preventing the progression of colon cancer but the mechanisms are not well understood. In the present study, we have investigated the possible role of AR in mediating the TNF-α-induced production of PGE2 in colon cancer Caco-2 cells. Our results show that inhibition of AR prevents TNF-α–induced up-regulation of inflammatory markers such as Cox-2 and PGE2 in Caco-2 cells, identifying a novel target for preventing colon inflammation that may lead to carcinogenesis.
2. Materials and Methods
2.1. Materials
Dulbecco's modified Eagle's medium (DMEM), Opti-MEM, phosphate-buffered saline (PBS), penicillin/streptomycin solution, trypsin, and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). Antibodies against Cox-1, Cox-2 and phospho PKC-β2 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Sorbinil and tolrestat were gifts from Pfizer and American Home Products, respectively. The stock solutions of AR inhibitors were made by dissolving in 25 % DMSO. Cox activity assay and PGE2 assay kits were obtained from Cayman Chemical Company (Ann Arbor, MI). All other reagents used were of analytical grade obtained from Sigma Chemical Co. (St. Louis, MO).
2.2. Cell Culture
Human colon cancer cell line, Caco-2 cells were obtained from American type culture collection (ATCC, Manassas, VA). Caco-2 cells were grown in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 5% CO2.
2.3. Measurement of Cytotoxicity and PKC Activity
Cell viability was determined by MTT assay and PKC activity was measured using the Promega-Sigma TECT™ PKC assay system (San Luis Obispo, CA) as described earlier [12].
2.4. PGE2 and Cyclooxygenase activity assay
Caco-2 cells were plated in 6 well plate at a density of 2×105 cells/well. After 24 hours, medium was replaced with serum-free medium with or without sorbinil or tolrestat (20 μM) followed by treatment with TNF-α (2nM), for another 24 h. The medium was collected from each well and analyzed for PGE2 by using an Enzyme Immuno Assay kit according to the manufacturer's instructions (Cayman Chemical Co., Inc. Ann Arbor, MI). For determination of Cox activity in Caco-2 cells, TNF-α–treated cells were collected and homogenized in cold buffer (4 °C), 0.1M Tris-HCl, pH 7.8 and 1mM EDTA, and Cox activity was measured in 96 well plate according to the manufacturer's instructions (Cayman Chemical Co., Ann Arbor, MI).
2.5. NF-κB-Dependent Reporter Secretory Alkaline Phosphatase (SEAP) Expression Assay
Caco-2 cells (1.5 × 105 cells/well) were plated in six-well plates and after attachment overnight, cells were serum-starved in Opti-MEM medium for 24 h with or without AR inhibitor, sorbinil or tolrestat (20 μM) and were transiently transfected with pNF-κB-SEAP construct or control plasmid pTALSEAP DNA (Clontech, Palo Alto, CA) using the lipofectamine plus reagent. After 6 h of transfection, cells were treated with TNF-α, HNE, GS-HNE or GS-DHN for 48 h in regular serum-free medium. The cell culture medium was then harvested and analyzed for SEAP activity according to manufacturer instructions (Clontech, Palo Alto, CA), using a 96-well chemiluminiscence plate reader and Kodak Image Station 2000R.
2.6. RT-PCR
Total RNA was isolated from Caco-2 cells by using RNaeasy micro isolation kit (Qiagen). Total RNA (1.0 μg) from each sample was reverse-transcribed with Omniscript and Sensiscript reverse transcriptase one-Step RT PCR system with HotStarTaq DNA polymerase (Qiagen, Valencia, CA) at 55°C for 30 min followed by PCR amplification. The oligonucleotide primer sequences used were: 5'-TGAAACCCACTCCAAACACAG-3' (sense) and 5'TCATCAGGCACAGGAGGAAG -3' (antisense) for Cox-2, 5'- GTTTGAGACCTT CAACACCCC -3' and 5'- GTGGCCATCTCCTGCTCGAAGTC -3' for β-actin. PCR reaction was carried out in a GeneAmp 2700 thermocycler (Applied Biosystems, Foster City, CA) under the following conditions: initial denaturation at 95°C for 15 min; 35 cycles of 94°C 30 s, 60°C 30 s, 72°C 1 min, and then 72 °C 5 min for final extension. PCR products were electrophoresed with 2% Agarose-1× TAE gels containing 0.5 μg/ml ethidium bromide.
2.7. Antisense Abolition of AR
Caco-2 cells were grown to 50–60% confluence in DMEM supplemented with 10% FBS and washed four times with Opti-MEM, 60 min before transfection with oligonucleotides. The cells were incubated with 2 μM AR antisense or scrambled control oligonucleotides using LipofectAMINE Plus (15 μg/ml) as the transfection reagent as suggested by the supplier [12]. Changes in the expression of AR were detected by Western blot analysis using anti-AR antibodies.
2.8. Preparation of GS-aldehydes esters
The cells permeable conjugates of glutathione ethyl ester with HNE (GS-HNE-ester) and the reduced form of the esterified glutathione-HNE conjugate (GS-DHN-ester) were prepared as described earlier [11]. Briefly, the radiolabeled [4-3H]-HNE was synthesized from the dimethylacetal of HNE which was oxidized to the 4-keto derivative using polymer-supported chromic acid. The resulting ketone was further reduced to the dimethylacetal of HNE by using tritiated NaBH4. The conjugate of glutathione reduced ethyl ester with HNE (GS-HNE-ester) was prepared by incubating 1μmol of [4- 3H] HNE (55000 cpm/nmol) with 5 μmol of GSH ethyl ester in 0.1 M potassium phosphate, pH 7.0 for 1h at room temperature. The reaction was monitored by following the consumption of HNE at 224 nm. The GS-HNE-ester conjugate was purified by reverse phase HPLC. For the synthesis of the reduced form of the esterified glutathione-HNE conjugate (GS-DHN-ester), 100 nmol of GS-HNE-ester was incubated with 300 nmol of NADPH and 100 μg aldose reductase in 0.1 M potassium phosphate, pH 6.0 for 3 hours at 37 °C. The reaction was monitored by following the consumption of NADPH at 340 nm. At the end of the incubation, the GS-DHN-ester conjugate was separated from GS-HNE-ester by reverse phase HPLC and confirmed by ESI/MS.
2.9. Statistical Analysis
Data are presented as mean ± SE and P values were determined by unpaired Student's t test. P values of <0.01 were considered significant.
3. Results
3.1. Inhibition of AR prevents Cox activity and PGE2 production
Treatment of serum-starved Caco-2 cells with TNF-α (2 nM) for 24 h increased the production of PGE2 and pretreatment with two structurally distinct AR inhibitors sorbinil or tolrestat prevented it (Fig. 1A). In the absence of TNF-α, the AR inhibitors alone did not affect PGE2 production. Although sorbinil and tolrestat are specific inhibitors of AR, their non-specificity in the biological system could not be excluded. Therefore, we examined whether abolition of AR by antisense oligonucleotides will have similar effects in colon cancer cells as AR inhibitors. Transient transfection of Caco-2 cells with AR antisense but not scrambled antisense oligonucleotides abolished AR protein by >95% (Fig.1B inset). Similar to inhibitors, antisense abolition of AR also significantly prevented the TNF-α-induced PGE2 production in Caco-2 cells (Fig.1B). Since biosynthesis of PGE2 from its precursor arachidonic acid is catalyzed by Cox enzymes, we next examined the effect of AR inhibition on TNF-α–induced Cox activity in Caco-2 cells. As shown in Fig.2A, treatment of Caco-2 cells with TNF-α significantly increased Cox activity and pre-treatment of Caco-2 cells with AR inhibitors, sorbinil or tolrestat significantly prevented the increase in Cox activity. This indicated that AR-dependent Cox activation is required for PGE2 production. Because Cox activity is dependent on Cox-1 (constitutive) and Cox-2 (inducible) isozymes, we next determined which Cox isozyme is affected by AR inhibition. Our immunoblot analysis showed that treatment of Caco-2 cells with TNF-α caused over-expression of Cox-2 enzyme which was inhibited significantly by AR inhibitors (Fig.2B). TNF-α as well as AR inhibitors had no effect on constitutive Cox-1 expression (Fig. 2C). Further, we determined the effect of AR inhibition on transcriptional activation of Cox-2 by quantifying its mRNA levels using RT-PCR. As shown in Fig.2E, treatment of Caco-2 cells with TNF-α (2 nM) significantly increased the mRNA levels of Cox-2 and sorbinil prevented it by ∼60 % suggesting that AR could regulate the transcriptional activation of Cox-2 gene.
Fig.1.
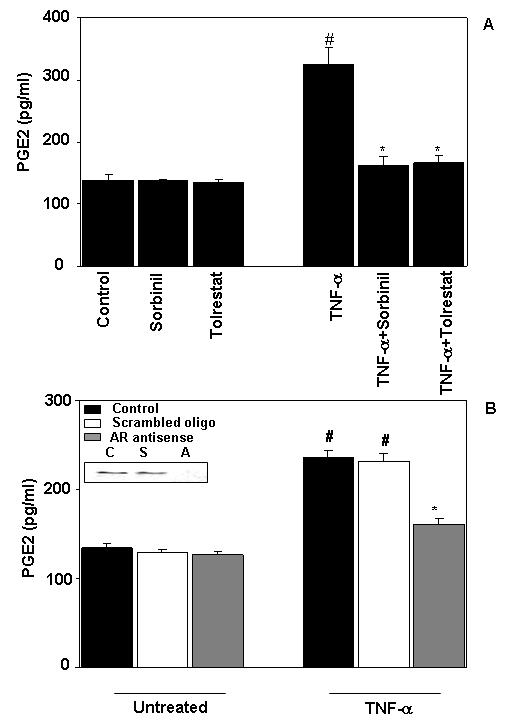
Inhibition/abolition of AR prevents TNF-α -induced PGE2 production in Caco-2 cells. (A) Growth-arrested Caco-2 cells were pre-incubated with sorbinil 20 μM or carriers for 24 h. (B) The cells were transfected with AR antisense or scrambled oligos. Subsequently, the cells in both (A) and (B) were stimulated with TNF-α (2 nM) for 24 h. The PGE2 released in the culture medium was determined by using monoclonal Enzyme Immuno Assay kit. The insert in Fig. 1B represents Western blot analysis of untransfected (c), scrambled (s) and AR antisense (a) oligo transfected cell extracts for determination of AR protein levels. Bars represent mean ± S.E. (n = 4); #, p < 0.001 compared to control cells, *, p < 0.001 compared to cells treated with TNF-α alone.
Fig.2.
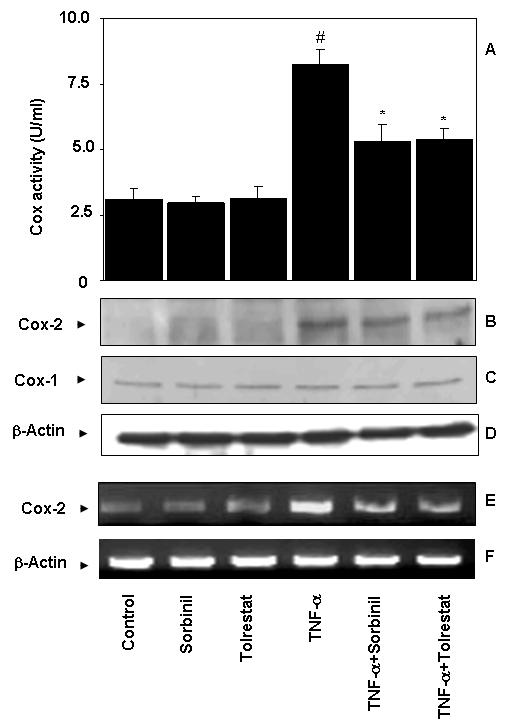
Inhibition of AR prevents TNF-α -induced Cox activity in Caco-2 cells. (A) Cox activity was determined by using Enzyme Immuno Assay kit. (B-C) Pooled cell extracts from 3 independent experimental sets were subjected to SDS-PAGE and Western blots were developed using antibodies against Cox-2 (B), Cox-1 (C) and GAPDH (D). (E) Cox-2 mRNA expression was determined by Qiagen RT-PCR (F) mRNA loading control, β-actin. One unit of Cox activity is defined as the amount of enzyme that will cause the oxidation of 1 nmol of TMPD per minute at 25 °C. Bars represent mean ± S.E. (n = 4); #, p < 0.001 compared to control cells, *, p < 0.01 compared to cells treated with TNF-α alone.
3.2. Attenuation of TNF-α-induced NF-κB activation by AR inhibitors
Since transcription of the Cox-2 gene is regulated by NF-κB [15], we next examined how inhibition of AR affects TNF-α-induced NF-κB activation in Caco-2 cells. As shown in the Fig.3, treatment of Caco-2 cells with TNF-α significantly induced NF-κB activation and sorbinil and tolrestat prevented it, whereas AR inhibitors alone did not affect the NF-κB-SEAP activity. Based on these results, we concluded that inhibition of AR could prevent TNF-α-induced activation of NF-κB in Caco-2 cells, which may transcriptionally activate Cox-2 gene expression.
Fig.3.
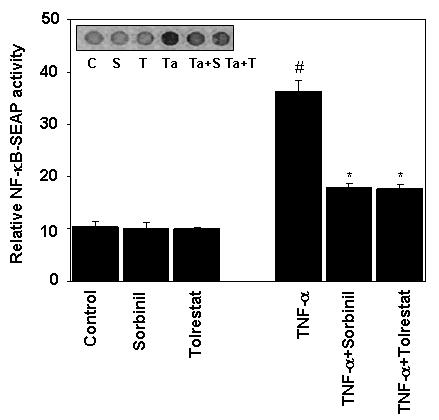
Inhibition of AR prevents TNF-α -induced NF-κB activation in Caco-2 cells. Growth-arrested Caco-2 cells were pre-incubated with or without sorbinil or tolrestat 20 μM for 24 h and transiently transfected with pNF-κB secretary alkaline phosphatase construct and control plasmid, pTALSEAP and measured alkaline phosphatase (SEAP) activity as described in methods. The Inset in D shows the chemiluminescence of SEAP detected with Kodak Image Station 2000R. Bars represent mean ± S.E. (n = 4); #, p < 0.001 as compared to control cells. *, p < 0.001 compared to cells treated with TNF-α alone.
3.3. Attenuation of AR prevents TNF-α -induced PKC Activation
Other than Cox-2 and PGE2, activation of PKC has been shown to mediate inflammation in colon cancer [16,17] and the activation of NF-κB also depends on activation of PKC. Therefore, we next examined the effect of AR inhibition on TNF-α-induced PKC activity in Caco-2 cells. As shown in Fig.4A, sorbinil or tolrestat alone did not affect PKC activity. Stimulation with TNF-α (2 nM) led to a significant increase in membrane-bound PKC activity and sorbinil and tolrestat significantly prevented it (Fig.4A). Since among the PKC isozymes, PKC-β2 is the major isozyme associated with progression of colon cancer [16,17], we next examined the effect of AR inhibition on TNF-α -induced PKC-β2 phosphorylation. Stimulation of Caco-2 cells with TNF-α led to a marked increase in the phosphorylation of PKC β2, whereas pretreatment of cells with sorbinil or tolrestat significantly prevented it (Fig.4B). These results indicate that AR is essential for the activation of PKC isozymes in TNF-α-induced colon cancer inflammation.
Fig.4.
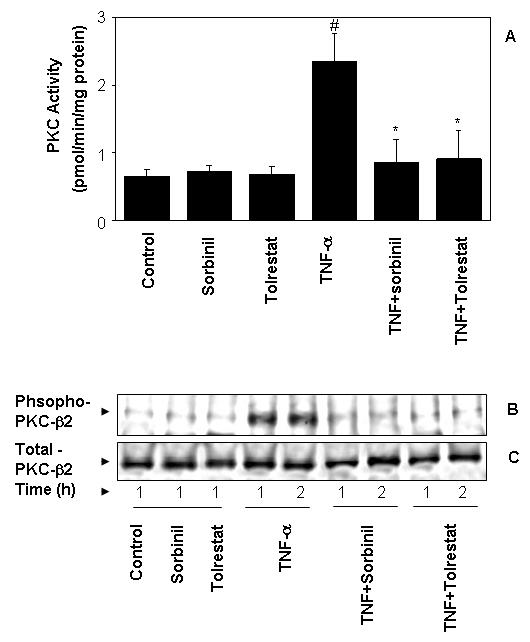
Inhibition of AR abrogates TNF-α- induced PKC activation. Quiescent Caco-2 cells were preincubated with 20 μM sorbinil for 24 h followed by stimulation with TNF-α (2 nM) for 2 h. (A) PKC activity was determined by using Promega's total PKC assay system. (B & C) The pooled cell extracts from 3 independent experimental sets were subjected to SDS-PAGE and Western blots were developed using antibodies, phospho-PKC-β2 (B) and nonphospho-PKC-β2 (C). Bars represent mean ± S.E. (n = 4); #, p < 0.001 as compared to control cells. *, p < 0.001 compared to cells treated with TNF-α alone.
3.4. Mitogenic effects of reduced glutathione-aldehyde conjugates (GS-DHN) in Caco-2 cells
To determine the mitogenic effect of AR-catalyzed product of glutathione lipid aldehyde such as GS-DHN, serum -starved Caco-2 cells were incubated in the absence or the presence of cell permeable esters of GS-HNE and GS-DHN without or with AR inhibitor, sorbinil. Both GS-HNE and GS-DHN (0.1 μM) increased Caco-2 cell proliferation as determined by MTT assay (Fig 5A). Inhibition of AR prevented the GS-HNE- but not GS-DHN -induced proliferation indicating that GS-DHN is insensitive to AR inhibition. Further, we have determined the effect of AR inhibition on GS-aldehydes –induced activation of NF-κB. Stimulation of Caco-2 cells with HNE, GS-HNE and GS-DHN resulted in the activation of NF-κB as determined by SEAP activity (Fig.5B). Inhibition of AR significantly inhibited the activation of NF-κB induced by HNE and GS-HNE but had no effect on GS-DHN-induced activation, suggesting that GS-DHN could be a signaling intermediate. Thus our results indicate that AR- catalyzed reaction products of GS-aldehydes could be involved in the activation of NF-κB, responsible for mitogenicity in Caco-2 cells.
Fig.5.
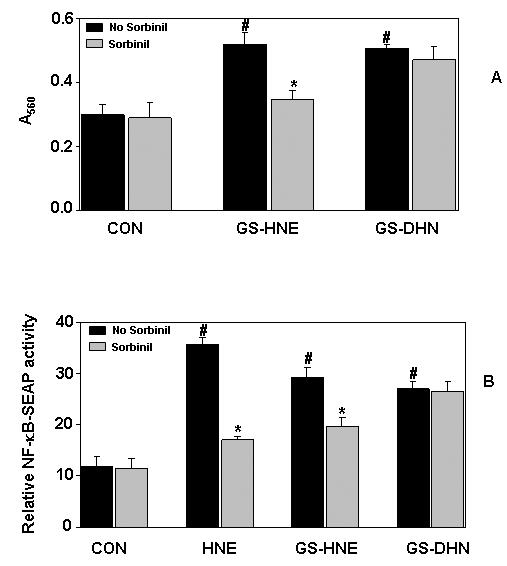
Inhibition of AR prevents lipid aldehyde-induced NF-κB activation and growth in Caco-2 cells. (A) Growth-arrested Caco-2 cells with or without sorbinil (20 μM) were incubated with 0.1 μM each of GS-HNE-ester and GS-DHN-ester for 24 h. The cell viability was determined by MTT assay. (B) The transiently transfected Caco-2 cells with pNF-κB secretary alkaline phosphatase reporter construct or control plasmid, pTALSEAP were incubated with 1 μM each of HNE, GS-HNE-ester or GS-DHN ester and alkaline phosphatase (SEAP) activity was measured as described in methods. Bars represent mean ± S.E. (n = 4); #, p < 0.001 as compared to control cells. *, p < 0.01 compared to cells treated with aldehydes alone.
4. Discussion
Deregulation of cytokines balance plays a pivotal role in the pathogenesis of IBDs such as Crohn's and ulcerative colitis [4,6,7]. Constant exposure of IECs to various agents such as bacteria, toxins and chemicals triggers mucosal immune response and causes increased expression and secretion of cytokines, chemokines, icosanoids etc [1-5]. One of the major stimulants of IECs is lipopolysaccharide from Gram negative bacterial infection which promotes inflammation due to increased transcription of cytokines [6,18,19]. Increased inflammatory cytokines secreted in Crohn's and ulcerative colitis are major risk factors of colon cancer [20]. Inflammatory cytokines such as TNF-α that induce Cox-2 enzyme which in turn catalyzes the formation of prostaglandins from arachidonic acid have been implicated in tumorigenesis. This is supported by the resistance of tumorigenesis in TNF-α knockout mice [21]. Our demonstration that inhibition of AR that reduces lipid aldehydes and lipid aldehyde glutathione conjugates to corresponding alcohols prevents the cytokines- and growth factors-induced increase in proinflammatory cytokines and chemokines and prevents inflammation provides a novel pharmacological approach to combat inflammation, commonly associated with colon carcinogenesis.
Numerous reports in various cell lines have shown that TNF-α alone or in combination with other agents such as IFN-γ can cause inflammatory response leading to cytotoxicity [22-24]. One of the major inflammatory responses TNF-α is the activation of Cox-2, which catalyzes the formation of prostaglandins from arachidonic acid. In colon epithelial cells, Cox-2 is activated by a number of stimuli such as growth factors and cytokines [5, 25-27]. Our results show that inhibition of AR by two structurally distinct inhibitors, sorbinil and tolrestat, and by AR antisense prevents TNF-α-induced increased expression of Cox-2 protein and mRNA and generation of PGE2 in Caco-2 cells. These observations suggest that AR is a cytokine responsive protein which may be involved in facilitating metabolic changes that accompany cytotoxicity in colon cancer cells. Recently, we have shown that inhibition of AR prevents growth factor- and bacterial lipopolysaccharide (LPS) –induced Cox-2 expression and PGE2 production in colon cancer cells [28], and in murine macrophages RAW264.7 [29]. The observations by other investigators in mice showed that colon26 adenocarcinoma cells-induced cachexia symptoms are significantly prevented by inhibiting AR by ponalrestat [30,31].
We and other investigators have shown that growth factors- and TNF-α -induced NF-κB activation in VSMC [12], VEC [13] and HLEC [9] is prevented by inhibiting AR. However, the observation that activation of PKC by phorbol ester is insensitive to sorbinil [12], indicates that AR is upstream of PKC and plays a pivotal role in redox signaling of PKC-NF-κB axis. Various reports show that activation of PKC is an obligatory step in promoting colon cancer [16,17]. Our earlier demonstration that AR-catalyzed product of GS-HNE, GS-DHN causes the activation of PKC isozymes such as PKC-β1, PKC-β2, PKC-δ in VSMC [11,14] and inhibition of AR prevents such effects indicates that inhibition of AR could be anti-inflammatory and chemopreventive. Further, we have shown earlier that AR activity is required for the synthesis of diacylglycerol, which is an obligatory cofactor for the activation of PKC isozymes [14]. These results indicate that both PKC and AR may be coordinately regulated by cytokines and growth factors-induced signals. Significant inhibition of TNF-α -induced NF-κB activation by sorbinil and tolrestat suggests that AR is required for the activation of NF-κB which transcribes various inflammatory markers including TNF-α and AR.
In conclusion, the present study demonstrates that AR mediates the inflammatory changes caused by TNF-α in Caco-2 cells and AR inhibitors could be excellent chemopreventive agents.
Acknowledgements
This work was supported in part by NIH grants GM 71036 (to KVR), and DK 36118 (to SKS).
Abbreviations
- AR
aldose reductase
- ARI
aldose reductase inhibitor
- Cox
cyclooxygenase
- HNE
4-hydroxy-trans-2-nonenal
- GSH
glutathione
- GS-HNE
glutathionyl-4-hydroxynonanal
- GS-DHN
glutathionyl-1,4-dihydroxynonane
- PGE2
prostaglandin E2
- MTT
[3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt]
- NF-κB
nuclear factor kappa binding protein
- PKC
Protein kinase C
- SEAP
Secretary alkaline phosphatase
- TNF-α
tumor necrosis factor-alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takahashi T, Takasuka N, Iigo M, Baba M, Nishino H, Tsuda H, Okuyama T. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci. 2004;95:448–453. doi: 10.1111/j.1349-7006.2004.tb03230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeWitt RC, Kudsk KA. The gut's role in metabolism, mucosal barrier function, and gut immunology. Infect Dis Clin North Am. 1999;13:465–481. doi: 10.1016/s0891-5520(05)70086-6. [DOI] [PubMed] [Google Scholar]
- 3.Demoule A, Divangahi M, Yahiaoui L, Danialou G, Gvozdic D, Labbe K, Bao W, Petrof BJ. Endotoxin triggers NF-{kappa}B-dependent upregulation of multiple pro-inflammatory genes in the diaphragm. Am J Respir Crit Care Med. 2006 doi: 10.1164/rccm.200509-1511OC. In Press. [DOI] [PubMed] [Google Scholar]
- 4.Panja A, Goldberg S, Eckmann L, Krishen P, Mayer L. The regulation and functional consequence of proinflammatory cytokine binding on human intestinal epithelial cells. J Immunol. 1998;161:3675–3684. [PubMed] [Google Scholar]
- 5.Weaver SA, Russo MP, Wright KL, Kolios G, Jobin C, Robertson DA, Ward SG. Regulatory role of phosphatidylinositol 3-kinase on TNF-alpha-induced cyclooxygenase 2 expression in colon epithelial cells. Gastroenterology. 2001;120:1117–1127. doi: 10.1053/gast.2001.23257. [DOI] [PubMed] [Google Scholar]
- 6.Courtois F, Seidman EG, Delvin E, Asselin C, Bernotti S, Ledoux M, Levy E. Membrane peroxidation by lipopolysaccharide and iron-ascorbate adversely affects Caco-2 cell function: beneficial role of butyricacid. Am J Clin Nutr. 2003;77:744–750. doi: 10.1093/ajcn/77.3.744. [DOI] [PubMed] [Google Scholar]
- 7.Jones SA, Butler RN, Sanderson IR, Wilson JW. The effect of specific caspase inhibitors on TNF-alpha and butyrate-induced apoptosis of intestinal epithelial cells. Exp Cell Res. 2004;292:29–39. doi: 10.1016/j.yexcr.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Swamy MV, Cooma I, Patlolla JM, Simi B, Reddy BS, Rao CV. Modulation of cyclooxygenase-2 activities by the combined action of celecoxib and decosahexaenoic acid: novel strategies for colon cancer prevention and treatment. Mol Cancer Ther. 2004;3:215–221. [PubMed] [Google Scholar]
- 9.Ramana KV, Friedrich B, Bhatnagar A, Srivastava SK. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB J. 2003;17:315–317. doi: 10.1096/fj.02-0568fje. [DOI] [PubMed] [Google Scholar]
- 10.Jobin C, Morteau O, Han DS, Balfour Sartor R. Specific NF-kappaB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1gene expression in HT-29 cells. Immunology. 1998;95:537–543. doi: 10.1046/j.1365-2567.1998.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J Biol Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 12.Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Aggarwal BB, Srivastava SK. Aldose reductase mediates mitogenic signaling in vascular smooth muscle cells. J. Biol. Chem. 2003;277:32063–32070. doi: 10.1074/jbc.M202126200. [DOI] [PubMed] [Google Scholar]
- 13.Ramana KV, Bhatnagar A, Srivastava SK. Aldose reductase regulates TNF-alpha-induced cell signaling and apoptosis in vascular endothelial cells. FEBS Lett. 2004;570:189–194. doi: 10.1016/j.febslet.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 14.Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes. 2005;54:818–829. doi: 10.2337/diabetes.54.3.818. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Zhao M, Rao R, Inoue H, Hao CM. C/EBP{beta} and its binding element are required for NF{kappa}B-induced COX2 expression following hypertonic stress. J Biol Chem. 2005;280:16354–16359. doi: 10.1074/jbc.M411134200. [DOI] [PubMed] [Google Scholar]
- 16.Gokmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 2001;61:1375–1381. [PubMed] [Google Scholar]
- 17.Rao CV, Simi B, Wynn TT, Garr K, Reddy BS. Modulating effect of amount and types of dietary fat on colonic mucosal phospholipase A2, phosphatidylinositol-specific phospholipase C activities, and cyclooxygenase metabolite formation during different stages of colon tumor promotion in male F344 rats. Cancer Res. 1996;56:532–537. [PubMed] [Google Scholar]
- 18.Ogle CK, Guo X, Hasselgren PO, Ogle JD, Alexander JW. The gut as a source of inflammatory cytokines after stimulation with endotoxin. Eur J Surg. 1997;163:45–51. [PubMed] [Google Scholar]
- 19.Witthoft T, Eckmann L, Kim JM, Kagnoff MF. Enteroinvasive bacteria directly activate expression of iNOS and NO production in human colon epithelial cells. Am J Physiol. 1998;275:G564–G571. doi: 10.1152/ajpgi.1998.275.3.G564. [DOI] [PubMed] [Google Scholar]
- 20.Gillen CD, Walmsley RS, Prior P, Andrews HA, Allan RN. Ulcerative colitis and Crohn's disease: a comparison of the colorectal cancer risk in extensive colitis. Gut. 1994;35:1590–1592. doi: 10.1136/gut.35.11.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suganuma M, Okabe S, Marino MW, Sakai A, Sueoka E, Fujiki H. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. 1999;59:4516–4518. [PubMed] [Google Scholar]
- 22.Kim DM, Koo SY, Jeon K, Kim MH, Lee J, Hong CY, Jeong S. Rapid induction of apoptosis by combination of flavopiridol and tumor necrosis factor (TNF)-alpha or TNF-related apoptosis-inducing ligand in human cancer cell lines. Cancer Res. 2003;63:621–626. [PubMed] [Google Scholar]
- 23.Farah M, Parhar K, Moussavi M, Eivemark S, Salh B. 5, 6-Dichlororibifuranosylbenzimidazole- and apigenin-induced sensitization of colon cancer cells to TNF-alpha-mediated apoptosis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G919–G928. doi: 10.1152/ajpgi.00205.2003. [DOI] [PubMed] [Google Scholar]
- 24.Lutz NW, Tome ME, Cozzone PJ. Early changes in glucose and phospholipid metabolism following apoptosis induction by IFN-gamma/TNF-alpha in HT-29 cells. FEBS Lett. 2003;544:123–128. doi: 10.1016/s0014-5793(03)00489-7. [DOI] [PubMed] [Google Scholar]
- 25.Han DS, Li F, Holt L, et al. Keratinocyte growth factor-2 (FGF-10) promotes healing of experimental small intestinal ulceration in rats. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1011–G1022. doi: 10.1152/ajpgi.2000.279.5.G1011. [DOI] [PubMed] [Google Scholar]
- 26.Liu W, Reinmuth N, Stoeltzing O, Parikh AA, Tellez C, Williams S, Jung YD, Fan F, Takeda A, Akagi M, Bar-Eli M, Gallick GE, Ellis LM. Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. Cancer Res. 2003;63:3632–3636. [PubMed] [Google Scholar]
- 27.Nakao S, Ogata Y, Yamamoto Y, Furuyama S, Sugiya H. Platelet-derived growth factor-induced arachidonic acid release for enhancement of prostaglandin E(2) synthesis in human gingival fibroblasts pretreated with interleukin-1beta. J. Cell Biochem. 2004;92:579–590. doi: 10.1002/jcb.20086. [DOI] [PubMed] [Google Scholar]
- 28.Tammali R, Ramana KV, Singhal SS, Awasthi S, Srivastava SK. Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin e2 production in human colon cancer cells. Cancer Res. 2006;66(19):9705–9713. doi: 10.1158/0008-5472.CAN-06-2105. [DOI] [PubMed] [Google Scholar]
- 29.Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. 2006;281(44):33019–3329. doi: 10.1074/jbc.M603819200. [DOI] [PubMed] [Google Scholar]
- 30.Kawamura I, Lacey E, Yamamoto N, Sakai F, Takeshita S, Inami M, Nishigaki F, Naoe Y, Tsujimoto S, Manda T, Shimomura K, Goto T. Ponalrestat, an aldose reductase inhibitor, inhibits cachexia syndrome induced by colon26 adenocarcinoma in mice. Anticancer Res. 1999;19:4105–4111. [PubMed] [Google Scholar]
- 31.Kawamura I, Lacey E, Inami M, Nishigaki F, Naoe Y, Tsujimoto S, Manda T, Goto T. Ponalrestat, an aldose reductase inhibitor, inhibits cachexia syndrome in nude mice bearing human melanomas G361 and SEKI. Anticancer Res. 1999;19:4091–4097. [PubMed] [Google Scholar]