

Abstract
Measles virus (MV), a morbillivirus that remains a significant human pathogen, can infect the central nervous system, resulting in rare but often fatal diseases, such as subacute sclerosing panencephalitis. Previous work demonstrated that MV was transmitted trans-synaptically, and that, while a cellular receptor for the hemagglutinin (H) protein was required for MV entry, it was dispensable for subsequent cell-to-cell spread. Here, we explored what role the other envelope protein, fusion (F), played in trans-synaptic transport. We made the following observations: 1) MV-F expression in infected neurons was similar to that seen in infected fibroblasts; 2) fusion inhibitory peptide (FIP), an inhibitor of MV fusion, prevented both infection and spread in primary neurons; 3) Substance P, a neurotransmitter with the same active site as FIP, also blocked neuronal MV spread; and 4) both genetic deletion and pharmacological inhibition of the Substance P receptor, neurokinin-1 (NK-1), reduced infection of susceptible mice. Together, these data implicate a role for NK-1 in MV CNS infection and spread, perhaps serving as a MVF receptor or co-receptor on neurons.
Keywords: Measles, Fusion inhibitory peptide, Substance P, neuron, subacute sclerosing panencephalitis, central nervous system, fusion, CD46, spread, neurokinin-1
Introduction
In rare cases, MV can invade and persist within the human central nervous system (CNS), leading to progressive and fatal neurological diseases, including subacute sclerosing panencephalitis (SSPE) (Anlar, 1997; Griffin and Bellini, 1996; Schneider-Schaulies et al., 1999). Two hallmarks of SSPE are the protracted period between initial infection and signs of disease (often years), and the inability to recover infectious virus from affected brain tissues, despite the extensive presence of MV RNA and protein (Katz, 1995; Payne, Baublis, and Itabashi, 1969). This is in contrast to the rapid and highly productive infections that occur in non-CNS tissues. The apparent differences in MV replication and spread within the human CNS implied that viral neurotransmission was substantively less cytopathic and productive than in non-neuronal tissues.
Mice are normally non-permissive for MV; thus, to study MV spread and pathogenesis in the CNS, a transgenic mouse model of neuronal infection was developed. These transgenic mice expressed CD46, the first identified human MV receptor (Dorig et al., 1993; Naniche et al., 1993), now thought to be the primary receptor for vaccine strains (Cattaneo, 2004), under the transcriptional control of the neuron-specific promoter, neuron-specific enolase (NSE)(Rall et al., 1997). Subsequent challenge of NSE-CD46 transgenic mice with the vaccine strain, MV-Edmonston, by either the intracerebral or intranasal route resulted in infection in all transgenic mice. While adult immunocompetent mice resolved the infection, both CD46+ neonates and immunodeficient (e.g., RAG knockout (KO)) CD46+ adults developed CNS disease resulting from unrestricted neuronal infection (Lawrence et al., 1999; Rall et al., 1997).
Using primary hippocampal neurons obtained from NSE-CD46+ embryos, we showed that MV neuronal infection was noncytopathic and did not result in release of extracellular progeny, despite extensive viral spread (Lawrence et al., 2000). Interestingly, neuron-to-neuron spread of MV occurred exclusively at the synapse, and did not require expression of CD46 (Lawrence et al., 2000; Oldstone et al., 1999; Rall et al., 1997), indicating that a hemagglutinin (H)-receptor interaction may not be required at the synaptic interface (Lawrence et al., 2000).
Based on this observation, we next wanted to establish what role, if any, the viral fusion protein played in trans-synaptic transport. While it is generally believed that H and F have interaction domains that coordinate receptor binding and fusion (Hu, Ray, and Compans, 1992), work by other laboratories has suggested that F may act independently of H, and may even govern target cell specificity. For example, in simian virus 5 (SV5), F promotes fusion in the absence of the hemagglutinin-neuraminidase (HN) protein, albeit less efficiently than when HN is present (Horvath et al., 1992; Ito et al., 1997). Moreover, SV5 F protein mutants that require SV5 HN for fusion at 37°C can fuse in an HN-independent manner at higher temperatures. Thus, fusion may require surmounting a thermodynamic threshold, which can be accomplished by a conformational change in the HN protein, increased temperature, or perhaps interaction of the F protein with its own cellular receptor (Dutch et al., 2001; Paterson, Russell, and Lamb, 2000). This final possibility has support in the literature, since the F2 fusion protein of respiratory syncytial virus (RSV), a Pneumovirus within the Paramyxovirus family, determines host cell tropism (Schendler et al., 2003), and recombinant RSV particles lacking the G protein remained viable and able to spread (Techaarpornkul, Barretto, and Peeples, 2001).
In this study, an inhibitor of paramyxovirus fusion, fusion inhibitory peptide (FIP) was used (Richardson and Choppin, 1983; Richardson, Scheid, and Choppin, 1980). The tri-peptide sequence of FIP (Phe-Phe-Gly) is identical to the active site of the neurotransmitter Substance P (Figure 4A), which can also inhibit MV spread in non-neuronal cultures (Harrowe, Mitsuhashi, and Payan, 1990; Schroeder, 1986). Substance P transduces signals to target cells by engagement of the neurotachykinin family of G protein-coupled neurotransmitter receptors, including neurokinin-1 (NK-1) (Gerard et al., 1991; Schaffer et al., 1998), a highly conserved protein expressed on diverse mammalian cells, including neurons, lymphocytes, smooth muscle cells and vascular endothelium (Pennefather et al., 2004). NK-1 expression is not ubiquitous within the CNS, though most central structures display some expression (Mantyh, 2002). In neurons, NK-1 is broadly distributed on the plasma membrane, though concentrated at the synapse (Bozic et al., 1996). Here, we show that both FIP and Substance P prevent MV spread in neurons, and that both genetic deletion and pharmacological inhibition of NK-1 also reduce infection of permissive mice. Together, these data indicate that fusion is a required step for viral transport across the synaptic cleft, and suggest that NK-1 plays a key role in this process, perhaps serving as a MV-F receptor.
Figure 4. Effect of neuropeptide transmitters on MV spread.
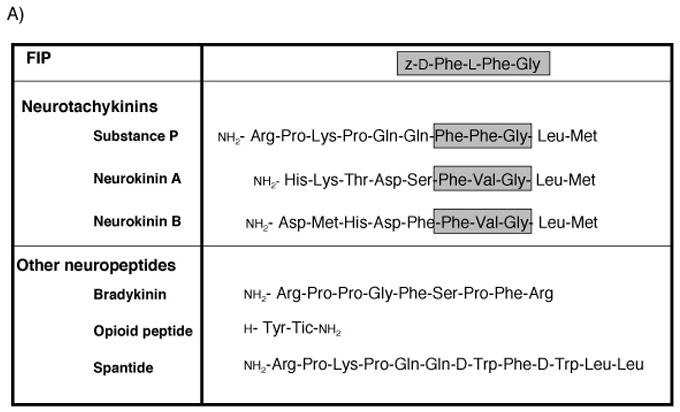
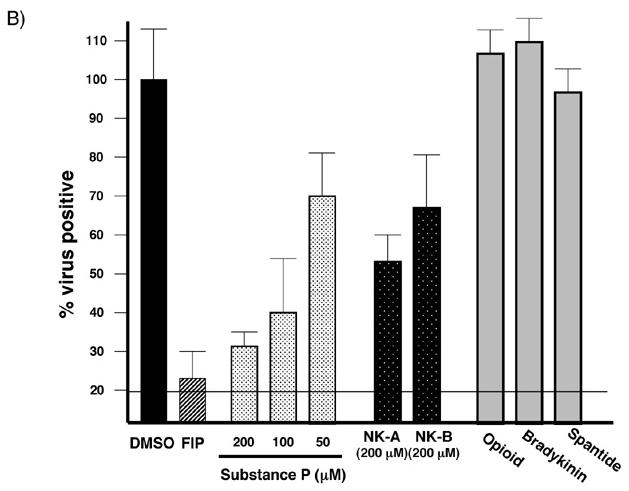
(A) Amino acid sequences of neuropeptides used. Gray boxes indicate active sites of the neurotachykinins and homology with FIP. (B) Inhibitory effect of each neuropeptide on MV spread. Neurons were infected with MV-Ed, and neuropeptides were added to a final concentration of 200 μM; for Substance P, three doses were tested: 200, 100 and 50 μM. Samples were collected 3 dpi and counted; values reflect the percent infection as compared with the DMSO control. The horizontal line indicates the initial level of infection after ∼1 round of replication (24 hpi). Because the peptides were added after infection, this represents the maximal achievable inhibitory effect. Shown is one representative experiment of five.
Results
Expression and distribution of the fusion protein in MV-infected primary neurons
We previously showed that inter-neuronal transmission of MV was trans-synaptic and CD46-independent (Lawrence et al., 2000). Because CD46-negative neurons were non-permissive for infection by extracellular virus, but could support infection when co-cultured with infected CD46+ neurons, we hypothesized that the MV-H protein, though needed for initial MV entry, may be dispensable for neuron-to-neuron trans-synaptic spread of MV.
Because both MV-H and F are typically involved in viral entry and spread, we next asked whether MV-F was translated and processed in infected neurons. Lysates were prepared from MV-infected or mock-infected neurons and Vero cells. Western blots using a rabbit antibody raised against the F carboxy terminus (Cathomen, Naim, and Cattaneo, 1998) were then performed on protein lysates equivalent to 105 infected cells. For the mock-infected controls, an amount of lysate corresponding to 105 cells was used. MV-F is synthesized as a precursor (F0) that is post-translationally cleaved into two subunits, F1 and F2. As shown in Figure 1, equivalent numbers of infected Vero fibroblasts and CD46+ neurons yielded both F0 and F1 fragments, though the amount of each present in neurons was consistently less. Despite slight differences in expression levels, cleavage of the F0 protein was efficient in both cell types. Note that the F2 fragment is not recognized by this antibody.
Figure 1. MV F expression in infected neurons.
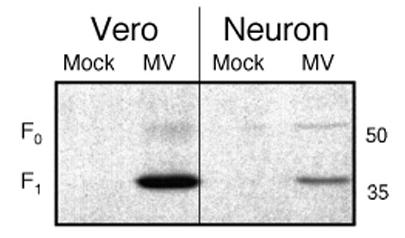
48 hpi with MV-Ed (MOI=3), primary CD46+ neurons and Vero cells were fixed for immunostaining or solubilized in protein sample buffer as described. The percent of infected cells was determined by immunochemistry, after which the protein lysate volume equivalent to 105 infected cells was then subjected to Western blot analysis using an anti-F antibody. Size standards are indicated at right. Shown is one representative experiment of three.
Fusion inhibitory peptide blocks MV spread in primary neurons
Fusion inhibitory peptide (FIP) is a synthetic tri-peptide (z-D-Phe-L-Phe-Gly) that prevents fusion by multiple viruses, though its strongest activity is against MV (Norrby, 1971; Richardson and Choppin, 1983; Richardson, Scheid, and Choppin, 1980). To determine if FIP blocked MV spread in neurons, primary neurons obtained from NSE-CD46+ embryonic hippocampi, and control Vero fibroblasts were infected with MV-Ed using a range of MOIs from 0.1-3, followed by addition of FIP at 6 hr post-infection (hpi). Cells were then collected 1-3 dpi and immunostained for the presence of MV antigens. One representative experiment of three using an MOI=0.1 and FIP at 200 μM is shown in Figure 2. In Vero cells without FIP, MV spread rapidly, resulting in extensive infection and syncytia (Figure 2A). As expected, addition of FIP after infection prevented viral spread and syncytia formation, and the monolayer remained intact (Figure 2B). Because FIP was added 6 hpi, individual MV-positive cells were expected.
Figure 2. Fusion inhibitory peptide prevents MV spread in primary neurons.
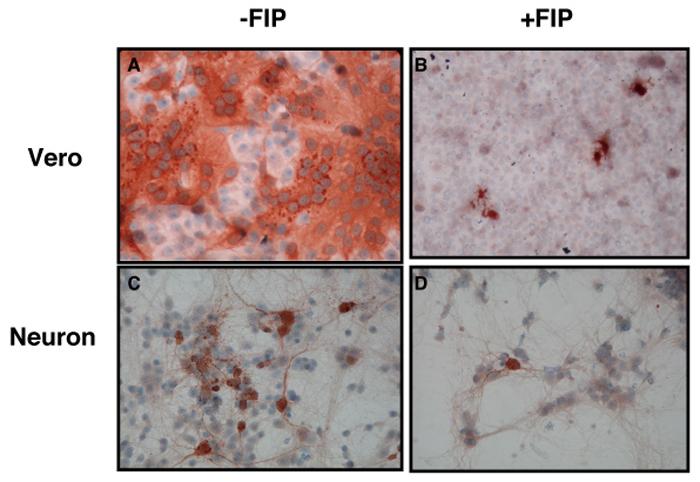
Vero cells and CD46+ neurons were plated onto coverslips as described, and infected with MV-Ed, MOI=0.1. Six hpi, FIP was added to achieve a final concentration of 200 μM, and slips were fixed and immunostained 3 days thereafter. (A) and (B): Vero cells; (C) and (D): neurons. (A) and (C): DMSO added; (B) and (D): FIP added. Original magnification = 400X. Shown are fields from one representative experiment of three at this virus and dose concentration. No differences were noted using MOIs ranging from 0.1-3.
Similarly, addition of FIP after primary infection blocked the trans-synaptic neuronal transmission of MV (Figure 2D) that was otherwise observed in untreated cultures (Figure 2C). Identical results were obtained when infection and FIP addition were separated by 1 hr, or when other MOIs were used (data not shown). As shown in the Table, the effect of FIP addition on MV spread was dose-dependent for both cell types, with >50% inhibition occurring at 50 μM. The inhibitory effect of FIP was specific for MV, as identical experiments with two other neurotropic RNA viruses, lymphocytic choriomeningitis virus (LCMV) and poliovirus (PV), showed no differences in infection levels in the presence of FIP (data not shown).
Table.
The ability of FIP to prevent MV spread is dose-dependenta.
| FIP concentration (μM) |
MV infection (%)b |
|
|---|---|---|
| Veros | Neurons | |
| 200 | 12 (+/−8) | 4 (+/−3) |
| 100 | 17 (+/−11) | 9 (+/− 5) |
| 50 | 42 (+/−18) | 14 (+/−4) |
| 5 | 92 (+/−6) | 15 (+/− 5) |
| DMSO | 87 (+/−11) | 28 (+/− 10) |
Veros and CD46+ primary neuron cultures were infected with MV-Ed at an MOI=3. FIP (at the indicated concentrations) or the diluent, DMSO, was then added 6 hpi, and cells were collected 2 dpi, immunostained and counted.
Values represent the percent of infected cells per field, viewed at 100X magnification, +/− standard deviation. A total of 10 random fields were counted from each of 3 independent experiments.
FIP blocks MV infection of primary neurons
The initial characterization of paramyxovirus hydrophobic fusion inhibitors showed that both de novo infection and subsequent cell-to-cell spread were prevented (Norrby, 1971; Richardson and Choppin, 1983; Richardson, Scheid, and Choppin, 1980). In our hands, however, addition of FIP prior to viral challenge did not prevent infection of Vero cells (Figure 3D), though subsequent viral spread was blocked (Figure 3E and F). The suppressive effect of FIP was reversible: when FIP was washed out of the cultures at 3 dpi, MV rebounded rapidly, resulting in the same cytopathicity and syncytia formation as observed in untreated cultures (compare Figure 3G and 3C). In contrast, pretreatment of CD46+ neurons with FIP completely blocked infection (Figure 3K-M), and, as expected, FIP washout did not result in MV re-emergence (Figure 3N). The inability of FIP to block infection of Veros could be due to cell-specific differences in receptor density, distribution (or utilization) of putative fusion receptors (discussed below), or access of FIP to cell surfaces. Taken together, these data indicate that FIP can prevent both infection and spread in primary CD46+ mouse hippocampal neurons.
Figure 3. FIP prevents de novo infection in neurons.
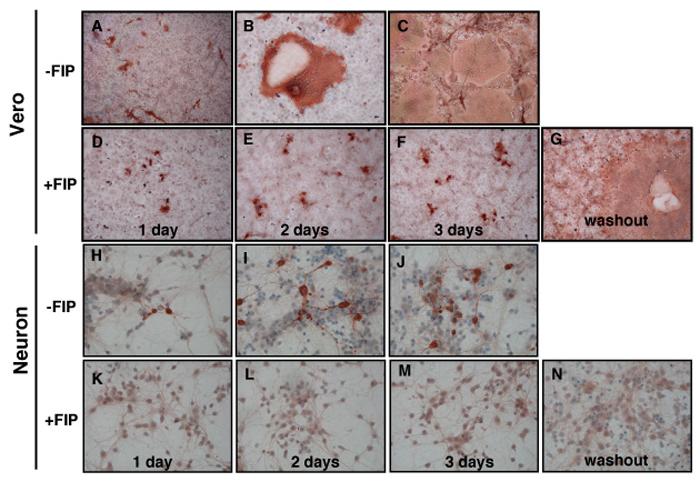
Vero cells (A-G) and primary CD46+ neurons (H-N) were plated on coverslips as described. FIP (200 μM) (D-G; K-N), or an equivalent volume of DMSO (A-C; H-J) were added and, after a 1 hr incubation, cells were infected with MV-Ed, MOI=3. Coverslips were fixed and immunostained 1-3 dpi. For the washout experiments (G and N), at 3 dpi the cells were washed and replaced with fresh media without FIP. Samples were then collected 48 hours post-FIP removal, equivalent to 5 dpi. Representative fields from one of four independent experiments are shown. Original magnification = 100X.
Inhibition of measles virus spread by neurotransmitters
The Phe-X-Gly sequence of FIP is also the active site of the neurotachykinin family of peptide neurotransmitters that include Substance P, Neurokinin A and Neurokinin B (Figure 4A). For Substance P, the central residue of the motif is a phenylalanine (as in FIP), while for Neurokinin A and B it is a valine. In addition, for FIP, the central Phe residue is the L-isomer, whereas for the neurokinins, it is the D-isomer.
It was previously shown that Substance P could inhibit MV spread in non-neuronal cultures (Harrowe, Mitsuhashi, and Payan, 1990; Schroeder, 1986). To determine if the neurotachykinins block MV trans-neuronal spread in our model system, we performed inhibition experiments identical to those described above with FIP, using the three neurotachykinins, two other non-neurotachykinin peptide transmitters (bradykinin and opioid receptor antagonist peptide), and a Substance P antagonist, spantide, which does not contain the Phe-X-Gly motif. Peptides were added at the indicated concentrations 6 hpi. Cells were collected at 3 dpi and immunostained for MV. The number of infected cells was expressed relative to DMSO treated cultures, which were normalized to 100%. One representative experiment of five is shown in Figure 4B. Note that, as in Figure 2, the neuropeptides were added after infection, so that some infected cells were expected in all cultures. The infection baseline at 1 dpi is indicated by the solid line, approximately 20%. For these studies, virus yields were not used as a measure of activity, as no extracellular virus is released from infected primary neurons (Lawrence et al., 2000).
All members of the neurotachykinin family inhibited MV transmission in a dose-dependent manner, although Substance P showed greater potency than NK-A or NK-B at the same concentrations (Figure 4B, and data not shown). Non-neurotachykinin peptides had no effect on MV transmission. Moreover, spantide, a Substance P antagonist that lacked the Phe-Phe-Gly motif and that presumably does not bind to the same site on NK-1 as Substance P, also had no effect on MV spread, underscoring the potential importance of this motif in MV-mediated fusion.
The Substance P receptor, neurokinin-1, contributes to MV trans-neuronal spread
We next asked if the cellular receptors for the neurotachykinin peptides were required for MV spread, with particular emphasis on NK-1, the predominant Substance P receptor. Two strategies were used: genetic knockout mice (Figure 5) and pharmacological NK-1 inhibitors (Figure 6).
Figure 5. Infection in neurokinin-1 knockout neonatal mice.
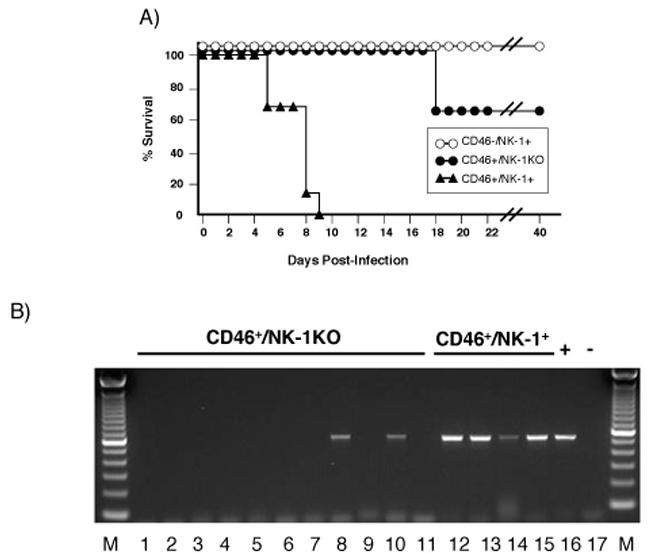
(A) Survival curves following MV challenge in CD46+/NK-1+ neonates (black triangles, n=11); CD46-/NK-1+ neonates (white circles, n=20); and CD46+/NK-1 KO neonates (black circles, n=12). Shown is one representative experiment of three. NSE-CD46+ neonates were chosen because these mice are permissive for MV infection and develop CNS disease within 1 week post-challenge (Lawrence et al., 1999). (B) RT-PCR analysis for MV-nucleoprotein of total brain RNA, harvested from neonatal CD46+/NK-1+ and CD46+/NK-1 KO mice at 6 dpi with MV-Ed, as described in Materials and Methods.
Figure 6. Weight loss, morbidity and MV infection levels in untreated and aprepitant-treated NSE-CD46+/RAG-2 KO mice.
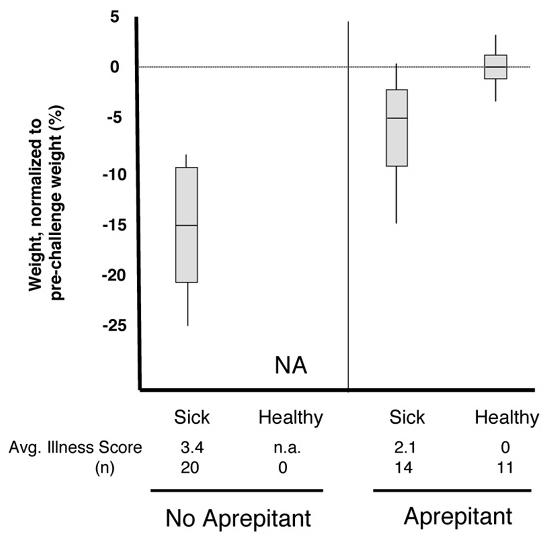
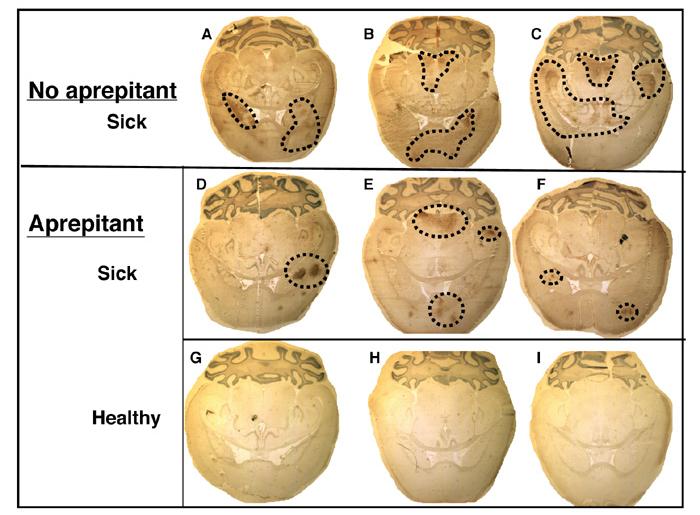
(A) Mice were infected as described and administered aprepitant orally one day prior to infection (double dose), one day after infection, and every other day thereafter. Mice were monitored daily, and scored from 0-4, based on the defined criteria (see Materials and Methods). Average values (obtained between days 6 and 10) are shown. Mice were also weighed on the day before infection to establish individual baselines, and every other day thereafter. Maximal weight changes are shown as percent weight loss or gain. (B) Mice were infected as described, and collected between 10-12 days post-infection. Shown are immunohistochemical results from untreated (a-c) and aprepitant-treated (d-i) mice. (d-f): sick animals at collection; (g-i): healthy animals at collection. Original magnification=20X.
NK-1 knockout (NK-1 KO) mice (Bozic et al., 1996) were intercrossed with NSE-CD46+ homozygous mice to obtain NSE-CD46+/NK-1 KO mice. As reported in the original characterization, NK-1 KO mice show no apparent developmental abnormalities with regard to lifespan, fertility, fecundity, or brain anatomy (Bozic, et al., 1996). All mice were genotyped for CD46 and NK-1 status, as described in Materials and Methods. Because neonatal CD46+ mice (that are also NK-1+) succumb to MV-mediated CNS disease following viral challenge (Lawrence et al., 1999; Rall et al., 1997), we asked what impact genetic deletion of NK-1 would have on MV neuronal infection and resultant pathogenesis in the neonatal model. As previously shown, MV-infected control CD46+/NK-1+ neonates (black triangles) developed symptoms within 4-8 dpi and died 1-2 days thereafter (Figure 5A). In contrast, CD46+/NK-1 KO neonates (black circles) either showed no signs of illness through adulthood, or developed illness at a much later time (18 dpi in 35% of CD46+/NK-1 KO neonates vs. 5-9 dpi in 100% of CD46+/NK-1+ neonates)(Figure 5A). None of the surviving CD46+/NK-1 KO mice showed evidence of MV CNS infection by RT-PCR analysis; in contrast, MV antigens were observed in all moribund CD46+/NK-1 KO mice collected at 18 dpi (data not shown). As expected, CD46-/NK-1+ mice (white circles) were non-permissive for infection and showed no signs of illness or infection at any timepoint, indicating that NK-1 alone cannot mediate MV entry.
To ascertain the extent of MV infection in NK-1 expressing and non-expressing neonates collected at the same timepoint post-infection, neonatal CD46+/NK-1+ and CD46+/NK-1 KO mice were challenged as described, and brains from all mice were collected 6 dpi, the approximate peak of infection within the CNS (Lawrence et al., 1999). Detection of the MV-N gene by RT-PCR analysis of total brain RNA showed evidence of replication in 4/4 (100%) CD46+/NK-1+ mice, whereas only 2/11 (18%) CD46+/NK-1 KO mice were positive (Figure 5B). These data suggest that, even when CD46 is present, expression of NK-1 is required for infection in the majority of mice. However, while NK-1 influences both the number of mice infected and the kinetics of infection, it is not obligatory, as some NK-1 KO neonates were susceptible to infection and neuropathogenesis.
As an alternative strategy to ascertain what role NK-1 played in MV infection of neurons, the high-affinity pharmacological NK-1 antagonist, aprepitant, currently used clinically as an anti-emetic (Emend®), was employed. Unlike the genetic knockout studies described above, neonates could not be used for the aprepitant studies because of the technical obstacle of oral drug delivery to neonatal mice. Thus, CD46+ adult mice on a RAG-2 KO background were used: these mice, which lack mature T and B cells, cannot resolve a MV challenge and virtually 100% become moribund between 10-14 dpi (Lawrence et al., 1999). As shown in Figure 6A, all MV-inoculated CD46+/RAG-2 KO mice that did not receive aprepitant showed an average weight loss of 15% and an average illness score of 3.4 (obtained between days 6-10) prior to sacrifice. In contrast, aprepitant-treated CD46+/RAG-2 KO mice segregated into two populations: those that developed neuropathology, and those that did not. In the representative experiment shown, 14/25 mice developed a milder illness than non-aprepitant treated controls (2.1 vs. 3.4), and weight loss in these mice was less, averaging 5%. Importantly, 11/25 MV-challenged mice treated with aprepitant showed no signs of illness or weight loss. RT-PCR analysis of purified total brain RNA confirmed the absence of MV-N RNA in brains of healthy aprepitant-treated mice (data not shown).
When brains were collected from aprepitant-treated and untreated, MV-infected CD46+/RAG-2 KO mice between 10-12 dpi and immunostained, mice without aprepitant showed characteristic extensive and unrestricted infection of the CNS (Figure 6B (a-c)), whereas sick mice that were treated with aprepitant had less viral infection that seemed to be more focally restricted within the parenchyma (Figure 6B (d-f)). No evidence of MV infection within the CNS was observed in healthy mice that received aprepitant (Figure 6B (g-i)). Taken together, the results from both the drug and knockout experiments indicate that NK-1, a receptor for the neurotransmitter Substance P, plays a crucial role in MV spread. We hypothesize that NK-1 may be a docking receptor for the MV-F protein, facilitating cell-to-cell MV transmission.
Discussion
Our previous studies demonstrated that the mechanism of MV transmission in neurons differed dramatically from that in non-neuronal cells (Lawrence et al., 2000). In fibroblasts, MV was highly cytolytic and resulted in robust release of infectious particles. In contrast, no virus-mediated lysis of infected primary hippocampal neurons was detected, nor could extracellular virus be recovered, despite extensive viral replication and spread. Electron microscopy studies revealed both a block in the ability of MV to bud from the neuronal plasma membrane as well as the presence of viral RNPs at synaptic junctions, indicating that MV adopts a trans-synaptic mode of spread in neurons (Lawrence et al., 2000). Interestingly, trans-synaptic transmission was CD46-independent, a characteristic that appears to be unique to neurons, as CD46 was required for viral spread in fibroblast co-cultures (Makhortova, et. al, manuscript in press).
A synthetic tripeptide, FIP, that blocks MV fusion (Norrby, 1971; Payan et al., 1984; Richardson and Choppin, 1983), inhibited both MV infection and spread in primary neurons expressing one of the human receptors, CD46. The mechanism by which FIP prevents fusion is not known, but may either be due to direct interference of the amino terminus of the F1 subunit with the target cell membrane, or to preventing the exposure of the fusogenic peptide (Norrby, 1971; Richardson and Choppin, 1983; Kelsey, 1991).
The similarity of sequence between FIP and the neurotransmitter Substance P, coupled with previous reports that Substance P could interfere with MV replication in nonneuronal cells (Schroeder, 1986; Richardson, Scheid, and Choppin, 1980) prompted us to ask whether Substance P and other members of the neurotachykinin family could prevent viral translocation across the synaptic cleft. Indeed, all neurotachykinins blocked spread, a process that appeared to depend, at least in part, on the presence of the neurotachykinin receptor, NK-1. The majority of NK-1 deficient neonates, as well as NK-1+ adults mice treated with the specific antagonist, aprepitant, survived viral challenge without any symptoms of disease or viral replication in the CNS. Thus, we speculate that NK-1 may promote MV entry into CD46+ neurons by serving as a docking receptor for the MV-F protein, and that the ability of FIP and Substance P to block infection and spread may be due to competitive inhibition with the NK-1 receptor, a hypothesis currently under study in our laboratory.
The apparent ability of NK-1 to mediate viral spread at the neuronal synapse in the absence of an H receptor may be a unique attribute of this specialized membrane. MV-Ed trans-synaptic spread can occur in the absence of CD46, and primary mouse neurons do not express the other identified MV receptor, SLAM/CD150w. While these data indicate that trans-synaptic spread may not require a receptor for the H protein, we cannot rule out the possibility that a protein expressed at the post-synaptic membrane may serve as a unique H-receptor in neurons, specifically facilitating neuron-to-neuron transport across synapses. However, given the similarity in sequence and activity of Substance P and the fusion inhibitor FIP, we favor the idea that F, through binding to neurokinin receptors, facilitates transport of MV RNPs across the synapse in the absence of a need for H. Importantly, NK-1 is expressed in all mammals and on virtually all cells, including the Vero fibroblasts used as controls in our experiments (data not shown). Thus, we are also currently testing whether NK-1 is a necessary co-factor for MV entry into all permissive cells. Moreover, given that these studies were performed with MV-Edmonston, a vaccine strain, it will be important to address what role NK-1 plays in entry and spread of wild type isolates.
While our data implicate an important role for NK-1 in MV spread in neurons, some infected mice with deleted or impaired NK-1 did become infected, and neither FIP nor Substance P were able to fully ablate MV spread in primary neurons. This inability could be explained by: a) an insufficient concentration of these inhibitors at the synaptic membrane; b) a rapid off-rate of Substance P from NK-1 (Sarntinoranont, Iadarola, and Morrison, 2003) and/or c) the potential for MV-F to utilize multiple neurokinin receptors, including NK-2 and NK-3, for entry. Because all three neurotachykinins inhibited MV spread to some degree (Figure 4B), and each serves as the ligand for three different neurokinin receptors, it remains possible that the other neurokinin receptors, NK-2 and NK-3, may serve as surrogates for MV entry under circumstances in which NK-1 is not present or active.
A recent paper provides additional support for the fusion-tachykinin relationship. The F protein of bovine respiratory syncytial virus (BRSV), a member of the Paramyxovirus family, is post-translationally cleaved twice, unlike MV-F, which is cleaved once. The small peptide that is released from BRSV-F cleavage is converted to a biologically active tachykinin, called virokinin (Zimmer et al., 2003). This soluble, virus-encoded peptide interacts with NK-1 and triggers the same downstream pathways as the bona fide ligand, Substance P. Thus, fusion proteins or post-translationally cleaved products derived from the fusion proteins of multiple Paramyxoviruses appear to interact with neurokinin receptors, though the benefit such an interaction confers to the virus is not yet understood.
Finally, these data may have implications for MV pathogenesis in the CNS. In the NSE-CD46 mouse model of neuronal MV infection, CNS disease occurs in infected, immunodeficient animals in the absence of neuronal death (Patterson et al., 2002), suggesting that CNS disease in this mouse model is due to neuronal dysfunction rather than overt cell loss. We hypothesize that MV-mediated fusion of neuronal synapses could promote neuropathology through a number of possible mechanisms, including constitutive neurotransmitter signaling or loss of synaptic integrity. Continued use of this model system will allow us to determine how receptor interactions may contribute to neuropathology, and may identify novel candidates for antiviral therapies to prevent or limit MV-induced neuropathology in humans.
Materials and Methods
Cells and virus
Vero fibroblasts were maintained in Dulbecco's modified Eagle medium (DMEM)(Life Technologies), supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 100 U/ml penicillin, and 100 ng/ml streptomycin. Primary hippocampal neurons were prepared from day e15-e16 embryonic mice, as described (Banker and Goslin, 1991; Pasick, Kalicharran, and Dales, 1994; Rall, Mucke, and Oldstone, 1995), except that the cells were maintained in Neurobasal medium (Life Technologies), containing 4 □g/ml glutamate, in the absence of an astrocyte feeder layer. All neurons were obtained from NSE-CD46+ embryonic brain tissues, except for the control poliovirus study, in which neurons were isolated from PVR+ transgenic mice (a gift from Raul Andino, UCSF). All cells were maintained at 37°C in a humidified incubator with 5% CO2.
Measles virus (MV; Edmonston strain) was purchased from American Type Cell Collection (ATCC) and was passaged and titered in Vero fibroblasts, as was poliovirus (PV; Mahoney strain), a gift from Luis Sigal (Fox Chase Cancer Center). Lymphocytic choriomeningitis virus (LCMV; Armstrong strain), a gift from Michael Oldstone (The Scripps Research Institute, La Jolla, CA) was passaged on BHK-21 fibroblasts and titered on Vero fibroblasts.
Western Blot analysis
Hippocampal neurons were plated onto poly-lysine coated 6-well tissue culture dishes, and were infected 72 hr post-plating with MV-Ed at a multiplicity of infection (MOI)=0.1-3. Vero fibroblasts (4 hr post-plating) were infected identically. Three days later, infected and mock-infected cell lysates were collected by scraping cells in 1X protein buffer, followed by a 20 sec sonication. Samples were stored at −70°C and boiled for 5 min prior to loading. Proteins were separated using 8% SDS-PAGE and transferred to a nylon membrane. Rabbit polyclonal antiserum directed against the MV-F cytoplasmic tail (a gift from Roberto Cattaneo, Mayo Clinic, Rochester, MN; (Cathomen, Naim, and Cattaneo, 1998)) was used as the primary antibody (diluted 1:1000 in 10% milk), followed by incubation with a peroxidase-conjugated anti-rabbit IgG secondary antibody (diluted 1:2000; Vector Laboratories). Bands were visualized using chemiluminescence (ECL; Dupont NEN).
Immunohistochemistry
Coverslip-bound cells or cryosectioned, 10 micron-thick horizontal brain sections were fixed with ice-cold methanol/acetone (1:1) for 10 min, dried, and rehydrated in PBS. Samples were blocked with 1% (vol/vol) normal goat serum in PBS, followed by avidin and biotin blocking (Vector Laboratories). A human antiserum (Keller, a gift from Michael Oldstone) was used at a dilution of 1:2000. Appropriate secondary antibodies conjugated to biotin were used at a 1:300 dilution. A streptavidin-peroxidase conjugate (ABC Elite; Vector) was then added, followed by visualization with diaminobenzidene (DAB; 0.7 mg/ml in 60 mM Tris; Sigma) and H2O2 (1.6 mg/ml). Cells and sections were counterstained with hematoxylin and mounted with an aqueous mounting medium. As controls, uninfected cells or brains, samples from which the primary antibody was omitted, or samples using an irrelevant, isotype-matched primary antibody were included.
Inhibitor studies in cell culture
To determine the impact of various reagents on viral infection, primary neurons or control Vero cells were first treated with the reagents at the indicated concentrations for 1 hr, followed by infection in the presence of the putative inhibitor. Following a 1 hr incubation with virus, the inoculum was removed, and the cells were gently washed with warmed PBS (pH 7.4). Thereafter, the appropriate medium, containing fresh inhibitor, was added. Coverslips were then fixed at the indicated day post-infection (dpi).
To explore the impact of these reagents on viral spread, the process was reversed: cells were infected for either 1 or 6 hr, followed by addition of the reagents thereafter at the indicated concentrations. All reagents were obtained from Bachem, and were dissolved in DMSO or 1N acetic acid. Reagents used include the following: fusion inhibitory peptide (FIP; z-D-Phe-L-Phe-Gly-oh); the neurotachykinins Substance P (h-Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-nh2), neurokinin A (h-His-Lys-Thr-Asp-Ser-Phe-Val-Gly-Leu-Met-nh2) and neurokinin B (h-Asp-Met-His-Asp-Phe-Phe-Val-Gly-Leu-Met-nh2); bradykinin (h-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-oh); the opioid receptor antagonist, H-Tyr-L-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (h-Tyr-Tic-nh2); and the Substance P antagonist, Spantide (h-D-Arg-Pro-Lys-Pro-Gln-Gln-D-Trp-Phe-D-Trp-Leu-Leu-nh2). Control cultures were incubated with an equivalent volume of DMSO or 1N acetic acid and coverslips were harvested and stained for MV as indicated above.
Challenge experiments in mice
Adult mice between 6-10 weeks of age and neonatal mice between 2-3 days of age were used. Neurokinin-1 knockout (KO) mice (H-2b) were obtained from Norma Gerard (Harvard University) (Bozic et al., 1996), and intercrossed for 8 generations with NSE-CD46 transgenic mice (H-2b). For all mice, genotype verification for CD46, NK-1 and haplotype was performed on DNA isolated from tail biopsies or on blood samples collected from the retro-orbital sinus. To confirm CD46 status, standard PCR (30 cycles) was performed using the following primers: upstream: CGGTC GCTAC CATTA CCAGT; downstream: CCCCC TGAAC CTGAA ACATA. To confirm NK-1 KO status, two primer pairs were used, one pair to verify the absence of the targeted region of the NK-1 gene (upstream: TTGAG TCCTG CTACT CAGGA; downstream: ACTTT TACTA GCTGC GCTGC), and the other to confirm the presence of the neomycin resistance gene (upstream: TCAGC GCAGG GGCGC CCGGT TCTTT; downstream: ATCGA CAAGA CCGGC TTCCA TCCGA).
Neonatal CD46+/NK-1 KO mice (less than 3 days of age) were challenged intracerebrally with 104 PFU MV-Ed, delivered in a volume of 10 μL along the midline using a 27 gauge needle. Infected pups were monitored daily, and animals showing signs of virus-induced illness (maternal rejection, absence of weight gain) were euthanized.
For the aprepitant (Emend®, Merck Pharmaceuticals) studies in adult mice, the same dose recommended for humans (80 mg/kg), adjusted for body weight, was given to adult CD46+ mice on a RAG-2 KO background (Lawrence et al., 1999). The drug was suspended in PBS, and delivered in a volume of 200 μl via oral gavage every other day following viral challenge. The day prior to infection, a double dose of the drug was administered. Animal health was monitored daily, and mice were weighed and assessed every other day following viral challenge. For the health assessment, mice were scored based on the following subjective scale: 0=healthy; 1=slight ruffling, hunching, or lack of mobility; 2=any two of these symptoms or severe appearance of one; 3=all three symptoms, or severe incidence of two; 4=moribund. All mouse experiments were reviewed and approved by the Fox Chase Cancer Center IACUC.
Tissue harvesting, histology, and RNA isolation and analysis
From some mice, brain tissues were collected for either IHC or RNA analysis. For IHC studies, tissues were immersed in OCT embedding compound, quick-frozen in a dry ice/isopentane bath, and stored at −80°C. Thereafter, 10 micron cryosections were obtained, and stored at −80°C. Sections were thawed, fixed in ice-cold 95% ethanol, and immunostained as described above.
To isolate RNA from infected tissues, brains were snap-frozen in liquid nitrogen, and homogenized in TriReagent (Sigma). Thereafter, RNA was purified and quality-tested by gel electrophoresis. RNA was then subjected to RT-PCR using SuperScript II reverse transcriptase (Invitrogen), followed by amplification using MV-specific primers for nucleoprotein (upstream: ACTTA GGAGC AAAGT GATTG CCT; downstream: AACAA CACGG AACCT CTGCG G). Amplicons were visualized by electrophoresis on a 1.5% agarose gel.
Acknowledgements
G.R. was supported by NIH grants MH56951, NS40500, and a grant from the Kirby Foundation; C.E.P. was supported by NIH F32 NS11100.
The authors gratefully acknowledge Roberto Cattaneo (Mayo Clinic, Rochester MN), Denis Gerlier (Universite Claude Bernard, Lyon), Raul Andino (University of California, San Francisco), and Michael B. A. Oldstone (The Scripps Research Institute, La Jolla CA) for providing reagents and mice, and Christine Matullo, R. Wesley Rose, William Mason and Luis Sigal for constructive comments.
Abbreviations
- MV
measles virus
- CNS
central nervous system
- dpi
days post-infection
- hpi
hours post-infection
- NK-1
neurokinin-1
- F
fusion
- H
hemagglutinin
- NSE
neuron-specific enolase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anlar B. SSPE: diagnosis and drug treatment options. CNS Drugs. 1997;7:111–120. doi: 10.2165/00023210-199707020-00003. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Culturing nerve cells. MIT Press; Cambridge: 1991. [Google Scholar]
- Bozic CR, Lu B, Hopken UE, Gerard G, Gerard NP. Neurogenic amplification of immune complex inflammation. Science. 1996;273:1722–1725. doi: 10.1126/science.273.5282.1722. [DOI] [PubMed] [Google Scholar]
- Cathomen T, Naim HY, Cattaneo R. Measles virus with altered envelope protein cytoplasmic tails gain fusion competence. J . Virology. 1998;72:1224–1234. doi: 10.1128/jvi.72.2.1224-1234.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R. Four viruses, two bacteria and one receptor: membrane co-factor protein (CD46) as pathogens' magnet. J. Virol. 2004;78:4385–4388. doi: 10.1128/JVI.78.9.4385-4388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- Dutch RE, Hagglund RN, Nagel MA, Peterson RG, Lamb RA. Paramyxovirus fusion protein: a conformational change on cleavage activation. Virology. 2001;281:138–150. doi: 10.1006/viro.2000.0817. [DOI] [PubMed] [Google Scholar]
- Gerard NP, Garraway LA, Eddy RL, Shows TB, Ijima H, Paquet JL, Gerard C. Human substance P receptor (NK-1): organization of the gene, chromosome localization and functional expression of cDNA clones. Biochemistry. 1991;30:10640–10646. doi: 10.1021/bi00108a006. [DOI] [PubMed] [Google Scholar]
- Griffin DE, Bellini WJ. Measles virus. In: Knipe DM, Fields BN, Howley PM, et al., editors. Fields Virology. Lippincott-Raven; Philadelphia, PA: 1996. pp. 1267–1312. [Google Scholar]
- Harrowe G, Mitsuhashi M, Payan DG. Measles virus-Substance P receptor interactions: possible novel mechanism of viral fusion. Journal of Clinical Investigation. 1990;85:1324–1327. doi: 10.1172/JCI114571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol. 1992;66:4564–4569. doi: 10.1128/jvi.66.7.4564-4569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XL, Ray R, Compans RW. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 1992;66:1528–1534. doi: 10.1128/jvi.66.3.1528-1534.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Nishio M, Kawano M, Kusagawa S, Komada H, Ito Y, Tsurudome M. Role of a single amino acid at the amino terminus of the simian virus 5 F2 subunit in syncytium formation. J. Virol. 1997;71:9855–9858. doi: 10.1128/jvi.71.12.9855-9858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M. Clinical spectrum of measles. Current Topics in Micro. and Immunol. 1995:1–10. doi: 10.1007/978-3-642-78621-1_1. [DOI] [PubMed] [Google Scholar]
- Kelsey DR, Flanagan TD, Young JE, Yeagle PL. Inhibition of Sendai virus fusion with phospholipid vesicles and human erythrocyte membranes by hydrophobic peptides. Virology. 1991;182:690–702. doi: 10.1016/0042-6822(91)90610-n. [DOI] [PubMed] [Google Scholar]
- Lawrence DMP, Patterson CE, Gales TL, D'Orazio JL, Vaughn MM, Rall GF. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation or extracellular virus production. J. Virology. 2000;74:1908–1918. doi: 10.1128/jvi.74.4.1908-1918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence DMP, Vaughn MM, Belman AR, Cole JS, Rall GF. Immune response-mediated protection of adult but not neonatal mice from neuron-restricted measles virus infection and CNS disease. J. Virol. 1999;73:1795–1801. doi: 10.1128/jvi.73.3.1795-1801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW. Neurobiology of substance P and the NK1 receptor. J. Clin. Psychiatry. 2002;63:6–10. [PubMed] [Google Scholar]
- Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrby E. The effect of a carbobenzoxy tripeptide on the biological activities of measles virus. Virology. 1971;44:599–608. doi: 10.1016/0042-6822(71)90374-6. [DOI] [PubMed] [Google Scholar]
- Oldstone MBA, Lewicki H, Thomas D, Tishon A, Dales S, Patterson J, Manchester M, Homann D, Naniche D, Holz A. Measles virus infection in a transgenic model: virus-induced immunosuppression and central nervous system disease. Cell. 1999;98:629–640. doi: 10.1016/s0092-8674(00)80050-1. [DOI] [PubMed] [Google Scholar]
- Pasick JM, Kalicharran K, Dales S. Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J. Virol. 1994;68:2915–2928. doi: 10.1128/jvi.68.5.2915-2928.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson RG, Russell CJ, Lamb RA. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology. 2000;270:17–30. doi: 10.1006/viro.2000.0267. [DOI] [PubMed] [Google Scholar]
- Patterson CE, Lawrence DM, Echols LA, Rall GF. Immune-mediated protection from measles virus-induced central nervous system disease is noncytolytic and gamma-interferon dependent. J. Virol. 2002;76:4497–4506. doi: 10.1128/JVI.76.9.4497-4506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payan DG, Brewster DR, Missirian-Bastian A, Goetzl EA. Substance P recognition by a subset of human T lymphocytes. J. Clin. Investigation. 1984;74:1532–1539. doi: 10.1172/JCI111567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne FE, Baublis JV, Itabashi HH. Isolation of measles virus from cell cultures of brain from a patient with subacute sclerosing panencephalitis. N. Engl. J. Med. 1969;281:585–589. doi: 10.1056/NEJM196909112811103. [DOI] [PubMed] [Google Scholar]
- Pennefather JN, Lecci A, Candenas ML, Patak E, Pinto FM, Maggi CA. Tachykinins and tachykinin receptors: a growing family. Life Science. 2004;74:1445–1463. doi: 10.1016/j.lfs.2003.09.039. [DOI] [PubMed] [Google Scholar]
- Rall GF, Manchester M, Daniels LR, Callahan EM, Belman AR, Oldstone MBA. A transgenic mouse model for measles virus infection of the brain. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4659–4663. doi: 10.1073/pnas.94.9.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall GF, Mucke L, Oldstone MBA. Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex-expressing neurons in vivo. J. Exp. Med. 1995;182:1201–1212. doi: 10.1084/jem.182.5.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Choppin PW. Oligopeptides that specifically inhibit membrane fusion by paramyxoviruses: studies on the site of action. Virology. 1983;131:518–532. doi: 10.1016/0042-6822(83)90517-2. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Scheid A, Choppin PW. Specific inhibition of paramyxovirus replication by oligopeptides with amino acid sequences similar to those at the N-terminus of the F1 or HA2 viral polypeptides. Virology. 1980;105:205–222. doi: 10.1016/0042-6822(80)90168-3. [DOI] [PubMed] [Google Scholar]
- Sarntinoranont M, Iadarola MJ, Morrison PF. A kinetic analysis of substance P trafficking. J. Pharm. Sci. 2003;92:232–243. doi: 10.1002/jps.10280. [DOI] [PubMed] [Google Scholar]
- Schaffer M, Beiter T, Becker HD, Hunt TK. Neuropeptides: mediators of inflammation and tissue repair? Archives of Surgery. 1998;133:1607–1116. doi: 10.1001/archsurg.133.10.1107. [DOI] [PubMed] [Google Scholar]
- Schendler J, Zimmer G, Herrler G, Conzelmann KK. Respiratory syncytial virus (RSV) fusion protein subunit F2, not attachment protein G, determines the specificity of RSV infection. J. Virol. 2003;77:4609–4616. doi: 10.1128/JVI.77.8.4609-4616.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Schaulies J, Niewiesk S, Schneider-Schaulies S, Meulen V. ter. Measles virus in the CNS: the role of viral and host factors for the establishment and maintenance of a persistent infection. J. Neuroimmunology. 1999;5:613–622. doi: 10.3109/13550289909021290. [DOI] [PubMed] [Google Scholar]
- Schroeder C. Substance P, a neuropeptide, inhibits measles virus replication in cell culture. Acta Virologica. 1986;30:432–436. [PubMed] [Google Scholar]
- Techaarpornkul S, Barretto N, Peeples ME. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 2001;75:6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer G, Rohn M, McGregor GP, Schemann M, Conzelmann KK, Herrler G. Virokinin, a bioactive peptide of the tachykinin family, is released from the fusion protein of BRSV. J. Biol. Chem. 2003;278:46854–46861. doi: 10.1074/jbc.M306949200. [DOI] [PubMed] [Google Scholar]