

Abstract
Tyrosine hydroxylase (TyrH) catalyzes the conversion of tyrosine to dihydroxyphenylalanine (DOPA), the rate-limiting step in the biosynthesis of dopamine. Four mutations in the TyrH gene have recently been described in cases of autosomal recessive DOPA-responsive dystonia (Swaans et al., Ann Hum Genet 2000;64:25–31). All four are predicted to result in changes in single amino acid residues in the catalytic domain of the protein: T245P, T283M, R306H, and T463M. To determine the effects of these mutations on the molecular properties of the enzyme, mutant proteins containing the individual single amino acid changes have been expressed in bacteria and purified. Only the T283M mutation results in a decrease in the enzyme kcat value, while the T245P enzyme has a slightly higher value than the wild-type enzyme. The only case in which a Km value for either tyrosine or tetrahydrobiopterin is perturbed is the T245P enzyme, for which the Km value for tyrosine has increased about 50%. In contrast to the minor effects of the mutations on enzyme activity, the stability is decreased significantly by the mutations. The R306H and T283M enzymes are the least stable, losing activity 30- and 50-fold more rapidly than the wild-type enzyme. The apparent Tm value for unfolding was decreased by 3.9, 8.2, and 7.2° for the T245P, R306H, and T463M enzymes, while the T283M enzyme was too unstable for measurement of a Tm value. The results establish that the physiological effects of the mutations are primarily due to the decreased stability of the mutant proteins rather than decreases in their intrinsic activities.
Keywords: tetrahydrobiopterin, protein stability, kinetics, human, Parkinsonism
INTRODUCTION
TyrH catalyzes the conversion of tyrosine to DOPA, using molecular oxygen and BH4 as the other substrates.1,2 The enzyme is a member of a family of nonheme iron-dependent aromatic amino acid hydroxylases. TyrH is made up of three functional domains, an N-terminal regulatory domain, a catalytic domain, and an oligomerization domain. In rat TyrH the N-terminal regulatory domain includes residues 2–164, the catalytic domain residues 165–455, and the oligomerization domain residues 456–498.3,4 There are four isoforms of the human enzyme; all arise from the same gene by differential splicing.5,6 The differences among the four isoforms are due to the size of an insertion after Met30, with isoform 1 containing no insertion and thus aligning with the rat enzyme, and isoform 4, the longest, containing 31 additional residues. There have been no reports to date of significant differences in the catalytic properties of the four human isoforms. The only structures presently available are of rat TyrH lacking the first 155 residues.7 This region of the rat enzyme is 93% identical to the corresponding region of the human enzyme.
TyrH is found in the brain and the adrenal medulla where it catalyzes the initial and rate-limiting step in the biosynthesis of the catecholamine neurotransmitters dopamine, norepinephrine, and epinephrine. These compounds are involved in the regulation of motor coordination, behavior, learning and memory, sleep-wake cycle regulation, endocrine and visceral functions, and arousal in adults. Complete loss of TyrH activity in an organism, as in the case of knock-out mice8 or Drosophila pale mutants,9 is lethal, directly demonstrating the physiological importance of the enzyme. Still, in the last few years, patients have been identified with altered catecholamine levels due to mutations in TyrH. The first example was diagnosed as a case of Segawa disease, a progressive Parkinson diseaselike dystonia that is responsive to therapy with DOPA.10-12 More recently, it has become apparent that the clinical manifestations of a defect in TyrH differ from Segawa disease,13 in that Segawa disease is typically a genetically dominant disease due to a defect in GTP cyclohydrolase (the rate-limiting enzyme in the biosynthesis of BH4).14 In the original series of cases, several patients showed the same mutation in TyrH, Q381K.11,12 Analysis of the effects of this mutation on the properties of recombinant enzyme showed an increase in the Km value for tyrosine of about sixfold and a decrease in the kcat value of about 60%.12 Recently, Swaans et al.15 described patients who displayed clinical symptoms in early childhood similar to those seen in other patients with TyrH deficiency, but responded well to treatment with DOPA in combination with the DOPA-decarboxylase inhibitors carbidopa or benserazide, allowing them to live a normal life. The patients formed two groups based upon the mutation. In one family, two patients were heterozygous for both the R306H and T463M mutations, with each parent exhibiting one such mutation and no symptoms. In the other, the patient similarly was heterozygous for two mutations, T245P and T283M, but DNA from the parents was not available. (For consistency, the numbering used here is based on human TyrH isoform 1. In their original reports Swaans et al.15 used the numbering for isoform 4, which contains an additional 31 residues after Met30, while Ludecke et al.11 used the numbering for isoform 1.)
Because TyrH is found in the adrenal glands and central nervous system, it is not possible to isolate the mutant enzymes from tissue and determine directly how the mutations affect the activity. Analysis of the effects of the mutations can only be done by analysis of purified recombinant enzymes. The effects of the four mutations identified by Swaans et al.15 on the properties of recombinant human TyrH are described herein.
MATERIALS AND METHODS
Materials
Tyrosine and DOPA were from Sigma. BH4 was from B. Schircks Laboratories.
Expression and Purification of Recombinant Proteins
Construction of human TyrH isoform 1 cDNA inserted into pET23d was as described in Sura et al.16 Plasmids for expression of the mutant proteins were constructed using the Stratagene QuikChange site-directed mutagenesis method with the following primers: T245P, 5′-GGCCTCTACGCCCCGCACGCCTGCGG-3′; T283M, 5′-CCTGAAGGAGCGCATGGGCTTCCAGCTGCG-3′; R306H, 5′-CCAGCCTGGCCTTCCACGTGTTCCAGTGCACC-3′; and T463M, 5′-CGACCCGTACATGCTGGCCATCGACG-3′. The mutated triplet is indicated in bold in each case. The T245P/T283M and R306H/T463M constructs were generated by introducing the T245P mutation into the T283M plasmid and the T463M mutation into the R306H plasmid, respectively, following the same protocol. For each construct, DNA sequencing of the entire coding region was performed to ensure that no other mutations were present. For expression, the plasmids containing the mutated genes were transformed into BL21DE star E. coli. Cell growth and protein purification were as previously described for wild-type rat TyrH,17,18 with the modifications for the mutant proteins that cells were grown overnight in Luria-Bertani broth with 1% glucose at 18°C after induction with 0.25 mM isopropyl β-D-thioglucanopyranoside, and nucleic acid precipitation was done with 0.5% (w/v) streptomycin sulfate instead of 2%.
Assays
The concentrations of wild-type and mutant TyrH were determined using an A2800.1% value of 1.0419 and a subunit molecular weight of 56,000. TyrH activity was measured using a colorimetric assay that measures the amount of DOPA produced.20 Standard conditions were 0.1 μM wild-type or mutant TyrH, 100 μg/mL catalase, 10 μM ferrous ammonium sulfate, 1 mM dithiothreitol, 50 mM HEPES, pH 7.0, 30°C. For determining the Km value for BH4, the concentration of tyrosine was constant at 30 μM and the concentration of BH4 was varied from 0 to 80 μM. The resulting steady-state kinetic data were fit directly to the Michaelis-Menten equation using the program Kaleidagraph (Synergy Software). For determining the Km value for tyrosine, the concentration of BH4 was fixed at 300 μM and the concentration of tyrosine was varied from 0 to 150 μM. Because substrate inhibition is seen with tyrosine as the varied substrate, the initial rate data obtained under these conditions were fit to Equation (1); here kcat is the rate of the reaction, [S] is the tyrosine concentration, Km the Michaelis-Menten constant for tyrosine, and Ki the inhibition constant for tyrosine:
| (1) |
To determine the effect of elevated temperature on the activities, the wild-type and mutant enzymes were incubated at 50°C in 100 mM KCl, 10% glycerol, and 50 mM HEPES, pH 7.0. At the times indicated in the figure legends, aliquots were removed and the residual TyrH activity measured using the standard assay. The residual activity as a function of time was fit to Equation (2), where vt is the residual activity at time t and v0 is the initial activity:
| (2) |
CD spectra were taken in 50 mM MOPS, pH 7.0, at 4°C using a 1 mm path-length. Unfolding as a function of temperature was followed by monitoring the CD signal at 228 nm of the individual proteins at 1 μM in 50 mM MOPS, pH 7.0, using an equilibration time at each temperature of 3 min, with increases of 2°C between. In the cases of the T283M and R306H enzymes the buffers also included 10% glycerol. The changes in the CD signal with temperature were analyzed using Equation (3).21 Here, the observed CD signal at each point in the unfolding experiment was ; N0, D0, aN, and aD corrected for changes in the baseline with temperature; and Tm was the apparent melting temperature:
| (3) |
RESULTS
Expression and Purification of Mutant Enzymes
To determine the effects of the individual amino acid changes implicated by the genetic studies on the molecular properties of TyrH, the T245P, T283M, R306H, and T463M mutations were incorporated into a pET23d-based plasmid for expression of the human TyrH isoform 1. Plasmids for expression of the T245P/T283M and R306H/T463M enzymes were also constructed. When cells containing the different plasmids were grown at 18°C, induction with IPTG yielded large amounts of recombinant protein in all cases. Whereas expression of the wild-type enzyme at 37°C yielded fully soluble protein, a significant fraction of each mutant protein was found in inclusion bodies even at the lower temperature (Fig. 1). Only the T245P and T463M enzymes were predominantly soluble. The T283M and R306H enzymes were mainly found in inclusion bodies but a small amount of soluble enzyme was produced. Accordingly, it was possible to purify all four single mutant proteins, although the yields of T283M and R306H TyrH were quite low. The buffers used during purification of TyrH routinely contained 10% glycerol, because the wild-type enzyme was very stable at 4°C under these conditions. The purified wild-type enzyme can be stored indefinitely at −80°C in the absence of glycerol; the T245P and T463M enzymes behaved similarly. In contrast, removal of glycerol from the T283M and R306H enzymes resulted in significant precipitation and loss of protein. For both proteins containing double mutations, all of the TyrH was insoluble and no enzyme could be purified. All four purified proteins containing a single mutation could be reconstituted with a stoichiometric amount of iron, similar to the wild-type enzyme (results not shown).
Fig. 1.
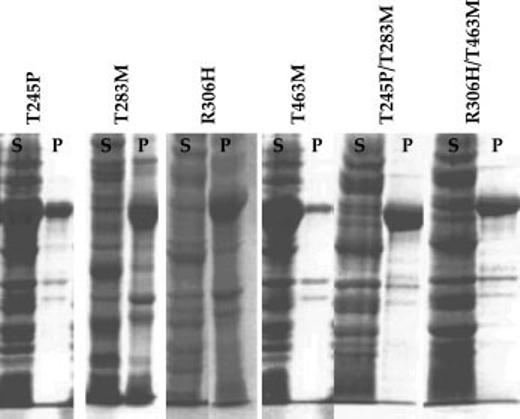
Analysis of the expression and solubility of TyrH mutant proteins by polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate. S and P indicate the soluble and insoluble fractions obtained by centrifugation of the cell lysates after sonication as described in Materials and Methods.
Kinetic Characterization of Mutant Enzymes
To determine if the mutations had any significant effect on the catalytic properties of these enzymes, the steady-state kinetic parameters were determined with tyrosine and BH4 as substrates. The wild-type enzyme shows substrate inhibition when tyrosine is varied with BH4 as the pterin substrate. The mutant enzymes similarly exhibited substrate inhibition, as illustrated in Figure 2 for the T283M enzyme. The kinetic parameters for the four mutant proteins are summarized in Table I. The Km and kcat values for the R306H and T463M enzymes are indistinguishable from the values for the wild-type enzyme. In contrast, the Km value for tyrosine, the Ki for substrate inhibition by tyrosine, and the kcat value were all elevated somewhat in the case of the T245P enzyme. The T283M enzyme was the only one for which a decreased kcat value was found, although all other parameters were unaffected in this case. TyrH is regulated in part by feedback inhibition by dopamine.22 The mutant enzymes bound dopamine as tightly as the wild-type enzyme (results not shown), consistent with the regulatory properties being unaffected by the mutations.
Fig. 2.
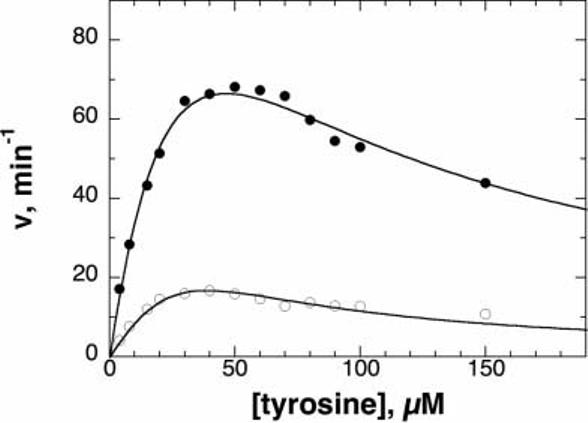
Steady state kinetics of wild-type (closed circles) and T283M (open circles) TyrH as a function of tyrosine concentration. Conditions: 300 μM BH4, 100 μg/mL catalase, 10 μM ferrous ammonium sulfate, 1 mM dithiothreitol, 50 mM HEPES, pH 7.0, 30°C. The lines are from fits of the data to Equation (1).
TABLE I.
Kinetic Parameters of Wild-Type and Mutant Tyrosine Hydroxylase
| Enzyme |
Ktyr (μM)a |
Kityr (μM)a |
KBH4 (μM)b |
kcat (min−1)a |
|---|---|---|---|---|
| Wild-type | 46 ± 1 | 46 ± 1 | 13 ± 2 | 200 ± 2 |
| T245P | 66 ± 5 | 73 ± 7 | 14 ± 1 | 321 ± 8 |
| T283M | 39 ± 2 | 37 ± 2 | 10 ± 1 | 48 ± 1 |
| R306H | 42 ± 3 | 48 ± 3 | 10 ± 1 | 221 ± 5 |
| T463M | 43 ± 3 | 44 ± 3 | 9 ± 1 | 232 ± 6 |
Conditions: 300 μM BH4, 0 to 150 μM tyrosine, 100 μg/mL catalase, 10 μM ferrous ammonium sulfate, 1 mM dithiothreitol, 50 mM HEPES, pH 7.0, 30°C.
Conditions: 30 μM tyrosine, 0 to 80 μM BH4, 100 μg/mL catalase, 10 μM ferrous ammonium sulfate, 1 mM dithiothreitol, 50 mM HEPES, pH 7.0, 30°C.
Stability of Mutant Proteins
The lack of significant change in the bulk of the kinetic parameters of the mutant proteins and the difficulty of obtaining recombinant enzymes in several cases suggested that the primary defect in the mutant enzymes was in the stability. The CD spectra of the mutant enzymes were determined at 4°C as a probe of their structural integrity. The spectra of the wild-type enzyme and the T245P and T463M proteins were obtained in the absence of glycerol and were identical [Fig. 3(A)]. The spectra of the T283M and R306H enzymes were taken in glycerol. Under these conditions, the spectrum of the R306H enzyme was also very similar to that of the wild-type enzyme, while the spectrum of the T283M enzyme was slightly perturbed [Fig. 3(B)]. These spectra are consistent with no significant change in the secondary structure of the T245P, R306H, and T463M enzymes at 4°C, while the T283M enzyme may be slightly unfolded under these conditions.
Fig. 3.
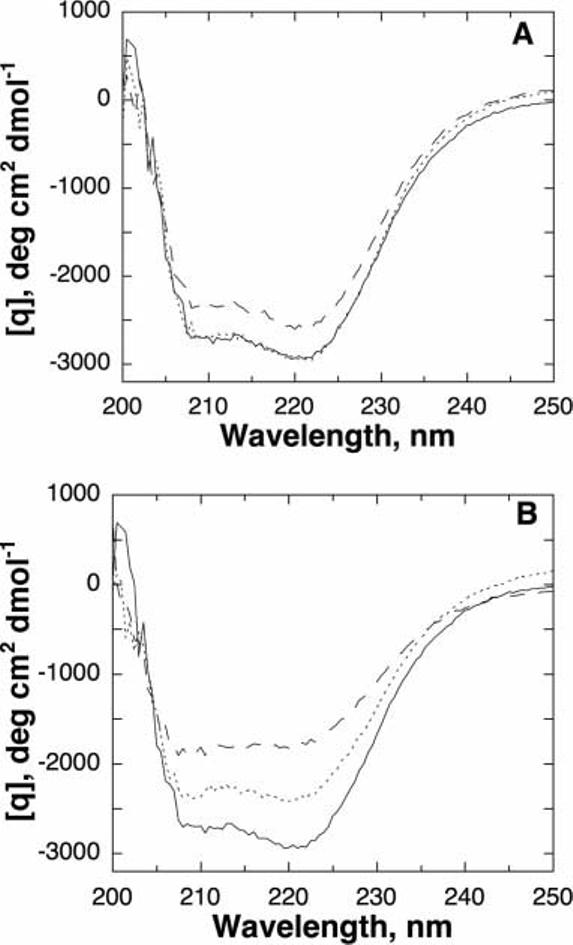
Circular dichroism spectra for wild-type and mutant TyrH. A: Wild-type (———), T245P (- - -), and T463M (· · ··) TyrH. B: Wild-type (———), T283M (- - -), and R306H (· · ··) TyrH. Conditions: 5 μM enzyme, 50 mM MOPS, pH 7.0, 4°C. For the spectra in (B), the buffer also contained 10% glycerol.
The effects of the mutations on the enzyme stability were determined by monitoring the loss of activity upon incubation at 50°C. The results are shown in Figure 4, and the rate constants for the loss of activity at this temperature are given in Table II. The wild-type enzyme was quite stable even at 50°C; after 90 min it still had 50% of the initial activity and after 4 h it still had 20%. The T245P enzyme was somewhat less stable, while the T283M, R306H, and T463M enzymes were much less stable, with the T283M and R306H enzymes losing at least half of the initial activity within 5 min at 50°C.
Fig. 4.
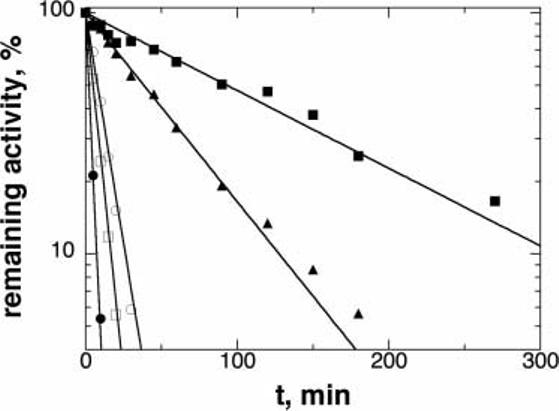
Stability of wild-type and mutant TyrH activity at 50°C. The proteins were incubated at 50°C in 100 mM KCl, 10% glycerol, 50 mM HEPES, pH 7.0; at the times indicated in the figure an aliquot was taken and the residual TyrH activity was determined as described in the Materials and Methods. Wild-type (closed squares), T245P (triangles), T283M (closed circles), R306H (open squares), and T463M (open circles) TyrH. The activity is expressed as percentage of the initial activity. The lines are from fits of the data to Equation (2).
TABLE II.
Effect of Mutations on the Stability of Tyrosine Hydroxylase
| Enzyme | Apparent Tme |
ΔTmf | kinacta (min−1) |
|---|---|---|---|
| Wild-type | 48.2 ± 0.3 | 0 | 0.0068 ± 0.0014 |
| (53.9 ± 1.0)b | |||
| T245P | 44.3 ± 0.1 | 3.9 ± 0.3 | 0.020 ± 0.001 |
| T283M | ndc | nd | 0.330 ± 0.026 |
| R306H | 45.8 ± 0.1b | 8.2 ± 1.1d | 0.142 ± 0.007 |
| T463M | 40.6 ± 0.1 | 7.7 ± 0.3 | 0.090 ± 0.003 |
First order rate constant for loss of activity at 50°C.
Buffer contained 10% glycerol.
nd, not determined.
Compared to wild-type enzyme in the presence of 10% glycerol.
Determined from a fit of the data to Equation (3).
Difference in apparent Tm from wild-type enzyme.
As an alternative measure of the effects of the mutations on the stability, the unfolding of the mutant enzymes with temperature was analyzed, using the CD signal at 228 nm to monitor the unfolding. The unfolding of TyrH is not reversible,23 so that these analyses do not yield true thermodynamic parameters for the equilibrium between the folded and unfolded forms. However, in the case of the wild-type enzyme and all but the T283M enzyme, the change in the CD signal with temperature could be described by Equation (3), which describes a thermal unfolding (Fig. 5), yielding apparent Tm values for unfolding. In the absence of glycerol, the T283M and R306H enzymes did not show a discrete unfolding midpoint temperature; however, addition of 10% glycerol stabilized the R306H enzyme sufficiently that a normal unfolding isotherm was seen [Fig. 5(B)]. This was not the case for the T283M enzyme; extending the analysis by beginning at 4°C did not result in a curve different from that shown in Figure 5(B). In the case of the other three enzymes, the apparent Tm values were significantly less than that for the wild-type (Table II), confirming that the mutations significantly destabilize TyrH.
Fig. 5.
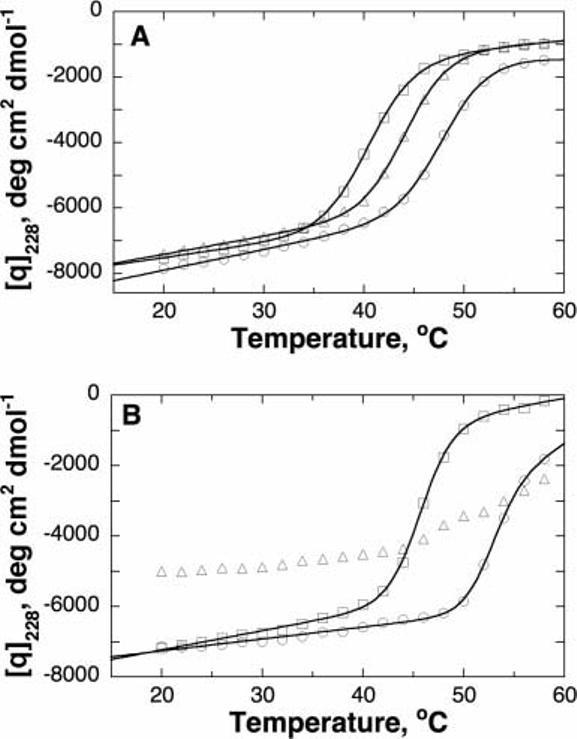
Thermal unfolding of wild-type and mutant TyrH as monitored by CD spectroscopy at 228 nm. A: Wild-type (circles), T245P (triangles), and T463M (squares) enzyme. B: Wild-type (circles), T283M (triangles), and R306H (squares) enzyme. Conditions: 1 μM enzyme, 50 mM MOPS, pH 7.0. For the spectra in (B), the buffer also contained 10% glycerol. The lines are from fits of the data to Equation (3).
DISCUSSION
In the present work we characterize four mutations in human TyrH described previously as causative of a DOPA-responsive dystonia.15 Deleterious point mutations would be expected to alter either the activity of the enzyme, due to decreased binding or catalysis, or its stability. Because Thr245, Thr283, Arg306, and Thr465 are conserved in TyrH, the mutations found in these residues were initially predicted to alter the catalytic activity of the enzyme.15 This would be similar to the effects of the Q381K mutation, the first naturally occurring mutation described in TyrH.11 In that case, the mutation resulted in a significant decrease in the activity of the enzyme due to a three- to fivefold decrease in the kcat value and a similar increase in the Km value for tyrosine.12 However, the kinetic parameters of the T245P, R306H, and T465M enzymes are indistinguishable from the wild-type values. Thus, these mutations have no significant effect on the binding of substrate or turnover. The kcat value is decreased by about fivefold in the case of the T283M enzyme, although the Km values are unaffected. While this suggests that this mutation does alter catalysis, the effects on stability are more drastic than the effects on catalysis. While the CD spectra of the mutant enzymes show that they are still folded correctly at 4°C, all four mutant proteins are less stable than the wild-type enzyme, irrespective of whether the stability is measured as the rate of loss of activity at elevated temperature or as the apparent Tm for thermal unfolding. T245P TyrH is the most stable protein, followed in order by the T465M, R306H, and T283M enzymes. Indeed, the T283M enzyme is sufficiently unstable that it readily denatures in the absence of glycerol even at 4°C.
Deficiencies in TyrH can result in mild or severe dystonia, with the mild form being responsive to treatment with DOPA.24 The patients with the mutations described here responded well to treatment with DOPA. The genetic analyses of the original patients established that each was heterozygous for two mutations, so that they would have two different TyrH genes, each with one mutation. The properties of the mutant proteins described here, normal catalytic activity but decreased stability, suggest that these patients would have low levels of active enzyme rather than a complete loss of activity; this provides a reasonable explanation for the mildness of their symptoms.
The three-dimensional structure of rat TyrH can be used to gain insight into the basis for the effects of these mutations.7,25 The available structures of the free enzyme and the dihydropterin bound enzyme lack the N-terminal 159 residues that make up the regulatory domain; none of the four residues analyzed here is in the missing regulatory domain. The active site of the enzyme is a 17 Å deep cleft with an iron atom at the bottom coordinated by two histidines and a glutamate. These four residues are not in the active site, so that they are not involved in the binding of the substrates. This is consistent with the lack of an effect of any mutation of the Km values for the substrates.
Thr245 is at the end of helix 3 of TyrH. The side-chain hydroxyl of Thr245 is 2.8 Å from the imidazole N1 of His246, the first residue in a short loop leading to helix 4, consistent with a good hydrogen bond. The only other moiety within 4 Å of the Thr245 hydroxyl is the backbone amide of Leu242 at 3.3 Å, more consistent with a van der Waals contact than a hydrogen bond [Fig. 6(A)]. When Thr245 is mutated to proline the hydrogen bonds involving the threonine hydroxyl are lost. Because Thr245 is at the end of a helix, the restricted side-chain of proline causes no backbone distortion, so that the greatest effect of the T245P mutation is due to the loss of the hydrogen bond with His246. While the contribution of a specific hydrogen bond to the stability of a protein varies, a typical value for the decrease in the ΔG for folding upon the loss of a hydrogen bond involving serine or threonine is 1 kcal/mol.26 The thermal denaturation of TyrH is not a reversible process, so that true ΔH values cannot be obtained from the data of Figure 5. However, a decrease in the ΔG for folding for a protein of 1 kcal/mol typically corresponds to a decrease in the Tm value of 3–4°.27,28 The decrease in the Tm value of 3.9°C for the T245P enzyme is fully consistent with the only significant disruption being the loss of the hydrogen bond to His246. Thr283 is found in a turn between helix 5 and strand 1, with its hydroxyl moiety 2.8 Å from the Leu339 backbone carbonyl [Fig. 6(B)]. The loss of this hydrogen bond would be expected to have an effect on the Tm value comparable to that of the T245P mutation. Instead, this is the most destabilizing mutation of the four analyzed here. In contrast to the T245P mutation, where the altered side-chain is readily accommodated without structural distortion, replacement of Thr283 by the much larger methionyl residue would be expected to cause alterations in the packing of neighboring residues. Replacement of small hydrophobic residues in protein cores with larger hydrophobic residues typically has a large destabilizing effect on a protein.29,30 Moreover, Leu339 is located in a turn at the end of helix 7; this helix also contains one of the metal ligands, His335, and the amide nitrogen of Leu339 forms the expected hydrogen bond to the backbone carbonyl of His335. The altered position of helix 7 required to accommodate the methionyl side-chain at residue 283 thus has the potential to alter the arrangement of a metal ligand. That this is indeed the case is supported by the decrease in the kcat value for the T283M enzyme. Thus, the large decrease in stability in the T283M enzyme can be attributed to both the loss of single hydrogen bond and distorted packing of helices in the core of the catalytic domain.
Fig. 6.
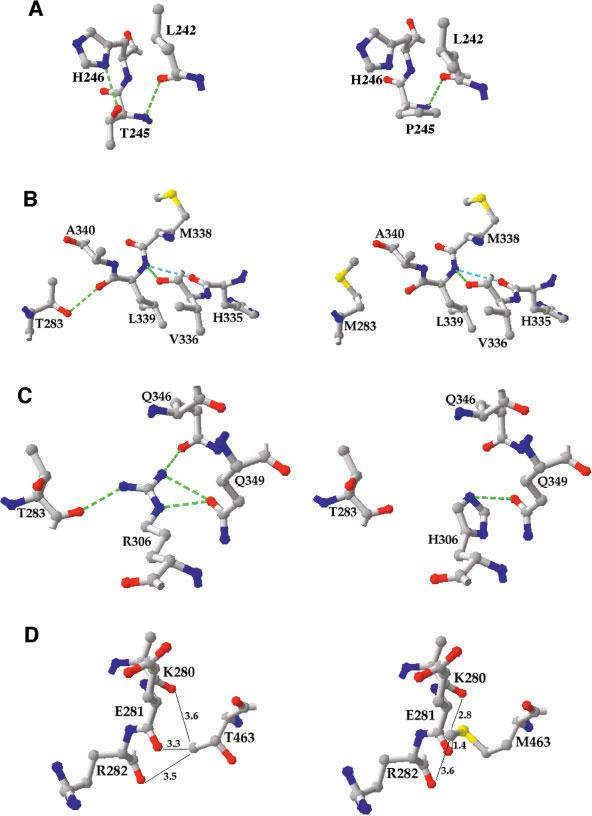
Interactions of the mutated residues in wild-type (left) and mutant (right) TyrH. A: Thr245; (B) Thr283; (C) Arg306; (D) Thr463. Hydrogen bonds are indicated by green dotted lines. In (D), the distances in angstrom from the side chain of Thr463 are given. The figure was generated using the PDB file 2TOH.
Mutation of Arg306 has the second greatest effect on stability of the four mutations analyzed here. The side-chain of Arg306 forms four hydrogen bonds, one with the Thr283 backbone carbonyl, and three with side-chains of residues in helix 8, Gln346, and Gln349 [Fig. 6(C)]. When arginine is mutated to histidine the interactions with Thr283 and Gln346 are lost. While it is possible to find an orientation in which the hydrogen bond with Gln349 remains, this would introduce a steric clash with the imidazole γ-carbon 2 [Fig. 6(C)], so this hydrogen bond is expected to be weaker than in the wild-type enzyme. The large decrease in the apparent Tm value of about 8 kcal/mol for the R306H enzyme is consistent with the loss of two to three hydrogen bonds.
The side-chain of Thr463 does not form a hydrogen bond with any residue in the same subunit. Instead, Thr463 is in a turn in the oligomerization domain responsible for the homotetrameric quaternary structure of TyrH. The methyl group of the Thr463 side-chain is in van der Waals contact with the backbone carbonyls of residues 280–282 in helix 5 of another subunit [Fig. 6(D)]. The larger methionine side-chain found in this mutant protein would clearly disrupt the packing between subunits, as was the case for the T283M mutation. However, in the case of Thr463 no hydrogen bond is lost, so that the decrease in stability is less when this residue is mutated to methionine.
The results presented here establish that four mutations that cause a mild form of DOPA-responsive dystonia can be attributed to the effects of the mutation on the stability of TyrH. Only one mutation results in a less active protein, but in that case the effect on stability is much greater than the effect on activity. The effects of these mutations contrast with the decrease in catalytic activity seen in the case of the previously characterized Q281K mutation.
ACKNOWLEDGMENTS
The authors thank Dr. J. Martin Scholtz and Abbas Razvi for their help with the CD studies.
Abbreviations
- BH4
tetrahydrobiopterin
- CD
circular dichroism
- DOPA
dihydroxyphenylalanine
- TyrH
tyrosine hydroxylase
Footnotes
Grant sponsor: National Parkinson Foundation-Parkinson's Disease Foundation Joint Research Grant Program; Grant sponsor: National Institutes of Health; Grant number: GM47291.
REFERENCES
- 1.Fitzpatrick PF. The tetrahydropterin-dependent amino acid hydroxylases. Ann Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 2.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daubner SC, Lohse DL, Fitzpatrick PF. Expression and characterization of catalytic and regulatory domains of rat tyrosine hydroxylase. Protein Sci. 1993;2:1452–1460. doi: 10.1002/pro.5560020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohse DL, Fitzpatrick PF. Identification of the intersubunit binding region in rat tyrosine hydroxylase. Biochem Biophys Res Commun. 1993;197:1543–1548. doi: 10.1006/bbrc.1993.2653. [DOI] [PubMed] [Google Scholar]
- 5.O'Malley KL, Anhalt MJ, Martin BM, Kelsoe JR, Winfield SL, Ginns EI. Isolation and characterization of the human tyrosine hydroxylase gene: Identification of 5′ alternative splice sites responsible for multiple mRNAs. Biochemistry. 1987;26:6910–6914. doi: 10.1021/bi00396a007. [DOI] [PubMed] [Google Scholar]
- 6.Grima B, Lamouroux A, Boni C, Julien J-F, Javoy-Agid F, Mallet J. A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature. 1987;326:707–711. doi: 10.1038/326707a0. [DOI] [PubMed] [Google Scholar]
- 7.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited diseases. Nature Struct Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Q-Y, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature. 1995;374:640–643. doi: 10.1038/374640a0. [DOI] [PubMed] [Google Scholar]
- 9.Neckameyer WS, Quinn WG. Isolation and characterization of the gene for Drosophila tyrosine hydroxylase. Neuron. 1989;2:1167–1175. doi: 10.1016/0896-6273(89)90183-9. [DOI] [PubMed] [Google Scholar]
- 10.Segawa M, Nomura Y. Hereditary progressive dystonia with marked diurnal fluctuation. Pathophysiological importance of the age of onset. Adv Neurol. 1993;60:568–576. [PubMed] [Google Scholar]
- 11.Ludecke B, Dworniczak B, Bartholome K. A point mutation in the tyrosine hydroxylase gene associated with Segawa's syndrome. Hum Genet. 1995;95:123–125. doi: 10.1007/BF00225091. [DOI] [PubMed] [Google Scholar]
- 12.Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet. 1995;4:1209–1212. doi: 10.1093/hmg/4.7.1209. [DOI] [PubMed] [Google Scholar]
- 13.Bräutigam C, Wevers RA, Jansen RJT, Smeitink JAM, De RijkVan Andel JF, Gabreëls FJM, Hoffmann GF. Biochemical hallmarks of tyrosine hydroxylase deficiency. Clin Chem Acta. 1998;44:1897–1904. [PubMed] [Google Scholar]
- 14.Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, Fujita K, Nagatsu T. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–242. doi: 10.1038/ng1194-236. [DOI] [PubMed] [Google Scholar]
- 15.Swaans RJM, Rondot P, Renier WO, Van Den Heuvel LPWJ. Steenbergen-Spanjers GCH, Wevers RA. Four novel mutations in the tyrosine hydroxylase gene in patients with infantile parkinsonism. Ann Hum Genet. 2000;64:25–31. doi: 10.1017/S0003480000007922. [DOI] [PubMed] [Google Scholar]
- 16.Sura G, Daubner SC, Fitzpatrick PF. Effects of phosphorylation by protein kinase A on binding of catecholamines to the human tyrosine hydroxylase isoforms. J Neurochem. 2004;90:970–978. doi: 10.1111/j.1471-4159.2004.02566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis HR, Daubner SC, Fitzpatrick PF. Mutation of serine 395 of tyrosine hydroxylase decouples oxygen-oxygen bond cleavage and tyrosine hydroxylation. Biochemistry. 2000;39:4174–4181. doi: 10.1021/bi9928546. [DOI] [PubMed] [Google Scholar]
- 18.Daubner SC, Lauriano C, Haycock JW, Fitzpatrick PF. Site-directed mutagenesis of Serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J Biol Chem. 1992;267:12639–12646. [PubMed] [Google Scholar]
- 19.Haavik J, Andersson KK, Petersson L, Flatmark T. Soluble tyrosine hydroxylase (tyrosine 3-monooxygenase) from bovine adrenal medulla: Large-scale purification and physicochemical properties. Biochim Biophys Acta. 1988;953:142–156. doi: 10.1016/0167-4838(88)90019-2. [DOI] [PubMed] [Google Scholar]
- 20.Fitzpatrick PF. The steady state kinetic mechanism of rat tyrosine hydroxylase. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 21.Nicholson EM, Scholtz JM. Conformational stability of the Escherichia coli HPr protein: test of the linear extrapolation method and a thermodynamic characterization of cold denaturation. Biochemistry. 1996;35:11369–11378. doi: 10.1021/bi960863y. [DOI] [PubMed] [Google Scholar]
- 22.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 23.Gahn LG, Roskoski R., Jr Thermal stability and CD analysis of rat tyrosine hydroxylase. Biochemistry. 1995;34:252–256. doi: 10.1021/bi00001a030. [DOI] [PubMed] [Google Scholar]
- 24.Furukawa Y. Genetics and biochemistry of dopa-responsive dystonia: significance of striatal tyrosine hydroxylase protein loss. Adv Neurol. 2003;91:401–410. [PubMed] [Google Scholar]
- 25.Goodwill KE, Sabatier C, Stevens RC. Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 Å resolution: self-hydroxylation of phe300 and the pterin-binding site. Biochemistry. 1998;37:13437–13445. doi: 10.1021/bi981462g. [DOI] [PubMed] [Google Scholar]
- 26.Myers JK, Pace CN. Hydrogen bonding stabilizes globular proteins. Biophys J. 1996;71:2033–2039. doi: 10.1016/S0006-3495(96)79401-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alber T, Dao-pin S, Wilson K, Wozniak JA, Cook SP, Matthews BW. Contributions of hydrogen bonds of Thr 157 to the thermodynamic stability of phage T4 lysozyme. Nature. 1987;330:41–46. doi: 10.1038/330041a0. [DOI] [PubMed] [Google Scholar]
- 28.Shirley BA, Stanssens P, Hahn U, Pace CN. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry. 1992;31:725–732. doi: 10.1021/bi00118a013. [DOI] [PubMed] [Google Scholar]
- 29.Gassner NC, Baase WA, Matthews BW. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc Natl Acad Sci USA. 1996;93:12155–12158. doi: 10.1073/pnas.93.22.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu R, Baase WA, Matthews BW. The introduction of strain and its effects on the structure and stability of T4 lysozyme. J Mol Biol. 2000;295:127–145. doi: 10.1006/jmbi.1999.3300. [DOI] [PubMed] [Google Scholar]