

Abstract
Background:
Chronic pain conditions may result from peripheral nerve injury, chronic peripheral inflammation, or sensory ganglia inflammation. However, inflammatory processes may also contribute to peripheral nerve injury responses. To isolate the contribution of local inflammation of sensory ganglia to chronic pain states, we previously developed a rat model in which long lasting pain is induced by inflaming sensory ganglia without injuring the neurons. This results in prolonged mechanical pain, local increases in pro-inflammatory cytokines, increased neuronal hyperexcitability and abnormal spontaneous activity.
Methods:
We used whole-cell patch clamp in acutely isolated small diameter neurons to determine how localized inflammation (3 – 5 days) of L4 and L5 ganglia altered voltage-gated K+ and Na+ currents.
Results:
Tetrodotoxin (TTX)-sensitive Na+ currents increased 2 to 3-fold in neurons from inflamed ganglia. TTX-resistant Na+ currents increased over 2-fold, but only in cells that bound IB4. These increases occurred without shifts in voltage dependence of activation and inactivation. The similar results are seen in models of peripheral inflammation, except for the large magnitudes. Unlike most pain models, localized inflammation increased rather than decreased voltage-gated K+ currents, due to increased amplitudes of the sustained (delayed rectifier) and fast-inactivating transient components. The overall effect in current-clamp experiments was an increase in excitability as indicated by decreased rheobase and lower action potential threshold.
Conclusions:
Neuronal inflammation per se, in the absence of nerve injury, causes large increases in Na channel density and enhanced excitability. The unusual finding of increased K+ current may reflect regulation of excitability in the face of such large increases in Na+ current.
Introduction
Animal models of chronic pain have generally been classified as either nerve injury or inflammation. Both nerve injury and inflammation can produce spontaneous pain, hyperalgesia and allodynia.1 Both are associated with increased excitability and ectopic spontaneous discharges of dorsal root ganglion (DRG) neurons.2-6 These changes in sensory neurons, along with altered information processing in the spinal cord or higher centers, are thought to contribute to increased pain sensation.7 However, the underlying ionic mechanisms mediating the increased neuronal excitability are thought to differ between nerve injury and chronic inflammation. For example, nerve injury generally results in overall downregulation of Na+ channels along with changes in the isoforms expressed, while chronic peripheral inflammation often results in upregulation of tetrodotoxin (TTX)-resistant Na+ channels8. Voltage-dependent K+ currents are downregulated in almost all nerve injury and peripheral inflammation models, but the details differ between different models.2,9-15
Recent work has suggested that the distinction between inflammation and nerve injury may not be complete. Nerve injury models often include some inflammation. For example, in peripheral nerve injury, macrophage infiltration, release of localized pro-inflammatory cytokines and their retrograde transport to the DRG have been shown to be important for hyperalgesia.16-18 Inflammatory processes and glial activation within the DRG have also been proposed to play important roles in some nerve injury models, though these processes have been more extensively studied in central nervous system and peripheral nerve (for review, 19-21).
To gain a better understanding of the inflammatory contributions to pathologic pain, we developed a rodent pain model in which the somata of sensory neurons are subjected to direct inflammatory stimulus by depositing a small drop of the immune activator zymosan into the epidural space in the vicinity of the DRG.22 Localized inflammation of the DRG (LID), in the absence of nerve trauma or damage, occurs in certain clinical pain states such as post herpetic neuralgias and some forms of low back pain such as following lumbar disc rupture. Material released from the nucleus pulposus is known to possess immunogenic and chemogenic capacities.23,24 However, we also view the LID model as one which allows us to study the effects of DRG inflammation per se, (i.e., in the absence of nerve damage or axotomy). This is of more general interest since inflammation is one component of most pain models including those based on nerve injury. Local inflammation results in bilateral mechanical hyperalgesia which begins by day 1, peaks at days 3-7, and lasts up to 28 days. LID also results in rapid macrophage infiltration and glial activation, increased levels of pro-inflammatory cytokines, and abnormal sprouting of sympathetic fibers in the DRG.22 Like other pain models, LID leads to increased excitability and spontaneous activity of the DRG cells, including C-cells. In the present study, we used patch clamp techniques on inflamed small-diameter DRG cells to investigate changes in Na+ and K+ channels that might contribute to the excitability changes observed in LID.
Materials and Methods
Young female Sprague-Dawley rats (body weight 100-150 g) were housed one or two per cage under a controlled diurnal cycle of 12 h light and 12 h dark with free access to water and food. The ambient environment was maintained at constant temperature (22 ± 0.5°C) and relative humidity (60-70%). All the surgical procedures and the experimental protocol were approved by the institutional animal care and use committees (IACUC) of the University of Cincinnati (Cincinnati, Ohio, USA).
In vivo localized inflammatory irritation of the DRG (LID)
Rats were anesthetized with isoflurane. The paraspinal muscles on the right side were separated surgically from the L4-L6 vertebrae as described previously.22,25 The superior articular and the transverse processes between L4 and L6 vertebrae were cleaned, the L5 and L4 spinal nerves that course through the intervertebral foramen were identified, and a small hole (0.49 mm in diameter) was drilled through the junction of transverse process and lamina over the L5 and L4 DRG. The drill bit had a limited length (1.5-2.0 mm) which could be finely adjusted during the surgery to ensure that the drill just barely penetrated the overlying bone without injuring the underlying ganglion. The location of the hole was determined according to the course of the L5or L4 spinal nerve and was approximately 2 mm from the inferior edge of the transverse process. Before the surgery, a 25-gauge needle (0.5 mm in diameter) was cut to a length less than the thickness of the transverse process (2-3 mm in length) and was forced into the hole in close proximity to the DRG but without contracting the ganglion. Then, 20 μl of freshly prepared zymosan (0.5 μg/μl; 10 μg total, in incomplete Freund's adjuvant) was slowly injected into the hole through the inserted needle. The needle with attached syringe was left in place for at least 3 min after the injection to ensure complete delivery of the solution and avoid possible extravasations outside the DRG site.
Acute culture of sensory neurons
DRG were isolated from the animals on post-operative day (POD) 3-6, a time period during which the behavioral measurements of mechanical hyperalgesia (von Frey test) and mechanical allodynia (cotton wisp test) have reached their maximum values and are not changing rapidly.22 The rats were deeply anesthetized by pentobarbital sodium (40 mg/kg, i.p.). The ipsilateral DRGs (L4 and L5) were isolated and carefully removed in ice-cold normal Ringer solution for cleaning. The solution contained (in mM) 130 NaCl, 3 KCl, 2 CaCl2, 2 MgSO4, 1.25 KH2PO4, 26.2 NaHCO3 and 10 Dextrose, and was bubbled with 95% O2 and 5% CO2. The pH of the solution was adjusted to 7.4 and the osmolarity was ∼290 - 310 mOsm. The connective tissue of the DRGs was dissolved by exposure to Ca2+-free normal Ringer solution containing 1.0% collagenase II (265 u/mg, Worthington Biochemical Corp., Lakewood, NJ, USA) for 30 min at 37°C followed by rinsing in normal Ringer solution for another 10 min. The DRG cells were then dissociated by trituration with a fire-polished Pasteur pipette and were plated onto poly-D-lysine coated glass coverslips in MEM-BS containing 10% heat-inactivated fetal bovine serum (FBS) and 1,000 U/ml each of penicillin and streptomycin. The DRG cells were incubated at 37°C (5% CO2 balance air) before recording.
Electrophysiological recording
After short-term culture (2 – 30 hours), coverslips were transferred to a recording chamber and DRG cells were visualized under differential interference contrast using an inverted microscope (IX71, Olympus America Inc., Center Valley, PA, USA). Whole cell voltage-clamp recordings of small DRG neurons (diameter 9 to 25 μm) were conducted at room temperature with an AxoPatch-200B amplifier (Axon Instruments, Foster City, CA, USA). Patch pipettes (2.5-4.0 MΩ) were fabricated from borosilicate glass (Sutter Instruments, Novato, CA). The recording chamber was continuously perfused at room temperature with oxygenated bath solution at a flow rate of 2 ml/min. Data were acquired on a Pentium IV computer with the Clampex 8 program (Axon Instruments, Foster City, CA, USA). The cell capacitance artifact was canceled by the nulling circuit of the recording amplifier. Ohmic leakage currents were subtracted offline based on the input resistance measured during voltage steps in the hyperpolarizing range where no voltage-gated currents were evoked. Voltage errors were minimized by using ≥80% series resistance compensation. The current was filtered at 5 kHz and sampled at 50 kHz. All chemicals were purchased from Sigma (Sigma Chemical Co., St. Louis, MO, USA) unless otherwise indicated.
K+ current and action potential parameters
Excitability measurements were made under current clamp conditions, followed by voltage clamp measurements of voltage activated K+ currents using the same internal solution. After obtaining the whole cell configuration, only neurons that had a stable membrane potential more than −45 mV were used for further study. Excitability measurements included the threshold current (rheobase), action potential (AP) threshold, resting membrane potential (Vm), AP maximum rising rate (mV/ms), AP maximum falling rate (mV/ms), amplitude and duration of afterhyperpolarization and input resistance. Vm was measured 1 min after a stable recording was obtained. Current pulses from −0.2 to 0.37 nA (80-ms pulse duration) were delivered in increments of 0.03 nA until one or more APs were evoked. The threshold current (rheobase) was defined as the minimum current required to evoke an AP. The AP voltage threshold was defined as the first point on the upstroke of an AP.26 The AP amplitude was measured between the peak and AP threshold level. The AP rising rate was defined as the maximum rising rate from baseline to the AP peak and the AP falling rate was defined as the maximum falling rate from the AP peak to the afterhyperpolarization valley peak.27 The afterhyperpolarization amplitude was measured between the maximum hyperpolarization and the final plateau voltage, and the afterhyperpolarization duration was measured at the voltage half way between these two points.28,29 The input resistance for each cell was obtained from the slope of a steady-state I-V plot in response to a series of hyperpolarizing currents of 80 ms duration, delivered in decreasing steps of 0.03 nA between 0 and −0.2 nA. Any cells without AP were excluded from the study. After measurement of the excitability parameters in current clamp mode, the amplifier was switched to whole cell voltage clamp mode for measurements of K+ currents. Total potassium current was measured by depolarizing voltage steps after a one-sec prepulse to −120 mV. The difference between the peak current and the current at the end of the pulse was used as a measure of the transient K+ current, and the current at the end of the pulse was used as a measure of the steady-state current. Voltage dependence of inactivation (h∞) was measured by test pulses to 20 mV following one second prepulses to voltages between −100 mV and 0 mV.
Na+ current measurement
For the sodium current study, we labeled cells with isolectin B4 (IB4) conjugated to FITC (1 μg/μl) for 30-60 min prior to the start of the recording session, in order to identify IB4-positive and IB4-negative neurons.30,31 This procedure has been previously shown not to affect the currents measured.32 Neurons showing a robust fluorescence signal were classified IB4-positive and those displaying no signal at all, IB4-negative. Any cells showing an intermediate or weak signal were excluded from the study. The fluorescence was not examined until after completion of current recordings from each cell; i.e., IB4 binding was not used as a criterion in selecting which cells to record. In some experiments, 100 nM TTX was added to the Na+ bath solution after currents were recorded in the absence of TTX. In most experiments the TTX-resistant currents were isolated from the TTX-sensitive currents by using a holding potential of −50 mV. Both TTX-resistant and TTX-sensitive current were activated by depolarizations from a holding potential of −80 mV. The amplitude of the TTX-sensitive current in each cell (Fig 6) was measured during depolarizations to −10 mV from −80 mV in these activation protocols, based on the distinct kinetic properties of the two currents. At this voltage, TTX-sensitive current was measured as the difference between the early peak current and the current 2 msec after the beginning of the depolarization, a time window in which the TTX-sensitive current essentially completely inactivates but TTX-resistant current changes little. Similar separation of the two components could sometimes be obtained by digitally subtracting the currents evoked from −50 mV (TTX-resistant) from the currents evoked from −80 mV (TTX-resistant plus TTX-sensitive). However, many cells especially from control animals had a very small TTX-sensitive component, and it was observed that in these cases the subtraction method gave difference currents that were dominated by residual TTX-resistant currents due to slight differences in inactivation of that current at −50 vs. −80, or due to small time-dependent changes. Hence the activation data for the TTX-sensitive current over the full voltage range (Fig. 6B) was obtained only from cells in which that was the predominant current and readily separated from the TTX-resistant current by the subtraction method.
Figure 6.
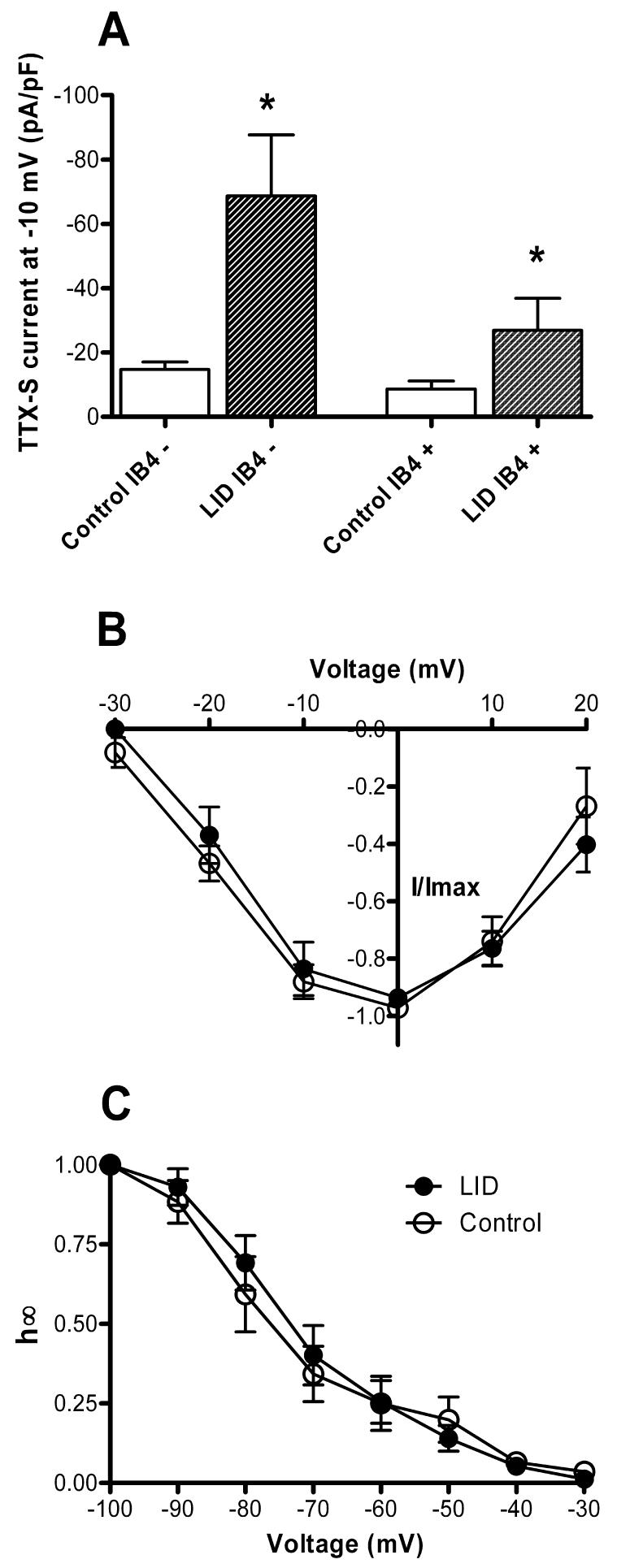
Effects of DRG inflammation on the TTX-sensitive Na+ current. A. Peak TTX-sensitive current density at −10 mV was significantly increased by LID in both IB4-positive and IB4-negative cells. In LID cells, the difference between IB-positive and IB4-negative cells approached significance (p = 0.06). B. Voltage dependence of activation, plotted as in Fig. 5-C, was not affected by inflammation. C. Voltage dependence of steady-state inactivation, measured as in Fig. 5-D, was similar before and after inflammation.
Solutions for recording potassium current and action potential
The normal bath solution for recording excitability parameters contained (in mM) 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose. The pH was adjusted to 7.4 with NaOH, and the osmolarity adjusted to ∼300 - 310 mOsm with sucrose. The bath solution for recording K+ currents contained (in mM) 130 Choline Cl, 5 KCl, 1 MgCl2, 2 CoCl2, 10 HEPES and 10 glucose. The pH was adjusted to 7.4 with Tris-base, and the osmolarity was adjusted to ∼300 - 310 mOsm with sucrose. The pipette solution contained (in mM) 140 KCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, 2 Mg adenosine triphosphate and 1 Li guanosine triphosphate. The pH was adjusted to 7.2 with Tris-base, and osmolarity to ∼290 - 300 mOsm with sucrose. Voltages were not corrected for liquid junction potentials, which were estimated to be <10 mV in all cases.
Solutions for recording sodium currents
Sodium currents were measured under a reduced sodium gradient to ensure better voltage control. The Na+ bath solution contained (in mM) 30 NaOH, 110 tetramethyl ammonium chloride, 5 tetraethyl ammonium chloride, 2 MgCl2, 1 CaCl2, 0.1 CdCl2, 10 HEPES, and 10 glucose. The pH was adjusted to 7.4 with HCl, and the osmolarity was adjusted to ∼300 - 310 mOsm with sucrose. The pipette solution contained (in mM) 140 CsCl, 5 NaOH, 2 MgCl2, 1 CaCl2, 11 EGTA, 10 HEPES, 2 Mg adenosine triphosphate and 1 Li guanosine triphosphate. The pH was adjusted to 7.2 with CsOH, and the osmolarity to ∼290 - 300 mOsm with sucrose.
Data analysis and statistics
Data were analyzed using pClampfit 8.2 (Axon Instruments, Foster City, CA, USA)) and Origin 7 (OriginLab Corp., Northampton, MA, USA). Currents were normalized by cell capacitance. Data are expressed as means ± standard error of the mean (SEM). Differences in proportion of cells binding IB4 were examined with Fisher's exact test. Statistical significance of differences between average values in LID and control neurons were analyzed by Student's t-test or, for data that were not normally distributed, the Mann-Whitney rank sum test, as indicated, using SigmaStat Software (Systat Software, Inc., San Jose, CA, USA). In the case of multiple comparisons over a voltage range for activation or inactivation data, the data were analyzed by two-way repeated measures ANOVA (RM ANOVA) using SigmaStat software, with pairwise multiple comparison (Holm-Sidak method) to determine at which voltages the differences between LID and control cells were significant if an overall effect of LID was observed. Significance was ascribed for p < 0.05. Any results found to be significant with this method of analysis were re-analyzed at individual voltages with two-way ANOVA using both condition (LID vs. control) and time-in-culture (short, 2.5 – 8 h, vs. long, 18 – 30 h) as factors, to ensure that differences between LID and control values were still significantly different after time-in-culture was accounted for. Average culture times did not differ between cells from control and LID animals in any of the experiments. Results presented were obtained from 3 to 7 different cultures for each condition (control and LID), as indicated. In order to further ensure that significant findings were not the results of spurious differences between cultures, we reevaluated the primary significant results by performing t-tests on the average values for each culture, instead of on the values for individual cells. Despite the much smaller N values in these comparisons, the following effects of localized inflammation still approached or attained significance (p values are in parentheses following each value): the increase in amplitude of the steady state component (0.055) and fast component (0.09) of the K current at −100 mV; the increase in TTX-sensitive current at −20 mV in IB4-negative cells (0.006) and IB4-positive cells (0.19); and the reduced action potential threshold (0.005), and rheobase (0.01), but not the increase in action potential rising rate ( 0.89) or falling rate (0.35).
3. Results
LID increases excitability
Membrane properties were recorded in sixty acutely isolated small-diameter DRG neurons using the current-clamp configuration of the whole-cell patch clamp. As summarized in Table 1, several measures of excitability were increased in cells isolated from animals 3 – 6 days after LID, compared to cells isolated from control animals. The mean rheobase current was significantly smaller, and the voltage threshold was significantly more negative, for LID than for control neurons. In addition, both the maximum AP rising rate and falling rate were significantly faster for LID than for control neurons, though this effect was less robust when differences between different batches of cells was examined (see Methods). No significant difference was found between control and LID neurons in cell capacitance, resting membrane potential, input resistance, AP amplitude, AP duration, afterhyperpolarization amplitude, and afterhyperpolarization duration (Table 1).
Table 1.
Membrane properties of isolated control and LID neurons measured in whole cell patch clamp experiments
| Cell and AP properties | Control | LID |
|---|---|---|
| Number of Cells | 31 | 29 |
| Diameter, μm | 20.1 ± 3.1 | 19.8 ± 0.6 |
| Cell Capacitance, pF | 19.1 ± 1.1 | 17.6 ± 1.6 |
| Rest Potential, mV | −49.8 ± 1.2 | −48.4 ± 1.1 |
| Input Resistance, MΩ | 636.4 ± 58.4 | 689.1 ± 47.2 |
| AP Rheobase, pA | 148.6 ± 18.5 | 54.6 ± 6.0** |
| AP Threshold, mV | −20.2 ± 1.2 | −28.0 ± 1.0** |
| AP Rising Rate, mV/ms | 111.6 ± 5.1 | 128.1 ± 5.2* |
| AP Falling Rate, mV/ms | 87.0 ± 3.4 | 97.2 ± 3.0* |
| AP Amplitude, mV | 75.5 ± 1.9 | 74.3 ± 2.7 |
| AP Duration, ms | 3.5 ± 0.4 | 3.3 ± 0.3 |
| AHP Amplitude, mV | 13.7 ± 1.7 | 13.5 ± 1.5 |
| AHP Duration, ms | 9.6 ± 1.0 | 10.6 ± 0.9 |
Values are mean ± SEM. AP, action potential; AHP, afterhyperpolarization. LID: localized inflammation of the dorsal root ganglion.
P < 0.05
P < 0.01 (Student's t-test, significant differences between control and LID).
Voltage-gated K+ currents increase after LID
Voltage-activated K+ currents were recorded as shown in Fig. 1. To measure total potassium current, the membrane was held at −60 mV, and voltage steps were applied in 10 mV increments to a value of +60 mV after a 1s prepulse to −120 mV. The total K+ current was comprised of a sustained current as measured at the end of the 500 msec pulse, and a transient component which decayed during a 500 msec depolarizing pulse. In comparison with control neurons, LID neurons had significantly increased density of both the transient (overall p value = 0.009) and the sustained components (overall p value <0.001). Two-way repeated measure ANOVA indicated that the effect of LID was significant at individual voltages above 10 – 20 mV (Fig. 1 C and D). These effects were due to increased amplitude of the current, without any change in the voltage dependence of activation.
Figure 1.
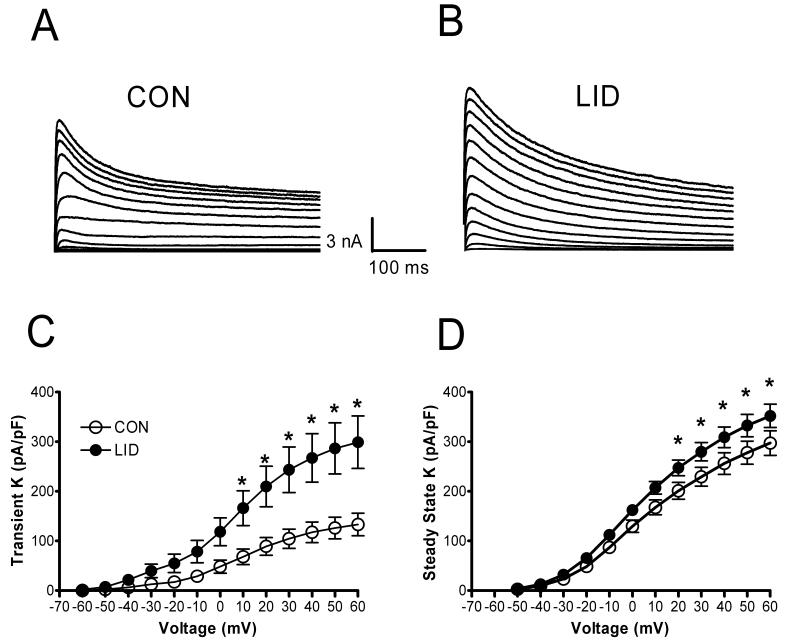
Voltage activated K+ currents increase after localized DRG inflammation. Currents shown were evoked by depolarizing pulses from −60 to + 60 mV. Examples are shown from a cell from a control animal (A) and from an animal after LID (B). The current density of both the transient component (C) and the value at the end of the pulse (D) were significantly increased in cells from LID animals. *, significant difference between LID and control cells (Two-Way RM ANOVA).
The voltage dependence of steady-state inactivation was also determined for the voltage activated K+ currents. The protocol consisted of a 1-s conditioning prepulse to potentials ranging between −100 and 0 mV followed by a depolarizing voltage step to a +20 mV test pulse for 1 second. The outward current at +20 mV could be well described in most cells as the sum of a sustained component and two exponentially decaying transient components whose time constants differed by an order of magnitude (Fig. 2). In five of 24 control cells and 3 of 26 LID cells, only a slow component was observed; in 3 of 26 LID cells only a fast component was observed. As shown in Fig. 2 B and C, the average values for the slow and fast time constants were unchanged by LID. The steady-state inactivation of each of these three components was studied individually by fitting the data as shown in Fig. 2. As shown in Fig. 3, the fast inactivating (Fig. 3B, E) component showed a simple increase in amplitude after LID (overall p value from two-way RM ANOVA = 0.0004), with no shift in the voltage dependence of inactivation (p = 0.124). Similar results were obtained for the steady-state component (Fig 3C, F): the increase in amplitude was significant (overall p value = 0.01), but the voltage dependence was not affected (p = 0.17). This analysis further confirms the increase in sustained current shown in figure 1D, since fitting the final steady-state value of the exponential decay should be less contaminated by the incompletely inactivated currents present at some voltages at the end of the 500 msec depolarizations shown in Fig. 1. Fig. 3 also demonstrates that the “steady-state” component as determined by fits to the current decay during 500 msec pulses can actually show some partial inactivation at more positive potentials, when measured with the 1 sec long prepulses used in this protocol.
Figure 2.
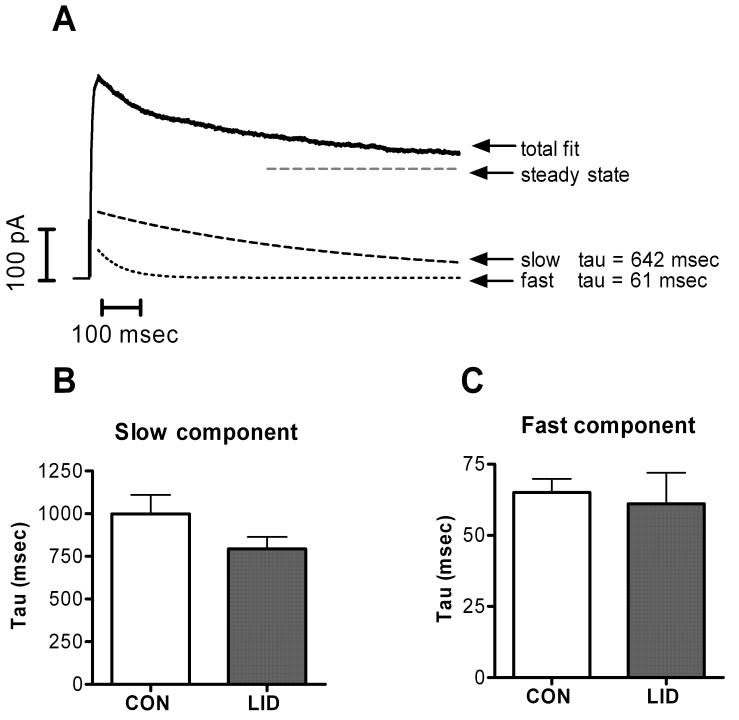
K+ currents decay with two exponential time constants. A. Sample trace showing current evoked during a step to +20 mV following a prepulse to −100 mV. The falling phase is fit with the sum of 2 exponentials plus a constant (steady-state) value. Individual components of the fit are also displayed. The average values of the slower time constant (B) and the faster time constant (C) were not significantly different after LID.
Figure 3.
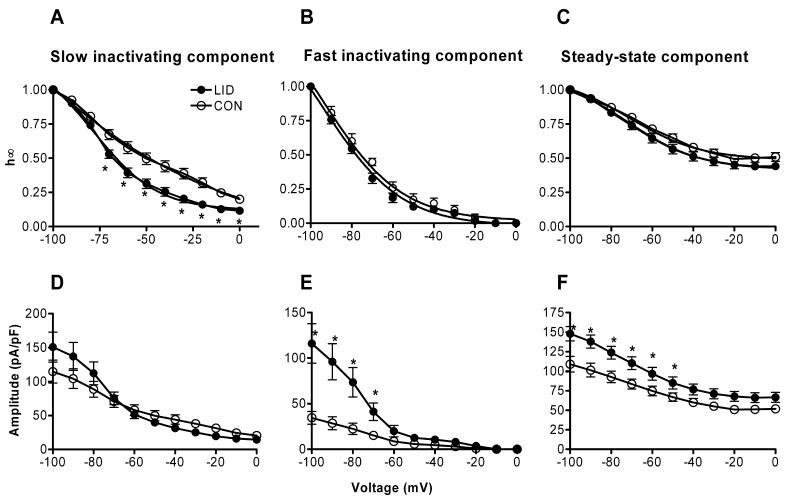
Effects of LID on the fitted 2 exponential fits to steady-state inactivation data. Currents during a pulse to +20 mV following a prepulse to the indicated voltage were fit with the sum of two exponentials plus a steady-state value, as shown in Fig. 2. The values of the amplitudes of each component are shown normalized to the value at −100 mV (top): A, slowly inactivating; B, rapidly inactivating; C, steady state component. The bottom panels show the absolute values of the fitted amplitudes D, slowly inactivating; E, rapidly inactivating; F, steady state component. *, significant difference between LID and control value (Two-Way RM ANOVA).
The effects on the slow inactivating component were more complex: although after LID there was a tendency for an increase in the maximum amplitude of the current evoked from most negative potential (Fig. 3D) (unadjusted p value = 0.04), the overall effect of LID on amplitude was not significant (overall p = 0.98). In addition, there was also a leftward shift of the inactivation curve (Fig. 3D) (overall p value = 0.001), which would tend to offset any increases in maximum amplitude. Due to this shift, the slow inactivating current density evoked from the more physiologically relevant value of −50 mV was 49.7 ± 7.5 pA/pF in control neurons vs. 38.2 ± 5.2 pA/pF in LID neurons, a difference that was not statistically significant.
Voltage-gated Na+ currents
Because the increased K+ currents could not account for the increased excitability observed after LID, we next examined the effects of LID on voltage-gated Na+ currents. Because previous studies showed differences between Na currents of the IB4-positive and IB4-negative subclasses of small diameter DRG neurons,32-34 for this study we determined whether the cell bound IB4. The fraction of IB4-positive cells (0.5) did not change significantly with LID (p = 0.8, Fisher's exact test).
Examples of Na+ currents are shown in Fig. 4. As described in many previous studies, the Na+ current (in both control and LID cells) included TTX-sensitive and TTX-resistant components. The TTX-resistant currents were distinguished by their incomplete steady-state inactivation at a holding potential of −50 mV, and by the slower time course of their inactivation during depolarizing pulses. Both before and after LID, and in both IB4-positive and IB4-negative cells, it was possible to observe cells with predominantly TTX-sensitive or predominantly TTX-resistant current. However, both before and after LID, the ratio of TTX-sensitive to total Na current (measured at −10 mV) was significantly higher in IB4-negative cells than in IB4-positive cells. In control cells this fraction was 0.70 ± 0.08 in IB4-negative cells vs. 0.36 ± 0.09 in IB4-positive cells (p = 0.008, Mann-Whitney rank sum test, T = 309, T = 308). In LID cells, this fraction was 0.30 ± 0.08 in IB4-negative cells vs. 0.08 ± 0.03 in IB4-positive cells (p = 0.007, Mann-Whitney rank sum test, T = 309). The time constants for decay of the currents during a depolarizing pulse were not significantly altered by LID (Fig. 4 D, E).
Figure 4.
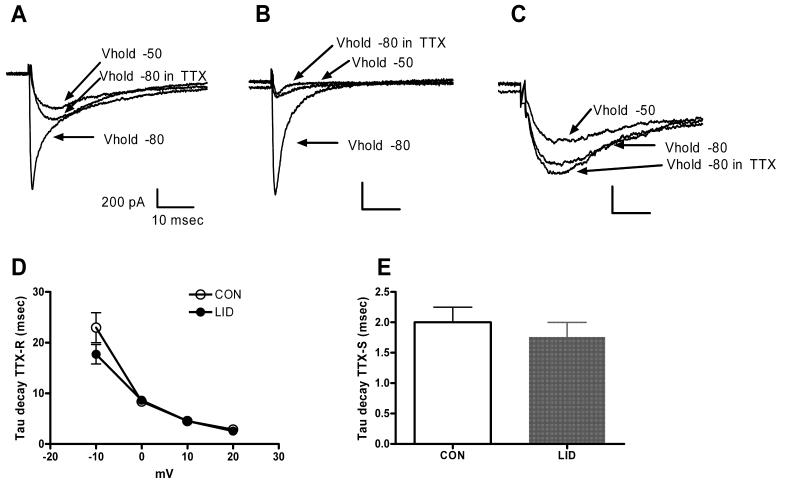
Na+ currents have TTX-sensitive and TTX-resistant components before and after LID. Currents shown are evoked by depolarization to −20 mV. A. Depolarization from −80 mV evokes a current with fast and slow inactivating components. The fast component is eliminated by TTX or by holding at −50 mV. Control cell, IB4-positive. B. Example of a cell with predominantly fast, TTX-sensitive current. Control cell, IB4-negative. C. Example of a cell with predominantly slow, TTX-resistant current. LID cell, IB4-negative. D. The time constant for decay of the TTX resistant current, measured during depolarization to the indicated voltage from a holding potential of −50 mV, was not significantly altered by LID. E. The time constant for decay of the TTX-sensitive component, measured during a depolarization to −20 mV from a prepulse potential of −100 mV, was not significantly altered by LID. Currents at −20 mV were generally well fit by the sum of two exponentials; the average tau shown is the average of the faster time constant in each fit. The faster decaying components were always blocked by TTX (n = 3 LID cells and 9 control cells.)
LID increases the amplitude of TTX-resistant Na+ currents
The amplitude of the TTX-resistant Na+ current, as measured from a holding potential of −50 mV which largely inactivates TTX-sensitive current, was significantly increased after LID. This increase was observed only in the IB4-positive cells (Fig. 5, A and B; overall p value = 0.017), where it was over 2-fold at 0 mV. In IB4-negative cells the current densities were not significantly different (p = 0.50). The increased amplitude occurred without any shift in the voltage dependence of activation (Fig. 5C; p = 0.54) or inactivation (Fig. 5D; p = 0.32). In figures 5C and 5D, the data from IB4-positive and IB4-negative cells have been combined as differences between the two cell-types were not observed for these parameters.
Figure 5.
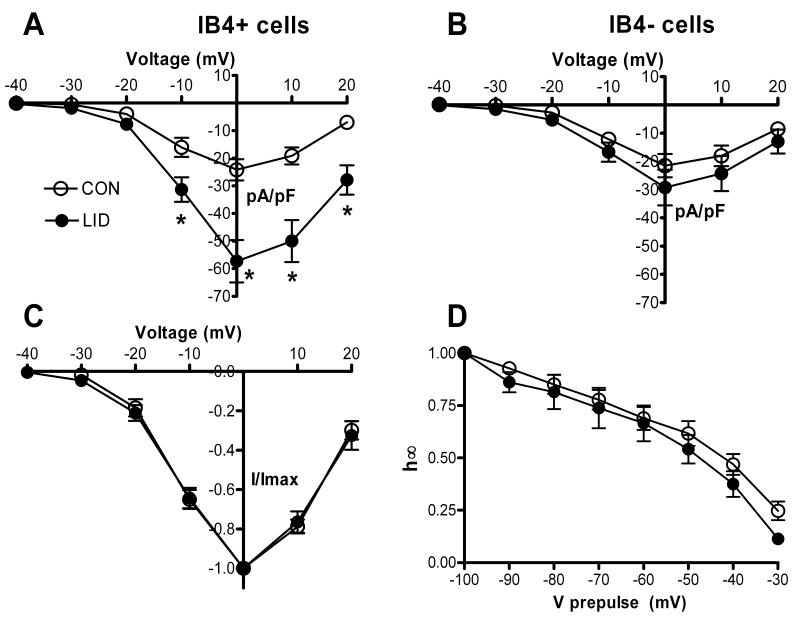
Effects of DRG inflammation on the TTX-resistant Na+ current. TTX-resistant current was isolated using a holding potential of −50 mV which largely inactivates the TTX-sensitive current. A. Peak TTX-resistant current density vs. voltage in IB4-positive cells. *, LID significantly different from control (Two-Way RM ANOVA). B. Peak TTX-resistant current density vs. voltage in IB4-negative cells. None of the differences was significant. C. Voltage dependence of activation, obtained by normalizing the data from each cell to the value at 0 mV. D. Steady-state inactivation vs. prepulse voltage. Currents were measured during a test pulse to −20 mV, and the TTX-sensitive and resistant currents were separated as described in methods. In C and D data from IB4-positive and IB4-negative cells are combined as there were no differences between the subgroup observed.
LID increases the amplitude of TTX-sensitive Na+ currents
The amplitude of the TTX-sensitive current, measured as described in Methods, was also markedly increased after LID (Fig. 6A). In contrast to the TTX-resistant current, the TTX-sensitive current markedly increased in both IB4-negative and IB4-positive cells. The voltage dependence of activation (Fig. 6 B) and inactivation (Fig. 6 C) were not significantly altered by LID. In Figures 6B and 6C, the data from IB4-positive and IB4-negative cells have been combined as differences between the two cell-types were not observed for these parameters.
Discussion
Excitability changes after LID
Our current clamp experiments demonstrated that several measures of excitability increased in small-diameter DRG neurons after localized inflammation. Hence, rheobase and action potential threshold were both reduced after LID. In addition, the rising and falling rates of the action potential increased. Increased excitability is a feature of virtually all animal models of chronic pain, whether they model nerve injury, peripheral inflammation, or compression of the DRG.35 It can be difficult to directly predict effects on excitability from voltage clamp measurements.36 However, the decreased rheobase and increased rising rate of the action potential seem at least qualitatively consistent with the observed increases in Na+ currents (particularly TTX-sensitive currents, which activate more quickly and at more negative potentials after LID). Although this study focuses on voltage-gated K+ and Na+ channels, it should be noted that other ion channels in sensory neurons may also play roles in chronic pain models, including the capsaicin receptor,37 hyperpolarization-activated cation channels,38 and voltage-gated calcium channels.39
Voltage-gated Na+ currents
The general characteristics of the TTX-sensitive and TTX-resistant Na+ currents described in this study are quite similar to those reported in many other patch clamp studies of small DRG neurons.40-43 As in most such studies, the TTX-resistant currents we observed had characteristics attributed to the Nav1.8 isoform. We did not routinely observe persistent Na+ currents that activate at very negative potentials and that are thought to be mediated by the Nav 1.9 channel, probably because of slow inactivation at the holding potentials used, and/or because the current may “washout” during whole cell patch clamp.44,45 In addition, the work of Murayama et al. 46 suggests that under our recording conditions (adenosine triphosphate in the pipette, Cl− rather and F− as the anion), the persistent current would have a very small magnitude.
Changes in Na+ channel expression or activity have been observed in other models of chronic pain. Nerve injury models generally lead to downregulation of TTX-resistant channels, with more complex effects on TTX-sensitive channels. For example, after sciatic nerve transection, there is an up-regulation of TTX-sensitive Nav 1.3 channels and a down-regulation of TTX-resistant Na+ channels and of some other isoforms of TTX-sensitive channels. In this case increased excitability is thought to result from redistribution of Na channels along the axon and into the neuroma, as well as from the lower threshold and higher firing frequencies permitted by the switch to the Nav 1.3 channel. Other nerve injury models such as spinal nerve ligation or chronic constriction injury also lead to reduced expression of TTX-resistant Na+ channels though the findings about TTX-sensitive currents are more variable (for review, see 8,47). In contrast, models of peripheral inflammation almost always lead to an upregulation of TTX-resistant current, although effects on the TTX-sensitive current are less consistent. In this regards, our results are perhaps most similar to those seen in the subcutaneous carrageenan injection model, in which both TTX-sensitive and TTX-resistant currents (the latter due to increased Nav 1.8 but not 1.9) are upregulated without shifts in voltage dependence of activation and inactivation.48,49 These results are quite similar to ours except that the magnitude of the changes was much larger in our experiments – e.g., 2 to 3-fold increase in TTX-sensitive current after LID vs. 40% increase after peripheral carrageenan, and 90% overall increase in TTX-resistant current after LID vs. 55% after peripheral carrageenan. This may be simply due to the fact that only a subset of L4 and L5 neurons should be affected by peripheral carrageenan, whereas all the neurons in the LID model experience inflammation. Effects of peripheral inflammation on the TTX-sensitive current are less consistent - particularly in models of visceral pain, there are examples of no increase or even decreases of TTX-sensitive current after inflammation 15,50,51 though most of these studies do report increases of varying magnitude of the TTX-resistant current. The reasons for these differences between these various models are not yet clear. One possible explanation is that the visceral pain studies looked at a longer time point – 7 to 10 days instead of 3 – 5 days in our study and the carrageenan studies; perhaps the effect on TTX-sensitive current is transient. Another possibility is that our study examined both cutaneous and visceral afferent neurons which are mixed in the L4 and L5 DRG; if the increase in TTX-sensitive current occurs only in cutaneous neurons, the differences between the above cited studies could be accounted for.
The changes in Na+ channels in nerve injury and inflammation models discussed above have in general been observed at the level of mRNA and protein as well as in functional studies, and are generally thought to reflect changes in channel expression. However, posttranslational modifications and alternative splicing have also been proposed as additional possible mechanisms.47 These mechanisms are distinct from the rapid, reversible increase in Na+ current caused by inflammatory mediators such as prostaglandins and bradykinin. Such effects, if present in the LID model in vivo, would not have been preserved in the acute culture methods used by this study and others cited above.
Small sensory neurons that are primarily nociceptors can be divided neurochemically into two populations: IB4-positive, primarily nonpeptidergic neurons, and IB4-negative peptidergic neurons. It has been shown that IB4-positive neurons depend on glial-derived neurotrophic factor, whereas IB4-negative neurons depend on nerve growth factor for survival during postnatal development.52 Furthermore, these two populations of nociceptors terminate in distinct regions of the superficial spinal cord. Our finding that the IB4-negative cells had a higher expression of TTX-sensitive current than did IB4-positive cells is in general agreement with previous studies. In our study, TTX-resistant currents with the characteristics of Nav1.8 were commonly observed in both IB4-positive and IB4-negative cells, also in general agreement with previous electrophysiological and protein or mRNA expression studies.32-34 In our study the increase in TTX-resistant current following DRG inflammation was confined to IB4-positive cells, suggesting that this may be an important variable to consider when conducting such studies.
Voltage-gated K+ currents
The voltage-gated K+ currents described in this study are quite similar to those reported in numerous other patch clamp studies of acutely isolated small neurons, which have described sustained (or delayed rectifier) currents and transient currents with fast inactivation (IA) and slow inactivation (ID) during a depolarizing test pulse.53-57 Calcium-activated K+ currents also present in these cells 58 were not examined in the present study due to the presence of EGTA in the recording pipette and Co2+ replacing Ca2+ in the extracellular solution.
A number of studies of voltage-gated K+ current changes in acutely isolated, small DRG neurons after peripheral inflammation or nerve injury have been reported, in different laboratories and animal models. These studies differ markedly from ours in that K+ currents were increased in our study, but are reduced in most studies involving either peripheral inflammation or nerve injury. A common finding is the functional reduction of IA or ID due to a reduced total conductance and/or to a leftward shift in steady-state inactivation that reduces channel availability at rest. This has been observed in several models of visceral inflammation as well as in the axotomy model of nerve injury. In some but not all of these models the sustained current is also reduced.2,9-15 In contrast, we observed a 36% increase in the maximum magnitude (i.e., that evoked from a holding potential of −100 mV) of the sustained current, with comparable and significant increases at −60 mV and −50, closer to the physiological resting potential. The fast inactivating component increased in maximum amplitude after LID, also in contrast to most other models. As in some other studies, we observed a leftward shift in steady state inactivation of the slow inactivating component, however, in effect this was apparently offset by small increases in magnitude such that there was no significant effect on this current at physiological resting potentials.
Our excitability data indicate that the overall effect of the increases in both K+ and Na+ currents after LID is to increase excitability, which is in agreement with other models. A possible explanation for the differences in K+ channel regulation between our study and other chronic pain models may lie in the magnitude of the Na+ current changes observed. Although our findings of increased Na+ current density are qualitatively similar to the results obtained in many models of peripheral inflammation, the magnitude of the changes is generally much larger in our LID model. It may be that basic intrinsic mechanisms regulating overall excitability of the neuron 59 dictate an increase in K+ currents to partially compensate for the very large increases in Na+ currents. In addition, the LID model may expose cells to different types or higher concentrations of the regulatory molecules such as cytokines and growth factors that may regulate the ion channels.22 In this vein it is interesting that 24 hour exposure to the pro-inflammatory cytokine IL-1β can increase TTX-sensitive Na+ currents by 67% in trigeminal nociceptors.60 In preliminary experiments we have also observed marked upregulation of both TTX-sensitive and TTX-resistant Na+ currents after overnight exposure to pro-inflammatory cytokines at concentrations similar to those observed in vivo after LID.
Summary
This study demonstrates that localized inflammatory irritation of lumbar DRG increases the magnitude of both voltage-gated K+ and Na+ currents. The effect on Na+ currents is similar to that seen in some models of peripheral inflammation, except in being particularly large, but the finding of increased K+ current is rather unique to this model. Measurements of action potential parameters in current clamp mode indicated that the overall effect was increased excitability. Thus these ion channel changes may help account for the pain and hyperalgesia associated with LID.
Acknowledgements
This work was supported in part by National Institute of Health (Bethesda, MD, USA) grants NS39568 and NS45594 (Jun-Ming Zhang), and University of Cincinnati Millennium Fund (Cincinnati, OH, USA).
Footnotes
Summary statement: Localized inflammation of the dorsal root ganglia in rat lead to increases in voltage-gated potassium and sodium current densities, and an overall increase in excitability, in acutely isolated small diameter sensory neurons.
Meeting presentation: Part of the work was presented on October 16, 2006 at the ASA annual meeting in Chicago, IL, USA.
References
- 1.Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- 2.Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. J Neurophysiol. 2001;85:644–658. doi: 10.1152/jn.2001.85.2.644. [DOI] [PubMed] [Google Scholar]
- 3.Kim YI, Na HS, Kim SH, Han HC, Yoon YW, Sung B, Nam HJ, Shin SL, Hong SK. Cell type-specific changes of the membrane properties of peripherally-axotomized dorsal root ganglion neurons in a rat model of neuropathic pain. Neuroscience. 1998;86:301–309. doi: 10.1016/s0306-4522(98)00022-0. [DOI] [PubMed] [Google Scholar]
- 4.Moore BA, Stewart TM, Hill C, Vanner SJ. TNBS ileitis evokes hyperexcitability and changes in ionic membrane properties of nociceptive DRG neurons. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1045–1051. doi: 10.1152/ajpgi.00406.2001. [DOI] [PubMed] [Google Scholar]
- 5.Song XJ, Zhang J-M, Hu SJ, LaMotte RH. Somata of nerve-injured sensory neurons exhibit enhanced responses to inflammatory mediators. Pain. 2003;104:701–709. doi: 10.1016/S0304-3959(03)00167-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J-M, Song XJ, LaMotte RH. Enhanced excitability of sensory neurons in rats with cutaneous hyperalgesia produced by chronic compression of the dorsal root ganglion. J Neurophysiol. 1999;82:3359–3366. doi: 10.1152/jn.1999.82.6.3359. [DOI] [PubMed] [Google Scholar]
- 7.Millan MJ. The induction of pain: An integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 8.Waxman SG, Cummins TR, Dib-Hajj S, Fjell J, Black JA. Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve. 1999;22:1177–1187. doi: 10.1002/(sici)1097-4598(199909)22:9<1177::aid-mus3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Takeda M, Tanimoto T, Ikeda M, Nasu M, Kadoi J, Yoshida S, Matsumoto S. Enhanced excitability of rat trigeminal root ganglion neurons via decrease in A-type potassium currents following temporomandibular joint inflammation. Neuroscience. 2006;138:621–630. doi: 10.1016/j.neuroscience.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 10.Dang K, Bielefeldt K, Gebhart GF. Gastric ulcers reduce A-type potassium currents in rat gastric sensory ganglion neurons. Am J Physiol Gastrointest Liver Physiol. 2004;286:G573–579. doi: 10.1152/ajpgi.00258.2003. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimura N, de Groat WC. Increased excitability of afferent neurons innervating rat urinary bladder after chronic bladder inflammation. J Neurosci. 1999;19:4644–4653. doi: 10.1523/JNEUROSCI.19-11-04644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang EK, Takimoto K, Hayashi Y, de Groat WC, Yoshimura N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience. 2004;123:867–874. doi: 10.1016/j.neuroscience.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Harriott AM, Dessem D, Gold MS. Inflammation increases the excitability of masseter muscle afferents. Neuroscience. 2006;141:433–442. doi: 10.1016/j.neuroscience.2006.03.049. [DOI] [PubMed] [Google Scholar]
- 14.Xu GY, Winston JH, Shenoy M, Yin H, Pasricha PJ. Enhanced excitability and suppression of A-type K+ current of pancreas-specific afferent neurons in a rat model of chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G424–431. doi: 10.1152/ajpgi.00560.2005. [DOI] [PubMed] [Google Scholar]
- 15.Stewart T, Beyak MJ, Vanner S. Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons. J Physiol. 2003;552:797–807. doi: 10.1113/jphysiol.2003.046409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taskinen HS, Roytta M. Increased expression of chemokines (MCP-1, MIP-1alpha, RANTES) after peripheral nerve transection. J Peripher Nerv Syst. 2000;5:75–81. doi: 10.1046/j.1529-8027.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Kleinschnitz C, Brinkhoff J, Zelenka M, Sommer C, Stoll G. The extent of cytokine induction in peripheral nerve lesions depends on the mode of injury and NMDA receptor signaling. J Neuroimmunol. 2004;149:77–83. doi: 10.1016/j.jneuroim.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- 20.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 21.Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Brain Res Rev. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Xie WR, Deng H, Li H, Bowen TL, Strong JA, Zhang J-M. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflammatory irritation of the spinal ganglia. Neuroscience. 2006;142:809–822. doi: 10.1016/j.neuroscience.2006.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi S, Yamashita T, Yokogushi K, Murakami T, Ohwada O, Sato N. Immunophenotypic analysis of the inflammatory infiltrates in herniated intervertebral discs. Spine. 2001;26:1209–1214. doi: 10.1097/00007632-200106010-00008. [DOI] [PubMed] [Google Scholar]
- 24.Satoh K, Konno S, Nishiyama K, Olmarker K, Kikuchi S. Presence and distribution of antigen-antibody complexes in the herniated nucleus pulposus. Spine. 1999;24:1980–1984. doi: 10.1097/00007632-199910010-00003. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J-M, Homma Y, Ackerman WE, Brull SJ. Topical application of acidic bupivacaine to the lumbar ganglion induces mechanical hyperalgesia in the rat. Anesth Analg. 2001;93:466–471. doi: 10.1097/00000539-200108000-00045. [DOI] [PubMed] [Google Scholar]
- 26.Anderson P, Storm J, Wheal HV. Thresholds of action potentials evoked by synapses on the dendrites of pyramidal cells in the rat hippocampus in vitro. J Physiol (Lond) 1987;383:509–526. doi: 10.1113/jphysiol.1987.sp016425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu K, Kocsis JD. Schwann cell engraftment into injured peripheral nerve prevents changes in action potential properties. J Neurophysiol. 2005;94:1519–1527. doi: 10.1152/jn.00107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J-M, Donnelly DF, Song XJ, Lamotte RH. Axotomy increases the excitability of dorsal root ganglion cells with unmyelinated axons. J Neurophysiol. 1997;78:2790–2794. doi: 10.1152/jn.1997.78.5.2790. [DOI] [PubMed] [Google Scholar]
- 29.Tan ZY, Donnelly DF, LaMotte RH. Effects of a chronic compression of the dorsal root ganglion on voltage-gated Na+ and K+ currents in cutaneous afferent neurons. J Neurophysiol. 2006;95:1115–1123. doi: 10.1152/jn.00830.2005. [DOI] [PubMed] [Google Scholar]
- 30.Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res Mol Brain Res. 1999;67:267–282. doi: 10.1016/s0169-328x(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 31.Rush AM, Craner MJ, Kageyama T, Dib-Hajj SD, Waxman SG, Ranscht B. Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive sodium channels in DRG neurons. Eur J Neurosci. 2005;22:39–49. doi: 10.1111/j.1460-9568.2005.04186.x. [DOI] [PubMed] [Google Scholar]
- 32.Wu ZZ, Pan HL. Tetrodotoxin-sensitive and -resistant Na+ channel currents in subsets of small sensory neurons of rats. Brain Res. 2004;1029:251–258. doi: 10.1016/j.brainres.2004.09.051. [DOI] [PubMed] [Google Scholar]
- 33.Ogata N, Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn J Pharmacol. 2002;88:365–377. doi: 10.1254/jjp.88.365. [DOI] [PubMed] [Google Scholar]
- 34.Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- 35.Devor M. Response of nerves to injury in relation to neuropathic pain. In: McMahon SB, Koltzenburg M, editors. Textbook of Pain. 5th Edition Elsevier; Churchill Livingstone: 2006. pp. 905–27. [Google Scholar]
- 36.Schild JH, Kunze DL. Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J Neurophysiol. 1997;78:3198–3209. doi: 10.1152/jn.1997.78.6.3198. [DOI] [PubMed] [Google Scholar]
- 37.Krause JE, Chenard BL, Cortright DN. Transient receptor potential ion channels as targets for the discovery of pain therapeutics. Curr Opin Investig Drugs. 2005;6:48–57. [PubMed] [Google Scholar]
- 38.Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. 2003;23:1169–1178. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao YQ. Voltage-gated calcium channels and pain. Pain. 2006;126:5–9. doi: 10.1016/j.pain.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 40.Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci. 1992;12:2104–2111. doi: 10.1523/JNEUROSCI.12-06-02104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592:283–297. doi: 10.1016/0006-8993(92)91687-a. [DOI] [PubMed] [Google Scholar]
- 43.Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19:RC43 (41–46). doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coste B, Osorio N, Padilla F, Crest M, Delmas P. Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol Cell Neurosci. 2004;26:123–134. doi: 10.1016/j.mcn.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Maruyama H, Yamamoto M, Matsutomi T, Zheng T, Nakata Y, Wood JN, Ogata N. Electrophysiological characterization of the tetrodotoxin-resistant Na+ channel, Na(v)1.9, in mouse dorsal root ganglion neurons. Pflugers Arch. 2004;449:76–87. doi: 10.1007/s00424-004-1315-0. [DOI] [PubMed] [Google Scholar]
- 47.Rogers M, Tang L, Madge DJ, Stevens EB. The role of sodium channels in neuropathic pain. Semin Cell Dev Biol. 2006;17:571–581. doi: 10.1016/j.semcdb.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka M, Cummins TR, Ishikawa K, Dib-Hajj SD, Black JA, Waxman SG. SNS Na+ channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. Neuroreport. 1998;9:967–972. doi: 10.1097/00001756-199804200-00003. [DOI] [PubMed] [Google Scholar]
- 49.Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain. 2004;108:237–247. doi: 10.1016/j.pain.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 50.Beyak MJ, Ramji N, Krol KM, Kawaja MD, Vanner SJ. Two TTX-resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis-induced hyperexcitability. Am J Physiol Gastrointest Liver Physiol. 2004;287:G845–855. doi: 10.1152/ajpgi.00154.2004. [DOI] [PubMed] [Google Scholar]
- 51.Bielefeldt K, Ozaki N, Gebhart GF. Mild gastritis alters voltage-sensitive sodium currents in gastric sensory neurons in rats. Gastroenterology. 2002;122:752–761. doi: 10.1053/gast.2002.31901. [DOI] [PubMed] [Google Scholar]
- 52.Molliver DC, Snider WD. Nerve growth factor receptor TrkA is down-regulated during postnatal development by a subset of dorsal root ganglion neurons. J Comp Neurol. 1997;381:428–438. doi: 10.1002/(sici)1096-9861(19970519)381:4<428::aid-cne3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 53.Everill B, Rizzo MA, Kocsis JD. Morphologically identified cutaneous afferent DRG neurons express three different potassium currents in varying proportions. J Neurophysiol. 1998;79:1814–1824. doi: 10.1152/jn.1998.79.4.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akins PT, McCleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993;56:759–769. doi: 10.1016/0306-4522(93)90372-m. [DOI] [PubMed] [Google Scholar]
- 55.Takeda M, Tanimoto T, Ikeda M, Kadoi J, Matsumoto S. Activaton of GABAB receptor inhibits the excitability of rat small diameter trigeminal root ganglion neurons. Neuroscience. 2004;123:491–505. doi: 10.1016/j.neuroscience.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Yoshida S, Matsumoto S. Effects of alpha-dendrotoxin on K+ currents and action potentials in tetrodotoxin-resistant adult rat trigeminal ganglion neurons. J Pharmacol Exp Ther. 2005;314:437–445. doi: 10.1124/jpet.105.084988. [DOI] [PubMed] [Google Scholar]
- 57.Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996;75:2629–2646. [Google Scholar]
- 58.Mongan LC, Hill MJ, Chen MX, Tate SN, Collins SD, Buckby L, Grubb BD. The distribution of small and intermediate conductance calcium-activated potassium channels in the rat sensory nervous system. Neuroscience. 2005;131:161–175. doi: 10.1016/j.neuroscience.2004.09.062. [DOI] [PubMed] [Google Scholar]
- 59.Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. Bioessays. 2002;24:1145–1154. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- 60.Liu L, Yang TM, Liedtke W, Simon SA. Chronic IL-1beta signaling potentiates voltage-dependent sodium currents in trigeminal nociceptive neurons. J Neurophysiol. 2006;95:1478–1490. doi: 10.1152/jn.00509.2005. [DOI] [PubMed] [Google Scholar]