

Abstract
The goal of this review is to present a rationale for use of exogenous H2O2, which has been demonstrated to have both toxicological and physiological signaling roles. Reasons for the use of exogenous application of non-toxic concentrations of H2O2 in model systems and caveats for interpretation of the data obtained will both be presented. Briefly, an argument for the cautious use of the addition of exogenous H2O2 is that, because of the permeability of cell membranes to this neutral small molecule, a concentration that is produced locally that is necessary for the physiological action can be mimicked. On the other hand, it must be recognized that the addition of an agent or its enzymatic generation in the media may produce reactions that may not normally occur because the total dose of H2O2 and the concentration of H2O2 in some cellular locations will exceed what is normally achieved even under a pathophysiological state. For this reason, this review will try to provide an unbiased balanced pros and cons analysis of this issue.
Keywords: Hydrogen peroxide, signal transduction, PTP1B, thioredoxin, kinetics, protein tyrosine phosphatase, ASK1, redox signaling
Chemistry
In a modern version of vitalism (the belief that life processes are exempt from the laws of physics and chemistry) there are members of the scientific community who deny the value of extrapolating in vitro experiments to biology and categorize the use of exogenous addition of agents in studies in cell culture as having little value in studying physiology. Certainly, there are differences between having the cell generate an agent and adding it as there are between exposing cells to a an agent in a stationary medium and delivery and removal of metabolites by circulation. Nonetheless, chemical principles apply to every biological reaction and rejection of model studies because they do not truly represent a physiological state ignores the advantage of situations in which variables can be better controlled and measurements made in real time. Obviously, not every reaction that occurs in model systems even under conditions that approximate physiologically relevant conditions will occur in vivo. For example, many reactions that can occur at pH 7 and 37 °C may be inherently too slow on a biological time scale to compete with a much faster enzymatic process to be significant. But, in some cases, as will be described here, addition of an exogenous compound is a reasonable way in which to approximate physiological generation of the compound. Thus, there needs to be a balance in extrapolating the information one can obtain from model systems, and from test tube enzymatic and non-enzymatic reactions as well to physiology and pathophysiology. This review will attempt to provide such a balanced view, using examples from the literature and from the author’s laboratory. Much of the conceptual framework, however, comes from principles that are found in physical chemistry textbooks.
Models of oxidative stress in cell culture
Endogenous generation
H2O2 can be generated directly in cells by some oxidoreductases, such as glucose oxidase [1], and the recently described DuOXs [2], which are isoforms of the NADPH oxidases. Most H2O2 production however, results from the dismutation of O2•− produced by most NADPH oxidases [2], leak from mitochondrial electron transport chain [3, 4], redox cycling of xenobiotic quinones [5] and several flavoproteins [1]. Which of these sources are involved in signaling is currently a matter of intense research and should be addressed in any study in which the effects of exogenous H2O2 are being extrapolated to cellular production of H2O2.
With the rapidly increasing interest in how H2O2 and other electrophiles participate in signaling, the question of how to obtain mechanistic data when the generation of the reactive species is very small is problematic. Ideally, the endogenous generation of the reactive species could be regulated and monitored. This method is however often limited by an inability to accurately measure production. When O2•− is generated on the external surface of the cell, such as occurs with phagocytes and many other cells types [6], accurate quantitative measurement can be made using superoxide dismutase inhibitable ferricytochrome c reduction and used to estimate H2O2 production that would result from non-enzymatic dismutation [7]. Extracellular H2O2 production can also be measured using horseradish peroxidase catalyzed oxidation of compounds that increase or decrease in fluorescence [8]. But, when hydrogen peroxide is generated inside the cell, measurement is often attempted using 2′,7′-dichlorofluorescin (DCFH) oxidation, which is at best an approximation and frequently erroneous because reactions that are not dependent upon H2O2 can also result in DCFH oxidation [9, 10]. Methods such as aminotriazole-dependent inactivation of catalase [11] and others [8] that have greater specificity and reliability are not as convenient as DCF fluorescence. This has produced an unfortunate proliferation of publications whose claims about the involvement of H2O2 are not as illuminating as the small amount of light emitted from DCF.
Exogenous addition
Although this review advocates the cautious use of exogenous addition of non-toxic concentrations of H2O2 or its generation by enzymatic systems such as glucose oxidase in the media, where feasible those studies should be coordinated with studies in which the physiological generation of H2O2 can be demonstrated directly or indirectly to produce the same response. As stated above however, it is not always possible to directly measure H2O2 production in cells. Indirect methods, such as overexpression of catalase or another peroxidase, is not always feasible and runs the risk of producing its own physiological effect as well. The advantage of the combining a variety of approaches is obvious as no one approach is without fault.
Clearly, the addition of H2O2 should be done within the bounds of what is reasonable to mimic either a physiological or pathophysiological process rather than to simply make sure something happens. Indeed, the latter situation, which the author named the “thermonuclear model of toxicology” almost two decades ago [12] has declined in the literature and is clearly not advocated here. In contrast, as recent reviews (see [6, 13–16] for example) have pointed out, H2O2 is clearly part of normal cell signaling that is involved in responses including cell replication, regulation of metabolism, specific gene induction, and apoptosis. In many instances these physiological phenomena have been first demonstrated using the exogenous addition of H2O2 and only later, and sometimes not, demonstrated with the endogenous generation of H2O2. While some nearly three-decade-old literature suggested the involvement of H2O2 in signaling by insulin [17], the current wave of interest largely stems from the demonstration that NF-κB and HIV-1 could be activated by the addition of micromolar H2O2 to T lymphocytes [18]. It was not until a few years later that the demonstration that macrophages could be stimulated to produce H2O2, which then activated NF-κB, provided one of the first clear demonstrations of actual physiological signaling by H2O2 [19]. Nonetheless, it was not until Lambeth and coworkers identified the NADPH oxidases from non-phagocytic cells [20], did the source of endogenous H2O2 have much consideration aside from the macrophage and other phagocytes. Indeed, a prominent review published that same year noted that many physiological responses seemed to involve H2O2 production but that the source was unknown [21]. So, not surprisingly, most studies of the role of H2O2 in signaling used exogenous addition of H2O2.
Today, studies using exogenous H2O2 are sometimes criticized because of the assertion that exogenous addition of H2O2 is always non-physiological. Some investigators have pointed out that environmental exposure, such as to cigarette smoke, can contain H2O2 at concentrations high enough to produce signaling events [22]. It has also been pointed out that H2O2 produced by phagocytes can be used in signaling to other cells, which is relevant in inflammation and other physiology [23]. Thus, there is precedent from biologically relevant conditions for the use of exogenous H2O2.
Oxidants can act as second messengers
Clearly, use of exogenous added or generated H2O2 will not precisely mimic every physiological situation in which H2O2 is involved but further consideration of how H2O2 acts in signaling should shed light on when such models are appropriate. This review focuses on H2O2 in signal transduction where it acts as a second messenger. Second messengers have five essential characteristics: (1) increases in their concentration occur either through enzymatic generation or regulated release into the cytosol from sites of higher concentration through channels; (2) decreases in their concentration occur through enzymatic degradation, or the restoration of the concentration gradients by the action of pumps, or through diffusion from the cell that may be enhanced by reaction or binding of the second messenger in another cell; (3) their intracellular concentration rises and falls within a short period; (4) they are specific in action; and (5) gradients of their concentration from their origin to where they are either degraded or sequestered determines where they are effective.
The real estate rule
The fifth characteristic above has been referred to as the real estate rule as location is the principle rule in that enterprise as well. Another way of looking at the characteristics that determine whether or not a second messenger is effective are how rapidly it is produced, how rapidly it is removed, and the concentration it must reach to alter the activity of its target effector. The latter is determined by the rate of association (or reaction) of the second messenger with the effector minus the rate of dissociation of the second messenger (or reversal of the reaction) in competition with binding (or reactions) of the second messenger with other proteins, including enzymes that reduce its concentration. As these reactions or associations are second order as they are dependent upon both effector and the second messenger concentrations, the higher the concentration of either, the more likely it is that their interaction will occur. Therefore, the closer an effector is to the site of second messenger synthesis (or release for second messengers such as calcium), the more likely it will interact with that second messenger. This is particularly important when the competing reactions are very rapid. Thus, for H2O2 to play a direct role in signaling, its target(s) must be localized near its site of production due to the high cellular activity and rate constants of glutathione peroxidase, catalase and other enzymes that eliminate H2O2. If H2O2 acts indirectly however, for example through the generation of a lipid peroxidation product, then the real estate rule does not necessarily apply. Nonetheless, this review focuses upon the direct action of H2O2 on signaling proteins.
Another aspect of the real estate analogy is timing. Just as the sale of a home in a great location will be affected by market fluctuations, addition of H2O2 prior to stimulation may produce changes that would normally occur later in the physiological process. For example, when a growth factor binds to its receptor it can activate the protein tyrosine kinase (PTK) activity of the receptor and downstream PTKs while also activating an NADPH oxidase that produces H2O2. The H2O2 may then transiently inactivate a protein tyrosine phosphatase (PTP), that opposes the action of the PTK. Thus, if H2O2 is added prior to stimulation the transient inactivation of the PTP may already be reversed or may continue too long to accurately mimic what happens during signaling. But, this problem is the same as that encountered with other agents used to mimic signaling such as a calcium ionophore that allows calcium entry from the media or dibutyryl cyclic AMP.
H2O2 is a second messenger
So how does H2O2 fit the role of a second messenger in terms of the five essential characteristics? (See section “Oxidants can act as second messengers” above for the characteristics). (1) H2O2 increases in concentration through enzymatic generation by some oxidoreductases and DuOXs and from dismutation of O2•− produced by other oxidoreductases; (2) H2O2 decreases through enzymatic degradation catalyzed by catalase, glutathione peroxidases and peroxiredoxins; (3) H2O2 concentration rises and falls within a short period from a steady state estimated as nanomolar [24]; (4) H2O2 is also specific in action as will be described in the following section; and (5) because of characteristics 1, 2 and 3, the gradient of H2O2 from its origin to where it is degraded is very steep. Indeed, because of the distribution of glutathione peroxidases and peroxiredoxins throughout the cell, H2O2 needs to react within a few molecular diameters of its site of production with its target effector.
Examples
Two recent studies from our lab represent the advantages and problems in using exogenous H2O2 in cell culture and in in vitro studies of signal transduction. The first, the activation of ASK1 through oxidation of Trx by H2O2, presents an example where there is indirect evidence to suggest that H2O2-dependent Trx oxidation occurs but where exogenous H2O2 produces an observable change in the redox state of Trx that is not observed during endogenous H2O2 production [25]. The second example presents a situation in which there is a clear mismatch between what is observed in vitro with purified protein tyrosine phosphatase 1B (PTP1B) and what happens in cells stimulated to produce H2O2 [26]. In both studies there was value in using exogenous H2O2 but only because the results were interpreted with caution.
ASK1 redox regulation through Trx oxidation
The mitogen activated protein kinase (MAPK) kinase kinase known as apoptosis signal-regulating kinase 1 (ASK1) activates the protein kinases, which are upstream of Jun N-terminal kinase (JNK) and p38 MAPK [27, 28]. TNF-α, Fas, endoplasmic reticulum stress, and exogenous H2O2, are all capable of activating ASK1 through phosphorylation and protein-protein interactions [29–31, 32 , 33–39]. In unstimulated cells, binding of the N-terminus of ASK1 to reduced Trx inhibits ASK1 activity [38, 40]. It has been demonstrated using addition of exogenous H2O2 to cells that Trx can be directly oxidized by H2O2 (or via catalysis by a peroxiredoxin) [38, 40, 41]. The oxidized form of Trx then dissociates from ASK1, which then homo-oligomerizes. Release of Trx also appears to be critical for the binding of ASK1 to other proteins that promote ASK1 oligomerization and increases its kinase activity [30, 36]. The oligomerized ASK1 then autophosphorylates at Thr845 resulting in further increased kinase activity [29].
In our recent study [25], we demonstrated that H2O2 generated by the ADP-stimulated respiratory burst in the rat alveolar macrophage NR8383 cell line, causes rapid Trx dissociation from ASK1, and the phosphorylation of ASK1 at Thr845. The only known mechanism for dissociation of ASK1 from Trx is the oxidation of Trx by H2O2 [38, 40]. The respiratory burst results in O2•− production by the phagocyte NADPH oxidase on the outer surface of the cell. There, O2•− and protons rapidly reacts to produce H2O2 and O2 (even without catalysis by superoxide dismutase the reaction rate is approximately 105 M−1s1 at pH 7 [42]). H2O2 can easily enter cells but its production on the outside allows the use of exogenously added catalase, which eliminates H2O2, to be used as a tool for demonstrating the involvement of H2O2 in signaling. So, as predicted, the addition of catalase prevented the dissociation of Trx from ASK1 as well as the phosphorylation of ASK1. Furthermore, the dissociation of Trx was transient, returning to control by 15 min after ADP stimulation. This suggested that the effect of the transient H2O2 production by the respiratory burst resulted in only transient Trx oxidation. Nonetheless, we did not observe the transient increase in Trx oxidation. But, this is where the use of exogenous H2O2 had its biggest impact. Adding a bolus of 100 μM H2O2, which is far more H2O2 than was produced by the ADP-stimulated cells, we only observed a transient increase in the oxidation of Trx. It seems unlikely that the inability to detect oxidized Trx in ADP-treated cells was due to a difference between the mechanism through which cell produced versus bolus H2O2 caused Trx dissociation. More likely, only the Trx that was associated with ASK1 and in close vicinity where H2O2 entered the cell following the respiratory burst was oxidized. Future studies may be able to test this hypothesis using techniques that have not yet been developed to localize changes in Trx oxidation. For now, the best evidence comes from the catalase inhibition of intact cellular H2O2-dependent ASK1 activation in combination with the results from exogenous addition of H2O2 to the cells.
PTP1B glutathionylation
One of the principal effectors for H2O2 signaling are the PTPs [43–49]. The reversible phosphorylation of tyrosyl residues in proteins play a key role in the various signaling pathways induced by the great number of stimuli that regulate cellular responses including growth, proliferation, differentiation and metabolism. Protein tyrosine phosphorylation is controlled through the coordinated actions of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). Initially it was thought that the PTPs could be a small number of proteins that serve a housekeeping function, but they are now recognized as a critical regulators of signal transduction under normal and pathophysiological condition [50, 51].
The first suggestion that PTP1B might by glutathionylated in vivo came from the indirect demonstration of a reversibly oxidized form of the protein upon stimulation of A431 cells with epidermal growth factor [46]. Stimulation of these cells or addition of exogenous H2O2 caused the loss of the ability to modify the active site cysteine with iodoacetate. After a few minutes, the ability to modify that cysteine returned suggesting the formation of a reversibly oxidized form of PTP1B, consistent with glutathionylation. Glutathionylation of PTP1B was directly demonstrated in vitro by treatment of isolated PTP1B with 25 mM GSSG [48]. Even with such a non-physiologically high concentration of GSSG the glutathionylated form of PTP1B was slowly formed. Our recently published results demonstrated that glutathionylation of PTP1B occurs in the alveolar macrophage cell line, NR8383, upon addition of exogenous H2O2, but more importantly upon stimulation of endogenous production of H2O2 through the ADP-stimulated respiratory burst led to the reversible glutathionylation of PTP1B in intact cells [26]. While there have been proposed mechanisms for how this occurs, it remains unclear which of these, if any, are responsible. As stated above, it is possible to observe very slow non-enzymatic glutathionylation of PTP1B with 25 mM GSSG in a test tube but such a concentration of GSSG would not be possible in vivo. We will consider here several possible mechanisms that may occur in NR8383 in response to generation of H2O2 through the respiratory burst leading to glutathionylation of PTP1B.
First we will consider the currently accepted (but actually improbable) hypothetical mechanism of non-enzymatic oxidation of PTP1B by H2O2. Unlike most cysteinyl residues in proteins, the active site residue cysteine of a PTP such as PTP1B has a low pKa and so is present as a thiolate anion (PS−). While protein thiols (PSH) are generally very slow to react with H2O2 without catalysis, thiolates in proteins (PS−) have been shown to react in reaction:
| <1> |
with rate constants varying from 10 to 105 M−1s−1 depending upon the surrounding amino acids and the conformation of the protein [52]. We have indicated the product as a sulfenate rather than sulfenic acid because sulfenic acids normally have a lower pKa than does the corresponding thiol [53]. Thus, we expect that if the cysteine is in the thiolate form, the oxidized cysteine would be in the sulfenate form. A reaction with GSH would then produce the glutathionylated form of the protein:
| <2> |
In the absence of GSH, it has been shown that the PSO− form of PTP1B can react with an amide from the protein backbone to form a sulfenyl-amide, which has been observed in crystals of isolated PTP1B treated with H2O2 [54, 55]:
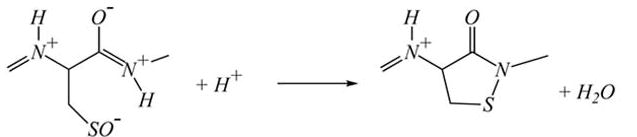 |
<3> |
These studies also suggest that the sulfenyl-amide form would react with GSH:
 |
<4> |
It should be kept in mind that formation of the sulfenic acid form, which was demonstrated by trapping with 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) [47] and formation of the sulfenyl-amide [54, 55] have only been observed with purified PTP1B and the latter only in the absence of GSH.
This elegant demonstration of protein chemistry is unfortunately an excellent example of where the caveat for extrapolation of in vitro results has not been paid sufficient attention. The big problems for this mechanism are kinetics and geometry. For PTP1B, a rate constant for H2O2 induced reversible inactivation of purified PTP1B in the absence of a reducing agent has been determined to be 9.1 M−1s−1 [47] while others have found slightly faster rates up to 43 M−1s−1 [56, 57]. From what we now know of the chemistry that occurs in vitro, this inactivation would involve reaction <1> followed by reaction <2>. Reaction <1> has a low rate constant that presents a problem in terms of how formation of the sulfenate or the sulfenyl-amide could occur in vivo. The intracellular steady state level of H2O2 has been estimated to be approximately 1 nM [58]. How much H2O2 concentration inside the cell increases during a respiratory burst when all the O2•− production occurs on the outside is unknown. But, for mechanism 1 to account for PTP1B oxidation, the source of H2O2 would have to produce a high concentration of H2O2 close to the PTP1B. This seems to be ruled out in the macrophage as our evidence thus far points to the classic plasma membrane NADPH oxidase of the macrophage (NOX2), which generates O2•− and hence H2O2 outside the cell, as responsible for PTP1B glutathionylation since catalase added to the outside of the cell abolishes glutathionylation. This means that the H2O2 would have to diffuse into the cell and the PTP1B be very close to that point of entry. Recent studies suggest that H2O2 diffusion into cells is limited and is actually modulated by diffusion through aquaporins [59] Thus, it seems quite unlikely that the non-enzymatic oxidation of PTP1B by H2O2 is involved in glutathionylation.
So, then how could glutathionylation occur? We have proposed that PTP1B is glutathionylated when NR8383 cells are stimulated to produce H2O2 by an as yet unidentified mechanism likely to involve an enzymatic process. In our published work [26], we showed that, although H2O2 was absolutely required for glutathionylation, PTP1B glutathionylation continued to increase for minutes after H2O2 production ended. Thus, some intermediate with a half life of minutes appeared to be involved. The rapid reaction of the sulfenyl-amide with GSH [54, 60], which is millimolar in NR8383 cells, would strongly suggest that the sulfenyl-amide would not account for that intermediate in the in vivo situation. Thus, the physiological mechanism of PTP1B glutathionylation remains unresolved. In this case, both the addition of exogenous H2O2 and stimulation of H2O2 production both transiently produce glutathionylated PTP1B, but this may be a situation in which a similar result occurs through different mechanisms.
Summary
Exogenous H2O2 may mimic signaling by endogenously produced H2O2 and has the same advantage as using any other membrane permeable second messenger, such as dibutyryl-cyclic AMP. The primary advantage is in verifying that the second messenger can do the signaling. The primary disadvantage is that the results can be misleading since H2O2 may do other things instead of or in addition to what the endogenously produced H2O2 can do. In order to assess the value, a combination of experimental approaches should be used and particular attention paid to the kinetics and concentration dependence of the reactions in which H2O2 is proposed to participate.
Acknowledgments
This work was supported by NIH grants HL37556, ES05511 and the California Tobacco-Related Diseases Research Program. The author thanks his colleagues, Drs. Jinah Choi, Hongqiao Zhang, Honglei Liu, and Alessandra Rinna for their comments and contributions to the studies from our laboratory.
Abbreviations
- ASK1
apoptosis signaling kinase 1
- DCF
2′,7′-dichlorofluorescein
- DCFH
2′,7′-dichlorofluorescin
- JNK
Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- PTP1B
protein tyrosine phosphatase 1B
- Trx
thioredoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schulman M, Sullivan PA. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochemical and Biophysical Research Communications. 1969;36:891. doi: 10.1016/0006-291x(69)90287-3. [DOI] [PubMed] [Google Scholar]
- 2.Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Letters. 1974;42:68. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 4.Forman HJ, Kennedy JA. Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochemical and Biophysical Research Communications. 1974;60:1044–1050. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 5.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. II. The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. J Biol Chem. 1970;245:1374–1377. [PubMed] [Google Scholar]
- 6.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 7.Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 8.Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res. 2001;89:224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- 9.Bonini MG, Rota C, Tomasi A, Mason RP. The oxidation of 2',7'-dichlorofluorescin to reactive oxygen species: a self-fulfilling prophesy? Free Radic Biol Med. 2006;40:968–975. doi: 10.1016/j.freeradbiomed.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 10.Jakubowski W, Bartosz G. 2,7-dichlorofluorescin oxidation and reactive oxygen species: what does it measure? Cell Biol Int. 2000;24:757–760. doi: 10.1006/cbir.2000.0556. [DOI] [PubMed] [Google Scholar]
- 11.Royall JA, Gwin PD, Parks DA, Freeman BA. Responses of vascular endothelial oxidant metabolism to lipopolysaccharide and tumor necrosis factor-alpha. Archives of Biochemistry and Biophysics. 1992;294:686–694. doi: 10.1016/0003-9861(92)90742-f. [DOI] [PubMed] [Google Scholar]
- 12.Forman HJ, Skelton DC, Loeb GA, Dorio RJ. Membrane permeability and oxidant induced injury. Basic Life Sci. 1988;49:523–530. doi: 10.1007/978-1-4684-5568-7_81. [DOI] [PubMed] [Google Scholar]
- 13.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 14.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Finkel T. Stem cell aging: what bleach can teach. Nat Med. 2006;12:383–384. doi: 10.1038/nm0406-383. [DOI] [PubMed] [Google Scholar]
- 16.Forman HJ, Fukuto J, Torres M. Signal transduction by reactive oxygen and nitrogen species : pathways and chemical principles. Dordrecht ; Boston: Kluwer Academic Publishers; 2003. [Google Scholar]
- 17.Mukherjee SP, Lane RH, Lynn WS. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem Pharmacol. 1978;27:2589–2594. doi: 10.1016/0006-2952(78)90332-5. [DOI] [PubMed] [Google Scholar]
- 18.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kB transcription factor and HIV-1. EMBO Journal. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaul N, Forman HJ. Activation of NF-κB by the respiratory burst of macrophages. Free Radical Biology & Medicine. 1996;21:401–405. doi: 10.1016/0891-5849(96)00178-5. [DOI] [PubMed] [Google Scholar]
- 20.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 21.Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- 22.Lemjabbar H, Li D, Gallup M, Sidhu S, Drori E, Basbaum C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulin. J Biol Chem. 2003;278:26202–26207. doi: 10.1074/jbc.M207018200. [DOI] [PubMed] [Google Scholar]
- 23.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 24.Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Zhang H, Iles KE, Rinna A, Merrill G, Yodoi J, Torres M, Forman HJ. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free Radic Res. 2006;40:865–874. doi: 10.1080/10715760600758514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda K, Hatai T, Hamazaki TS, Nishitoh H, Saitoh M, Ichijo H. Apoptosis signal-regulating kinase 1 (ASK1) induces neuronal differentiation and survival of PC12 cells. J Biol Chem. 2000;275:9805–9813. doi: 10.1074/jbc.275.13.9805. [DOI] [PubMed] [Google Scholar]
- 28.Wang XS, Diener K, Jannuzzi D, Trollinger D, Tan TH, Lichenstein H, Zukowski M, Yao Z. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J Biol Chem. 1996;271:31607–31611. doi: 10.1074/jbc.271.49.31607. [DOI] [PubMed] [Google Scholar]
- 29.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 30.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 31.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masters SC, Subramanian RR, Truong A, Yang H, Fujii K, Zhang H, Fu H. Survival-promoting functions of 14-3-3 proteins. Biochem Soc Trans. 2002;30:360–365. doi: 10.1042/bst0300360. [DOI] [PubMed] [Google Scholar]
- 33.Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, Park J, Eom SJ, Kim MJ, Chang TS, Choi SY, Shim J, Kim Y, Dong MS, Lee MJ, Kim SG, Ichijo H, Choi EJ. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:12749–12755. doi: 10.1074/jbc.M005561200. [DOI] [PubMed] [Google Scholar]
- 34.Dorion S, Lambert H, Landry J. Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S-transferase Mu from Ask1. J Biol Chem. 2002;277:30792–30797. doi: 10.1074/jbc.M203642200. [DOI] [PubMed] [Google Scholar]
- 35.Park HS, Cho SG, Kim CK, Hwang HS, Noh KT, Kim MS, Huh SH, Kim MJ, Ryoo K, Kim EK, Kang WJ, Lee JS, Seo JS, Ko YG, Kim S, Choi EJ. Heat shock protein hsp72 is a negative regulator of apoptosis signal-regulating kinase 1. Mol Cell Biol. 2002;22:7721–7730. doi: 10.1128/MCB.22.22.7721-7730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 37.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. Embo J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal- regulating kinase (ASK) 1. Embo J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 40.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 41.Song JJ, Lee YJ. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochemical Journal. 2003;373:845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klug D, Rabani J, Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- 43.Raugei G, Ramponi G, Chiarugi P. Low molecular weight protein tyrosine phosphatases: small, but smart. Cell and Molecular Life Sciences. 2002;59:941–949. doi: 10.1007/s00018-002-8481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Whorton AR. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys. 2003;410:269–279. doi: 10.1016/s0003-9861(02)00696-3. [DOI] [PubMed] [Google Scholar]
- 45.Chiarugi P, Fiaschi T, Taddei ML, Talini D, Giannoni E, Raugei G, Ramponi G. Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J Biol Chem. 2001;276:33478–33487. doi: 10.1074/jbc.M102302200. [DOI] [PubMed] [Google Scholar]
- 46.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 47.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 48.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 49.Krejsa CM, Nadler SG, Esselstyn JM, Kavanagh TJ, Ledbetter JA, Schieven GL. Role of oxidative stress in the action of vanadium phosphotyrosine phosphatase inhibitors. Redox independent activation of NF-κB. J Biol Chem. 1997;272:11541–11549. doi: 10.1074/jbc.272.17.11541. [DOI] [PubMed] [Google Scholar]
- 50.Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 51.Dube N, Tremblay ML. Beyond the metabolic function of PTP1B. Cell Cycle. 2004;3:550–553. [PubMed] [Google Scholar]
- 52.Stone JR. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch Biochem Biophys. 2004;422:119–124. doi: 10.1016/j.abb.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 53.Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 2002;348:122–136. doi: 10.1016/s0076-6879(02)48632-6. [DOI] [PubMed] [Google Scholar]
- 54.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 55.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 56.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 57.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 58.Boveris A, Cadenas E. Cellular sources and steady-state levels of reactive oxygen species. In: Clerch LB, Massaro DJ, editors. Oxygen, Gene Expression, and Cellular Function. New York: Marcel Dekker; 1997. pp. 1–25. [Google Scholar]
- 59.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 60.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for redox regulation of protein tyrosine phosphatase 1B (PTP1B) activity. J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]