

Abstract
Prodynorphins (PDYNs) from the African clawed frog (Xenopus leavis), originally described as ‘proxendorphins’, are novel members of the family of opioid-like precursor polypeptides and were recently discovered based on polymerase chain reaction (PCR) isolates from a Xenopus brain cDNA library. This amphibian prodynorphin was found in two isoforms, XenPDYN-A and XenPDYN-B, consisting of 247 and 279 amino acids, respectively. Each prepropeptide contains five potential opioid-like peptides, collectively named xendorphins. One of these, xendorphin B1 (XenPDYN-B sequence 96–111: YGGFIRKPDKYKFLNA), is a hexadecapeptide that displaced [3H]naloxone and the radiolabelled kappa opioid, [3H]dynorphin A (1–17), with nanomolar affinity from rat brain membranes. Using the acetic acid pain test, the present study examined the antinociceptive effects of spinally administered xendorphin B1 in amphibians. Xendorphin B1 produced a long-lasting and dose-dependent antinociceptive effect in the Northern grass frog (Rana pipiens) with an ED50 value of 44.5 nmol/frog. The antinociceptive effects of xendorphin B1 were significantly blocked by pretreatment with the non-selective opioid antagonist, naltrexone. This is the first report of the in vivo characterization of a non-mammalian prodynorphin-derived peptide and suggests that xendorphin peptides may play a role in the modulation of noxious information in vertebrates.
Keywords: opioid peptides, prodynorphin, frog, antinociception, Xenopus laevis, Rana pipiens
1. Introduction
Since the discovery of Met- and Leu-enkephalin more than 30 years ago [5], there are now four main families of natural opioid peptides isolated and characterized in vertebrates: enkephalins, endorphins, dynorphins and endomorphins [1–3;6;8;15;33]. Until now three precursor polypeptides for the endogenous opioids have been described in various species: proopiomelanocortin (POMC), proenkephalin (PENK) and prodynorphin (PDYN) [6;10;14], while the origin of the endomorphins is yet unknown.
Xendorphin B1 or ‘proxendorphin B (96–111)’ with the amino acid composition of YGGFIRKPDKYKFLNA is an opioid-like peptide determined from the predicted amino acid sequence of one of the proxendorphin polypeptides [16]. Proxendorphins A and B were cloned and sequenced from an African clawed frog (Xenopus leavis) brain cDNA library using degenerate oligonucleotides coding for the common N-terminal opioid peptide motif (YGGF) as a probe. Both proxendorphin A and B contain five opioid-like core peptides, each of them are marked by pairs of positively charged amino acids, such as Lys-Arg (KR) or Lys-Lys (KK). These dibasic repeats serve as cleavage sites for the processing endopeptidase enzymes, e.g., prohormone convertase. Three putative opioid peptides near the C-termini of the Xenopus’ propeptide isoforms are positionally and structurally homologous to the mammalian prodynorphins (PDYNs), hence the names of the frog precursors proxendorphin A and B should be changed into XenPDYN-A and XenPDYN-B, respectively. The two other deduced opioid sequences in Xenopus PDYNs are unique, because neither of them exist in mammalian PDYNs. The N-terminally located first potential opioid-like motifs from both XenPDYNs yield hexadecapeptides those were chemically synthesized and tested for displacement of [3H]naloxone using rat brain membrane homogenates. The most potent competitor, xendorphin B1, gave an apparent Ki value of 1.4 nM against [3H]naloxone and in further studies using type-selective opioid radioligands, displaced [3H]dynorphin A (1–17), a kappa opioid, more potently than selective mu or delta radioligands. Xendorphin B1 also stimulated the in vitro binding of [35S]GTPγS in rat and frog brain homogenates, suggesting an agonist-like interaction with the receptors [16].
Given that xendorphins were discovered using a Xenopus laevis brain cDNA library, it was reasonable to test xendorphin B1 for in vivo opioid action using an amphibian model. The acetic acid test in Northern grass frogs, Rana pipiens, is a suitable model with a background of opioid studies. Initial investigations of the antinociceptive effects of opioid administration in amphibians were conducted using non-selective opioid agonists, endogenous opioid peptides, and antagonists [19;26]. Tolerance to the analgesic effects of daily morphine administration in amphibians was documented [23] and stress-induced release of endogenous opioids was shown to produce antinociception that was potentiated by enkephalinase inhibitors [29]. The results of later studies examining the antinociception of selective mu, delta, or kappa opioid agonists administered by different routes in amphibians yielded an important finding: The relative antinociceptive potency of mu, delta, or kappa opioid agonists after systemic, intraspinal, or intracerebroventricular administration in amphibians was highly correlated to that observed in typical mammalian models and to the relative analgesic potency of opioid analgesics in human clinical studies [22;24;28]. This data established the amphibian model as a robust and predictive adjunct or alternative model for the testing of opioid analgesics [21].
The present study showed that xendorphin B1 produced a long-lasting and dose-dependent antinociceptive effect following spinal administration in amphibians. The log dose-response curve of the antinociceptive effect of xendorphin B1 gave an ED50 value of 44.5 nmol/frog with a relative potency of twenty times less than that of morphine, but equipotent to other opioid peptides administered by the spinal route in amphinbians. The antinociception produced by xendorphin B1 was blocked by pretreatment with the non-selective opioid antagonist, naltrexone. This is the first study examining the in vivo effects of a non-mammalian prodynorphin-derived peptide.
2. Materials and Methods
2.1. Animals
Handling and treating of animals were carried out accordingly with the NIH Guide for the Use and Care of Laboratory Animals. Northern grass frogs (also known as ‘leopard frog’), Rana pipiens (Sullivan, Nashville, TN) with a mean weight of 28 g were kept in groups of 48 in a flow-through, stainless steel enclosure at room temperature with running water after arrival. They were maintained with a 12-hour photoperiod and were fed live crickets twice a week. At least two days before experiments, animals were transferred to the laboratory and placed in individual plastic pans with an adequate amount of tap water. On the day of experimental study, frogs were randomly assigned to treatment groups and the water was adjusted to a depth such that the dorsal surface of the frog’s thigh was exposed for testing. Each animal was used in only one experiment.
2.2 Antinociceptive assay
The nociceptive threshold (NT) was determined by the acetic acid test as fully described previously [18]. Briefly, glacial acetic acid (17.5 M) was serially diluted to produce 11 concentrations. Code numbers were assigned to each dilution from 0 to 10 with the number 10 representing glacial acetic acid. Testing was performed by placing a single drop of the lowest concentration of acetic acid on the dorsal surface of a frog’s thigh with a Pasteur pipette and then proceeding with increasing concentrations on alternate hind limbs until the animal responded with a wiping response. The NT was defined as the code number of the acetic acid dilution that produces a vigorous wipe by the frog of the treated leg with either hindlimb. To prevent tissue damage, the acetic acid was washed off with a gentle stream of distilled water when the animal responded or if the animal failed to respond within 5 sec. Baseline NT was obtained 2 h after the water level was adjusted on the morning of the experiment and post-treatment NT at 1, 3 and 24 h after drug administration.
2.3 Drugs and administration
The hexadecapeptide, xendorphin B1 (YGGFIRKPDKYKFLNA) was synthesized by the solid-phase method (Advance Tech, M200) using tert-butoxy-carbonyl chemistry followed by high pressure liquid chromatography purification (HPLC) on a Vydac 218TP1010 column. Purified peptide was identified by electrospray ionization-mass spectrometry. The product was at least 98% pure as assessed by thin-layer chromatography and analytical RP-HPLC. Details of xendorphin peptide synthesis will be published elsewhere. Naltrexone hydrochloride was obtained from the National Institute on Drug Abuse, Drug Supply Program (kind assistance from Mr. Kevin Gormley of the Research Technology Branch). Drugs were mixed in physiological saline for frogs to give nmol/μl solutions of the peptide or free base. Xendorphin B1 or saline alone (control) was administered by intraspinal (i.s.) injection into the lumbar region of the spinal cord with a microsyringe fitted with a 26-gauge needle [26]. Injections were made percutaneously via the articulation between the seventh and eighth vertebrae, in a volume of 5 μl/animal. Naltrexone was administered by the systemic route, by injection into the dorsal lymph sacs, 60 min before i.s. xendorphin B1 at a dose of 100 nmol/g and a volume of 10 μl/g as previously described [22]. All treatment groups consisted of six animals per dose. Motor function was assessed by testing the animals for hindlimb withdrawal, corneal reflexes and their ability to right themselves. No animals showed untoward effects at the doses of xendorphin B1 used.
2.4 Data analysis and statistics
The raw NT data (code number of acetic acid solution) was converted to maximum percent effect (MPE) by the following formula:
Dose-response data was analyzed to give the ED50 and 95% confidence interval of xendorphin B1 using pharmacological software (PCS v. 4.0, MicroComputer Specialists, Philadelphia, PA). Dose-response curves were generated by taking the peak analgesic effects obtained for each animal at each dose and mean (and SEM values) plotted for construction of the curves. Opioid antagonist data were analyzed by a one-way ANOVA followed by the post-hoc Newman-Keuls test. Significant effects were considered at the P < 0.05 level.
3. Results
The time course of the antinociceptive effect of xendorphin B1 is shown in Fig. 1. Xendorphin B1 administered by the spinal route at 10, 30, or 100 nmol/frog produced a significant antinociceptive effect that lasted at least 3 hours. By 24 hours after administration, nociceptive thresholds returned to baseline levels.
Figure 1.
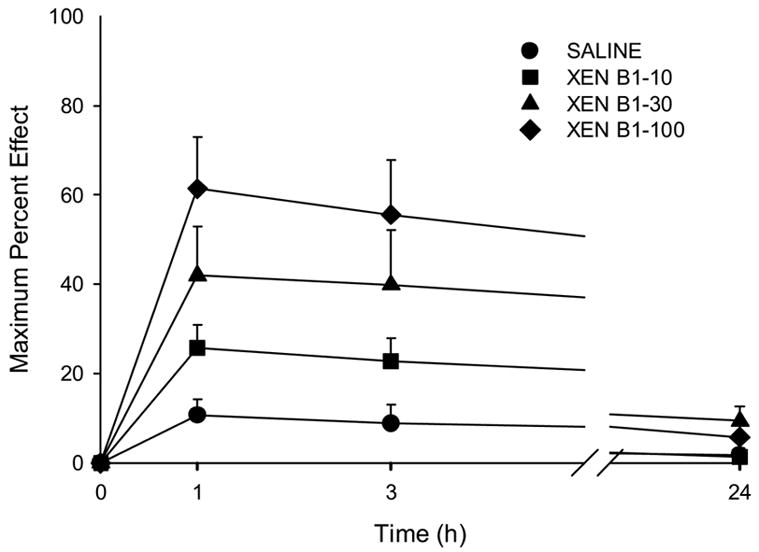
Time course of the antinociceptive effect of intraspinal xendorphin B1 (XEN B1) in amphibians. Doses used are given in the legend on the graph (in nmol/animal). Data points plotted as MPE +S.E.M (standard error of the mean). N=6 animals per dose and for the saline group.
A logarithmic dose-response curve of xendorphin B1 is shown in Fig. 2. Also plotted for comparison are the log dose-response curves of the selective mu opioid peptide [D-Ala2,NMePhe4,Gly5-ol]enkephalin (DAMGO) [4], the delta opioid cyclopeptide, c[D-Pen2-D-Pen5]enkephalin (DPDPE) [9], and the benzacetamide derivative kappa opioid compound, U-50488H [31] from previously published studies in amphibians [22]. Xendorphin B1 had a calculated ED50 value of 44.47 nmol/frog (95% confidence interval; 20.7–95.4) which yielded a relative potency of 0.05 compared to morphine (see Table 1) and about equipotent to other endogenous opioid peptides administered into the frog spinal cord [27]. As shown in Table 1, the slopes of the dose-response curves for all agents did not differ from each other.
Figure 2.
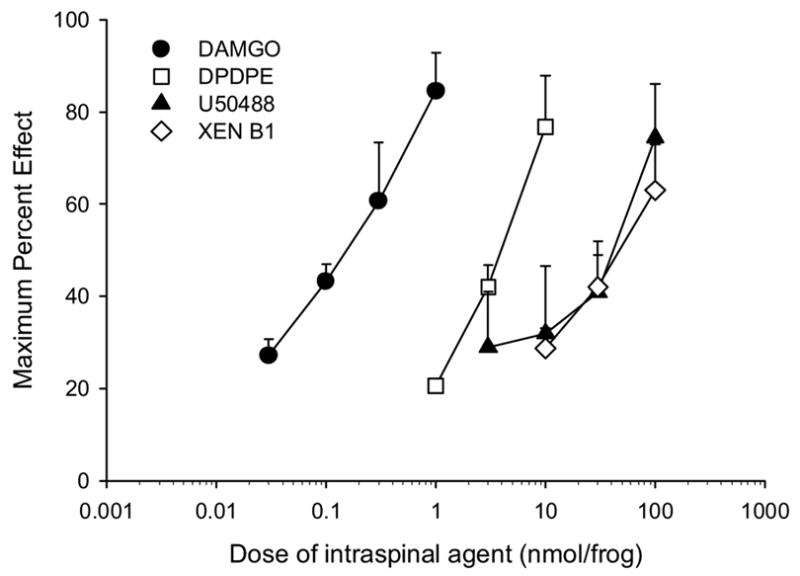
Log dose-response curves of the antinociceptive effect of intraspinal xendorphin B1 compared with the mu opioid DAMGO, the delta opioid, DPDPE, and the kappa opioid, U50488. Drug groups are denoted in the legend on the graph (XEN B1 is xendorphin B1). Data points are plotted as mean peak effect (MPE + S.E.M.) of individual animals grouped for each agent and dose. N= 6–8 animals per dose. Selective opioid agonist curves are from previous studies [22] and provided here for comparison.
TABLE 1.
Relative antinociceptive potency of xendorphin B1 compared to mu, delta, and kappa opioid agonists after intraspinal administration in amphibians. Xendorphin B1 data from present study, other data from previously published work [22] and are included for comparison.
| Opioid Agent | Class | ED50a | (95% C.I.)b | SLOPE | (95% C.I.) | R.P.c |
|---|---|---|---|---|---|---|
| DAMGO | Mu | 0.13 | (0.08–0.2) | 39.4 | (24.1–54.8) | 17.38 |
| Morphine | Mu | 2.26 | (1.7–3.0) | 48.4 | (37.2–59.5) | 1.00 |
| DPDPE | Delta0 | 3.29 | (2.3–4.7) | 56.2 | (35.3–77.0) | 0.69 |
| U50488 | Kappa | 36.82 | (19.3–70.2) | 36.7 | (16.8–56.5) | 0.06 |
| Xendorphin B1 | ? | 44.47 | (20.7–95.4) | 34.2 | (9.0–59.3) | 0.05 |
in nmol/animal.
95% confidence interval.
relative potency compared to morphine: ED50 morphine/ED50 agent.
The effect of naltrexone pretreatment on xendorphin antinociceptive effect is shown in Fig. 3. Treatment with saline or naltrexone alone did not generate a significant alteration of nociceptive thresholds. Xendorphin B1 (30 nmol/frog) produced a significant elevation of nociceptive thresholds. Naltrexone given at a dose of 100 nmol/g, s.c. one hour before the spinal administration of xendorphin B1 (30 nmol/frog) significantly blocked the antinociception produced by xendorphin B1 (one-way ANOVA followed by Newman-Keuls post-hoc test, P<0.01).
Figure 3.
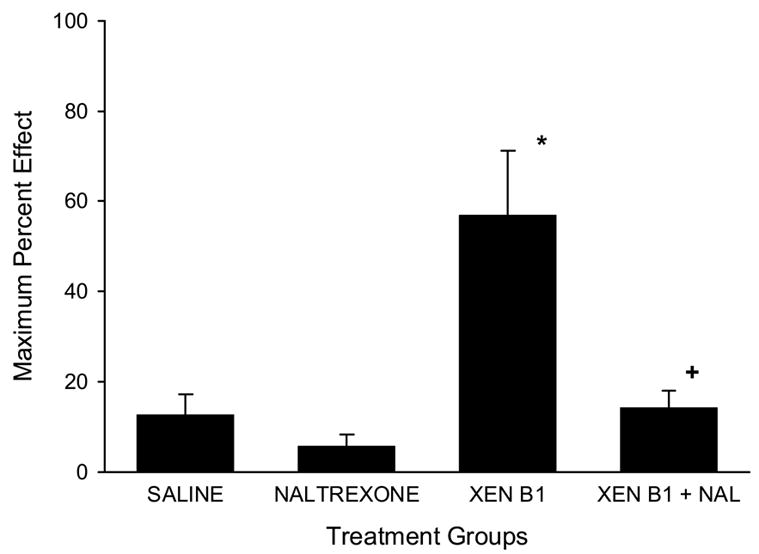
Naltrexone antagonism of the antinociceptive effects of intraspinal xendorphin B1. Time course of the antinociceptive effect of intraspinal xendorphin B1 in amphibians. Treatment group are denoted as Saline (5 μl per animal, i.s.), Naltrexone (100 nmol/g, s.c.), XEN B1 (xendorphin B1, 30 nmol/frog, i.s.) and XEN B1 + NAL (Naltrexone at100 nmol/g, s.c. given 1 h before xendorphin B1 at 30 nmol/frog, i.s). Data points plotted as maximum MPE (+S.E.M.) values across the time course. N=6 animals per treatment group. Asterisk (*) denotes significantly different from SAL group at P<0.01; plus sign (+) denotes significantly different than XEN B1 group at P<0.01 (one-way ANOVA followed by Newman-Keuls post-hoc test).
4. Discussion
Xendorphin B1 is the first opioid-like peptide predicted from cleavage sites in the Xenopus frog prodynorphin B polypeptide to be tested in an in vivo model for behavioral effects. XenPDYN-A and XenPDYN-B were isolated from Xenopus laevis brain cDNA by using a degenerate oligonucleotide probe corresponding to the common four amino acids found at the N-terminal of known endogenous opioid peptides (YGGF). Besides xendorphin 1 hexadecapeptides, there are four other potential opioid-like peptides containing within the precursor polypeptides: a big oligopeptide of 43 amino acids, and single copies of α-neo-endorphin, frog dynorphin-A and dynorphin-B [16]. Xendorphin B1 was chosen for initial in vivo studies based on its exceptional structure that is not represented in mammals, its high affinity in displacing the non-selective opioid antagonist [3H]naloxone from rat brain membranes and its agonist-like properties measured by sodium ion shift experiments and G-protein stimulation assays in vitro [17]. Xendorphin B1 was also highly potent in competing with [3H]dynorphin A (1–17), a kappa opioid radioligand [16].
In the present studies, xendorphin B1 was administered by the spinal route to unanaesthetized Northern grass frogs (Rana pipiens) and antinociceptive effects measured with the acetic acid test. The acetic acid test in amphibians is a well-documented algesiometric assay in this class of vertebrates (see Introduction). Xendorphin B1 administered by the spinal route in frogs produced a long-lasting antinociceptive effect that persisted for at least 3 hours (Fig. 1). This long-lasting antinociceptive effect is commonly observed with intraspinal injections of opioid peptides or opioid alkaloids in amphibians and is likely due to the low levels of re-distribution and/or drug biotransformation of opioids in this species [22]. There is much evidence for aminopeptidase activity in the nervous system of vertebrates which could account for xendorphin B1 degradation [7]. By 24 hours after spinal administration, nociceptive thresholds returned to normal which suggests that the behavioral measure used to determine the nociceptive thresholds (wiping response) was not confounded by untoward motor effects or tissue damage.
Xendorphin B1 produced a dose-dependent antinociceptive effect after spinal administration in amphibians (Fig. 2). The log dose-response curve of xendorphin B1 had an ED50 value of 44.5 nmol/frog (95% C.I.: 20.7–95.4) which was significantly less potent than DAMGO, morphine and DPDPE (see Table 1). The antinociceptive potency of xendorphin B1 did not differ from that of the kappa selective opioid, U-50488, nor other kappa opioids (CI-977, bremazocine, and nalorphine) tested after spinal administration in amphibians [22]. The slope of xendorphin B1 log dose-response curve was significantly positive (95% C.I. does not include zero) and did not differ from the other opioids. This shows that xendorphin produced a significant dose-dependent analgesic effect, however xendorphin B1 is a relatively weak antinociceptive agent in frog spinal cord, with about twenty times less potency than morphine. Xendorphin B1 potency was also similar to the analgesic potency of dynorphin tested after spinal administration in the amphibian model [27].
Naltrexone, a non-selective opioid antagonist, blocked the effect of a single dose of xendorphin B1 (Fig. 3). Naltrexone was used at the high dose of 100 nmol/g, s.c. given at 1 hour before the spinal administration of xendorphin B1 (30 nmol/frog, i.s.). This naltrexone pretreatment paradigm was previously shown to significantly block the antinociceptive effects of all mu, delta, and kappa opioids subsequently administered into the spinal cord [22]. This paper also demonstrated that the spinal analgesic action of morphine, and DAMGO, DPDPE, or U50488, was also blocked by the same naltrexone pre-treatment. Regardless of other actions that xendorphin B1 may have, the present data demonstrate that the antinociceptive effects of xendorphin B1 following spinal administration in amphibians is mediated by opioid receptors. In regard to the subtype of opioid receptor mediating xendorphin B1 action, the present data suggests a kappa opioid action of xendorphin B1 in producing antinociception based on the relative potency of xendorphin B1 compared to other kappa opioids [22] and dynorphin [27]. Additional evidence from previous studies of xendorphin B1 showed that it preferentially displaced the kappa opioid radioligand, [3H]dynorphin A (1–17), compared to the displacement of mu and delta opioid radioligands [16]. Further identification of the opioid type-selectivity of xendorphin B1 is needed, however, the most common highly-selective opioid antagonists used in mammalian studies, β-funaltrexamine (β-FNA, for mu) [32], naltrindole (NTI, for delta) [20], and nor-binaltorphimine (nor-BNI, for kappa) [30] did not show selectivity in blocking spinal opioids in initial behavioral studies with Rana pipiens [25]. Additionally, although radioligand binding data using amphibian brain and spinal cord homogenates clearly suggested that three types of opioid binding sites are present [11], the highly-selective opioid antagonists did not show differences in displacing [3H]naloxone [12;13]. Finally, mu, delta, and kappa opioid-like receptor proteins were cloned and sequenced from Rana pipiens brain and spinal cord mRNA, but bioinformatic analyses and preliminary characterization suggest that opioid receptors in amphibians are less type-selective than those in mammals [21].
The discovery of XenPDYNs and the in vivo characterization of xendorphin B1 suggest that this novel family of opioid-like precursors may have a role in modulating the transmission of noxious information in amphibians and perhaps other vertebrates. Further studies in amphibians and other vertebrate species are needed to clarify and elevate the status of prodynorphins to that of the established endogenous opioid peptide families.
Acknowledgments
This work was supported in part by research grants from the National Institutes of Health, NIDA 12448 (CWS), and from the National Office for Research and Technology (NKTH) RET-2004-DNT (SB, AB, GT), Budapest, Hungary.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akil H, Watson SJ, Young E, Lewis ME, Khatchaturian H. Endogenous opioids: biology and function. Ann Rev Neurosci. 1984;7:223–255. doi: 10.1146/annurev.ne.07.030184.001255. [DOI] [PubMed] [Google Scholar]
- 2.Bagosi Z, Jaszberenyi M, Bujdoso E, Szabo G, Telegdy G. The effects of endomorphins and diprotin A on striatal dopamine release induced by electrical stimulation-An in vitro superfusion study in rats. Neurochem Int. 2006;49:665–668. doi: 10.1016/j.neuint.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Cone RI, Goldstein A. A dynorphin-like opioid in the central nervous system of an amphibian. Proc Natl Acad Sci U S A. 1982;79:3345–3349. doi: 10.1073/pnas.79.10.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Handa BK, Lane AC, Lord JAH, Morgan BA, Rance MJ, Smith CFC. Analogues of beta-LPH 61–64 possessing selective agonist activity at mu opiate receptors. Eur J Pharmacol. 1981;70:531–540. doi: 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]
- 5.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258:577–579. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 6.Kakidani H, Furutani Y, Takahashi H, Noda M, Morimoto Y, Hirose T, Asai M, Inayama S, Nakanishi S, Numa S. Cloning and sequence analysis of cDNA for porcine beta-neo-endorphin/dynorphin precursor. Nature. 1982;298:245–249. doi: 10.1038/298245a0. [DOI] [PubMed] [Google Scholar]
- 7.Larrinaga G, Gil J, Meana JJ, Ruiz F, Callado LF, Irazusta J. Aminopeptidase activity in the postmortem brain of human heroin addicts. Neurochem Int. 2005;46:213–219. doi: 10.1016/j.neuint.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 9.Mosberg HI, Hurst R, Hruby VJ, Gee K, Yamamura HI, Galligan JJ, Burks TF. Bis-penicillamine enkephalins possess highly improved specificity toward delta opioid receptors. Proc Natl Acad Sci U S A. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakanishi S, Inoue A, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 11.Newman LC, Sands SS, Wallace DR, Stevens CW. Characterization of mu, kappa, and delta opioid binding in amphibian whole brain tissue homogenates. J Pharmacol Exp Ther. 2002;301:364–370. doi: 10.1124/jpet.301.1.364. [DOI] [PubMed] [Google Scholar]
- 12.Newman LC, Wallace DR, Stevens CW. Selective opioid receptor agonist and antagonist displacement of [3H]-naloxone binding in amphibian spinal cord. Brain Res. 2000;884:184–191. doi: 10.1016/s0006-8993(00)02967-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman LC, Wallace DR, Stevens CW. Selective opioid agonist and antagonist displacement of [3H]-naloxone binding in amphibian brain. Eur J Pharmacol. 2000;397:255–262. doi: 10.1016/s0014-2999(00)00265-x. [DOI] [PubMed] [Google Scholar]
- 14.Noda M, Furutani Y, Takahashi H, Toyosato M, Hirose T, Inayama S, Nakanishi S, Numa S. Cloning and sequence analysis of cDNA for bovine adrenal preproenkephalin. Nature. 1982;295:202–206. doi: 10.1038/295202a0. [DOI] [PubMed] [Google Scholar]
- 15.Noda M, Teranishi Y, Takahashi H, Toyosato M, Notake M, Nakanishi S, Numa S. Isolation and structural organization of the human preproenkephalin gene. Nature. 1982;297:431–434. doi: 10.1038/297431a0. [DOI] [PubMed] [Google Scholar]
- 16.Pattee P, Ilie AE, Benyhe S, Toth G, Borsodi A, Nagalla SR. Cloning and characterization of Xen-dorphin prohormone from Xenopus laevis: a new opioid-like prohormone distinct from proenkephalin and prodynorphin. J Biol Chem. 2003;278:53098–53104. doi: 10.1074/jbc.M306724200. [DOI] [PubMed] [Google Scholar]
- 17.Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science. 1973;182:1359–1361. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- 18.Pezalla PD. Morphine-induced analgesia and explosive motor behavior in an amphibian. Brain Res. 1983;273:297–305. doi: 10.1016/0006-8993(83)90854-5. [DOI] [PubMed] [Google Scholar]
- 19.Pezalla PD, Stevens CW. Behavioral effects of morphine, levorphanol, dextrorphan and naloxone in the frog Rana pipiens. Pharmacol Biochem Behav. 1984;21:213–217. doi: 10.1016/0091-3057(84)90217-x. [DOI] [PubMed] [Google Scholar]
- 20.Portoghese PS, Sultana M, Takemori AE. Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur J Pharm. 1988;146:185–186. doi: 10.1016/0014-2999(88)90502-x. [DOI] [PubMed] [Google Scholar]
- 21.Stevens CW. Opioid research in amphibians: a unique perspective on mechanisms of opioid analgesia and the evolution of opioid receptors. Reviews in Analgesia. 2003;7:122–136. [Google Scholar]
- 22.Stevens CW. Relative analgesic potency of mu, delta and kappa opioids after spinal administration in amphibians. J Pharmacol Exp Ther. 1996;276:440–448. [PubMed] [Google Scholar]
- 23.Stevens CW, Kirkendall K. Time course and magnitude of tolerance to the analgesic effects of systemic morphine in amphibians. Life Sci. 1993;52:PL111–116. doi: 10.1016/0024-3205(93)90097-m. [DOI] [PubMed] [Google Scholar]
- 24.Stevens CW, Klopp AJ, Facello JA. Analgesic potency of mu and kappa opioids after systemic administration in amphibians. J Pharmacol Exp Ther. 1994;269:1086–1093. [PubMed] [Google Scholar]
- 25.Stevens CW, Newman LC. Spinal administration of selective opioid antagonists in amphibians: evidence for an opioid unireceptor. Life Sci. 1999;64:PL125–PL130. doi: 10.1016/s0024-3205(99)00013-2. [DOI] [PubMed] [Google Scholar]
- 26.Stevens CW, Pezalla PD. A spinal site mediates opiate analgesia in frogs. Life Sci. 1983;33:2097–2103. doi: 10.1016/0024-3205(83)90333-8. [DOI] [PubMed] [Google Scholar]
- 27.Stevens CW, Pezalla PD, Yaksh TL. Spinal antinociceptive action of three representative opioid peptides in frogs. Brain Res. 1987;402:201–203. doi: 10.1016/0006-8993(87)91069-9. [DOI] [PubMed] [Google Scholar]
- 28.Stevens CW, Rothe KS. Supraspinal administration of opioids with selectivity for μ–,δ–, and κ–opioid receptors produces analgesia in amphibians. Eur J Pharmacol. 1997;331:15–21. doi: 10.1016/s0014-2999(97)01026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevens CW, Sangha S, Ogg BG. Analgesia produced by immobilization stress and an enkephalinase-inhibitor in amphibians. Pharmacol Biochem Behav. 1995;51:675–680. doi: 10.1016/0091-3057(94)00436-m. [DOI] [PubMed] [Google Scholar]
- 30.Takemori AE, Ho BY, Naeseth JS, Portoghese PS. Nor-binaltorphimine, a highly selective kappa-opioid antagonist in analgesic and receptor binding assays. J Pharmacol Exp Ther. 1988;246:255–258. [PubMed] [Google Scholar]
- 31.VonVoigtlander PF, Lahti RA, Ludens JH. U-50,488: A selective and structurally novel non-mu (kappa) opioid agonist. J Pharmacol Exp Ther. 1983;224:7–12. [PubMed] [Google Scholar]
- 32.Ward SJ, Portoghese PS, Takemori AE. Pharmacological profiles of beta-funaltrexamine (β-FNA) and beta-chlornaltrexamine (β-CNA) on the mouse vas deferens preparation. Eur J Pharmacol. 1982;80:377–384. doi: 10.1016/0014-2999(82)90083-8. [DOI] [PubMed] [Google Scholar]
- 33.Zadina JE, Hackler L, Ge LJ, Kastin AJ. A potent and selective endogenous agonist for the μ-opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]