

Abstract
Tyrosine hydroxylase (TyrH) is a pterin-dependent enzyme that catalyzes the hydroxylation of tyrosine to form dihydroxyphenylalanine. The oxidation state of the active site iron atom plays a central role in the regulation of the enzyme. The kinetics of reduction of ferric TyrH by several reductants were determined by anaerobic stopped-flow spectroscopy. Anaerobic rapid freeze–quench EPR confirmed that the change in the near-UV absorbance of TyrH upon adding reductant corresponded to iron reduction. Tetrahydrobiopterin reduces wild-type TyrH following a simple second-order mechanism with a rate constant of 2.8 ± 0.1 mM−1 s−1. 6-Methyltetrahydropterin reduces the ferric enzyme with a second-order rate constant of 6.1 ± 0.1 mM−1 s−1 and exhibits saturation kinetics. No EPR signal for a radical intermediate was detected. Ascorbate, glutathione, and 1,4-benzoquinone all reduce ferric TyrH, but much more slowly than tetrahydrobiopterin, suggesting that the pterin is a physiological reductant. E332A TyrH, which has an elevated Km for tetrahydropterin in the catalytic reaction, is reduced by tetrahydropterins with the same kinetic parameters as those of the wild-type enzyme, suggesting that BH4 does not bind in the catalytic conformation during the reduction. Oxidation of ferrous TyrH by molecular oxygen can be described as a single-step second-order reaction, with a rate constant of 210 mM−1 s−1. S40E TyrH, which mimics the phosphorylated state of the enzyme, has oxidation and reduction kinetics similar to those of the wild-type enzyme, suggesting that phosphorylation does not directly regulate the interconversion of the ferric and ferrous forms.
Tyrosine hydroxylase (TyrH)1 is a pterin-dependent monooxygenase that catalyzes the hydroxylation of tyrosine to form dihydroxyphenylalanine (DOPA) using a non-heme iron atom in the active site to activate molecular oxygen (1). Formation of DOPA is the first and rate-limiting step in the biosynthesis of the catecholamine neurotransmitters dopamine, epinephrine, and norepinephrine. Because of the important physiological role played by TyrH, its activity is regulated at the posttranslational level by phosphorylation of serine residues and by feedback inhibition by catecholamines. The redox state of the iron plays a central role in this regulation, as shown in Scheme 1 (2, 3). For catalysis, the active site iron must be ferrous (4); during catalysis it is oxidized to higher levels but returns to the ferrous form by the end of the catalytic cycle (5). However, in a side reaction independent of catalysis, oxygen can oxidize the ferrous iron to the inactive ferric form (2); the iron must then be reduced back to the ferrous form for DOPA formation to resume. The in vivo reductant for TyrH is unknown, although tetrahydrobiopterin (BH4) has been proposed to be the natural reductant due to its role as substrate for the enzyme (2). Oxidation of the ferrous enzyme to the ferric form results in an enzyme with high affinity for catecholamines (6). Consequently, if these products of the catecholamine bio-synthetic pathway accumulate in the cell, the ferric enzyme can be trapped as the inactive catecholamine complex. Consistent with such a model, when TyrH is isolated from eukaryotic sources, the protein contains a mixture of catecholamines bound to the ferric iron atom (7). To regain activity, this bound catecholamine must be released from TyrH. However, dissociation of dopamine and other catecholamines is extremely slow, such that the inhibition is effectively irreversible (6). Phosphorylation of TyrH at Ser40 increases the rate constant for dissociation for dopamine and the other catecholamines by 3 orders of magnitude, effectively relieving the inhibition and forming the free ferric enzyme which can then be reduced to the active ferrous form (8, 9). A requirement for reductive activation has also been demonstrated for phenylalanine hydroxylase (PheH) (10-12), but its role in regulation of that enzyme is not clear.
Scheme 1.

While the effects of phosphorylation and iron redox state on catecholamine binding to TyrH have been characterized, the kinetics and mechanism of interconversion of ferric and ferrous TyrH have yet to be examined. To probe the physiological viability of the mechanism of Scheme 1, the kinetics and specificity of the reduction and oxidation of the iron in TyrH have been analyzed.
MATERIALS AND METHODS
Materials
Tetrahydrobiopterin, 6-methyltetrahydropterin, and 5-methyltetrahydrobiopterin were purchased from B. Schircks Laboratories (Jona, Switzerland). Catalase and superoxide dismutase were from Roche Diagnostics Corp. (Indianapolis, IN). All other reagents were of the highest purity commercially available.
Enzyme Preparation and Purification
Wild-type and mutant proteins were overexpressed in Escherichia coli as previously described (13, 14). Purification of the enzymes was performed according to previously published methods (13) with the substitution of an AKTA FPLC with a 5 mL Hi-TRAP heparin column for the heparin–Sepharose column. After purification, the enzyme was precipitated with 50% ammonium sulfate and dissolved in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl. To obtain apoenzyme, the ammonium sulfate pellet was dissolved in the same buffer containing 1 mM EDTA and incubated on ice for 30 min. The enzyme was then dialyzed against the same buffer to remove excess EDTA. This produced enzyme containing less than 0.1 iron/monomer. To obtain enzyme containing a stoichiometric amount of Fe(III), 1.2 equiv of ferrous ammonium sulfate was added to the redissolved ammonium sulfate pellet lacking EDTA, and the solution was incubated on ice for 30 min. The enzyme was then dialyzed against the same buffer to remove excess iron, yielding enzyme containing 0.85–1 equiv of iron/monomer. To obtain ferrous enzyme, apoenzyme was placed in a flask sealed with a rubber septum, and the solution was made anaerobic by alternately incubating under argon and vacuum for 1 h. Using a gastight syringe, 0.9 molar equiv of Fe(II) (10 mM ferrous ammonium sulfate in 1 mM HCl) was then added to the enzyme. Iron stoichiometries were determined by atomic absorption on a Perkin-Elmer AAnaylst 700 (15).
Kinetics of Enzyme Reduction
Enzyme (100–250 μM in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl) was mixed with various concentrations of reductant at 20 °C in an Applied Photophysics SX-18MV stopped-flow spectrophotometer. Enzyme reduction was monitored by following the decrease in absorbance at various wavelengths between 350 and 410 nm. Tetrahydropterins were dissolved in 2 mM HCl, 10% glycerol, and 0.1 M KCl. For reactions under anaerobic conditions, the spectrophotometer was filled with a solution of 5 mM glucose, 10 μg/mL glucose oxidase, and 200 mM Hepes, pH 7.5, for at least 1 h beforehand. Oxygen was removed from solutions containing enzyme by alternately incubating under argon and vacuum for 1 h. Solutions containing reductants were sparged with argon for at least 15 min.
Reactions involving glutathione were carried out in an anaerobic cuvette using a Hewlett-Packard model 8452A diode array spectrophotometer. TyrH (100 μM) in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl was placed in the cuvette with a glutathione solution (1 mM) in the sidearm. The cuvette was then sealed, and its contents were made anaerobic. The contents were then mixed, and the reaction was followed at 350 nm.
Kinetics of Enzyme Oxidation
An anaerobic solution of ferrous TyrH (100–250 μM in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl) was mixed with various concentrations of oxygen dissolved in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl at 20 °C in the stopped-flow spectrophotometer. Enzyme oxidation was monitored by following the increase in absorbance at 350 nm. Solutions containing various amounts of dissolved oxygen were made using a modified MaxBlend medical oxygen blender (Maxtec) to mix oxygen and nitrogen to give the desired concentration of oxygen. The mixed gas was then bubbled through a solution containing 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl for at least 15 min.
Rapid Freeze–Quench Experiments
The freeze–quench experiments were performed as described by Ballou (16) using a rapid-quench apparatus from Update Instrument, Inc. (Madison, WI). The solutions of pterin and TyrH were prepared inside an anaerobic chamber from Vacuum Atmospheres (Hawthorne, CA) with an O2 tension of 2 ppm. The rapid-quench syringes were filled with 2.0 mL of each of the solutions and moved outside the chamber, where they were coupled to a rapid mixer and aging hose. The rapidly mixed reactions were freeze–quenched into liquid isopentane at −130 °C. After packing the resulting snow powder, the EPR spectra were recorded at 7 K using a Bruker EMX instrument (Billerica, MA) and a cryostat from Oxford Instruments Inc. (Concord, MA). The temperature was varied from 6 to 100 K and the power from 0.1 to 10 mM when analyzing for a radical signal. Samples were handled and stored in liquid nitrogen.
HPLC-Based Assay for Determining Pterin Stoichiometry
Standard conditions were 50 mM Hepes, pH 7.5, 100 μM 6-MPH4 or BH4, and 50 μM ferric TyrH. The reaction was initiated by adding the tetrahydropterin. After 5–60 s, the reaction was quenched by adding an aliquot (40 μL) to 0.36 mL of 1% trifluoroacetic acid. After this solution was filtered through a 0.2 μm filter, 50 μL was injected onto a Phenomenex Gemini C18 column (250 × 4.6 mm) with 1% trifluoroacetic acid as the mobile phase. The products were eluted isocratically with a flow rate of 1.5 mL/min and quantified by comparison with authentic standards (2).
Detection of Quinonoid Dihydropterin
Ferric TyrH (50 μM) was added to a solution containing 80 mM Hepes, pH 7.5, and 3 units of sheep dihydropteridine reductase in the bottom of an anaerobic cuvette. NADH (150 μM) was added into one sidearm of the cuvette, and 6-MPH4 (100 μM) was placed in the other. This solution was made anaerobic if necessary. The NADH was then mixed into the solution, and the background rate of NADH consumption was determined by following the reaction at 340 nm. After the background rate was determined, the 6-MPH4 was added to the solution.
Data Analyses
Single-wavelength data were analyzed using the program KaleidaGraph (Synergy Software) and eq 1, which describes a single-exponential process; here, At is the absorbance at any time, A∞ is the final absorbance, A is the amplitude of the phase, and k is the rate constant for the phase. In the case of simple second-order kinetics, the observed rate constants (kobs) were replotted vs reductant concentration to extract the second-order rate constant (k2) from a linear fit. To analyze data for reductants which displayed saturation kinetics, the data were fit to eq 2, where klim is the limiting first-order rate constant for reduction at saturating reductant concentrations, [R] is the concentration of the reductant, and Kd is the apparent dissociation constant for the reductant. The programs Ksim (17) and DynaFit (18) were used for kinetic simulations.
| (1) |
| (2) |
RESULTS
Kinetics of Reduction of Tyrosine Hydroxylase by Tetrahydropterins
The near-UV–visible absorbance spectrum of ferric TyrH exhibits a broad shoulder extending from the ultraviolet to about 500 nm (Figure 1), due to a ligand to metal charge-transfer interaction. The intensity of this absorbance decreases in the ferrous enzyme (Figure 1), providing a spectral signal for monitoring reduction of the enzyme. In preliminary experiments, the absorbance changes between 300 and 700 nm upon mixing 6-MPH4 with the ferric enzyme were followed by photodiode-array stopped-flow spectroscopy (results not shown). In the absence of oxygen, there was a monotonic decrease in absorbance, with a maximal change at about 375 nm. In the presence of oxygen this phase was followed by a much slower (∼400-fold) increase in absorbance; the changes in absorbance in the second phase were consistent with formation of quinonoid dihydropterin (q-PH2) by autoxidation. To avoid complications from the second phase, all analyses of the kinetics of reduction of the enzyme were carried out anaerobically.
Figure 1.
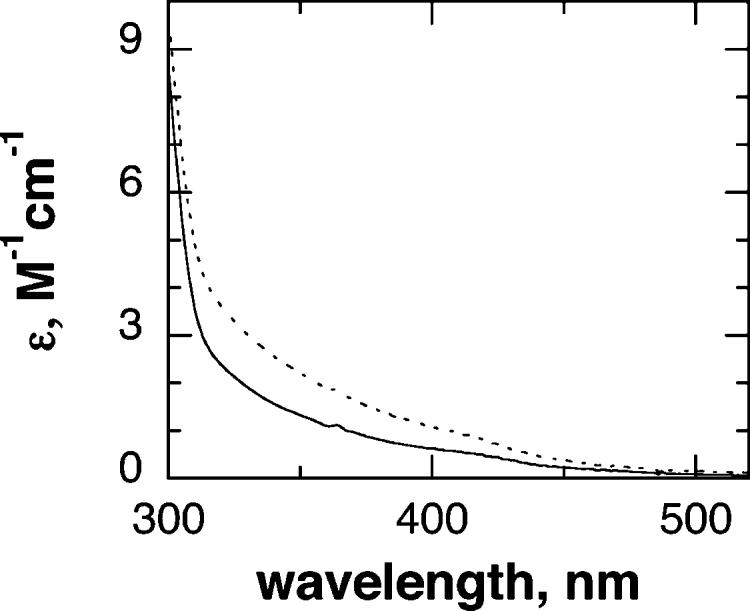
Spectra of ferrous (solid line) and ferric (dashed line) tyrosine hydroxylase.
Figure 2 shows representative absorbance changes at 375 nm when ferric TyrH is mixed with an excess of 6-MPH4 under anaerobic conditions. The changes in absorbance with time could be fit to eq 1, which describes a single first-order reaction. The observed rate constant for reduction increased with the concentration of 6-MPH4, as shown in Figure 3A. The concentration dependence of the observed rate constant fit well to eq 2, which describes a rapid initial binding event characterized by no significant absorbance change followed by an irreversible monophasic decrease in absorbance due to iron reduction. The absorbance changes at 375 nm when BH4 was mixed with ferric TyrH could similarly be described well by a single exponential decay (results not shown). However, in this case the observed rate constant varied directly with the concentration of the tetrahydropterin (Figure 3B), consistent with a simple irreversible second-order reaction.2 The kinetic parameters for reduction of TyrH by both 6-MPH4 and BH4 are given in Table 1.
Figure 2.
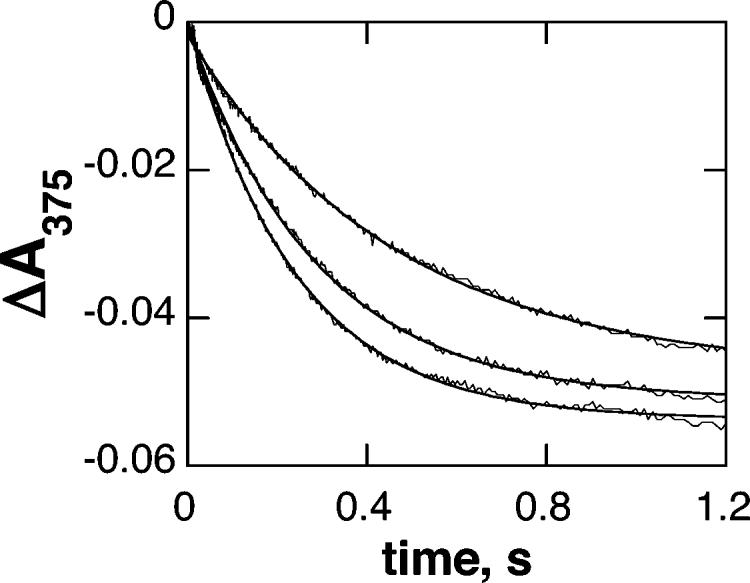
Time course of reduction of tyrosine hydroxylase by 6-MPH4. TyrH (100 μM) was mixed with 0.5, 1, or 2 mM 6-MPH4 at 20 °C. The solid lines are from fits of the data to eq 1.
Figure 3.
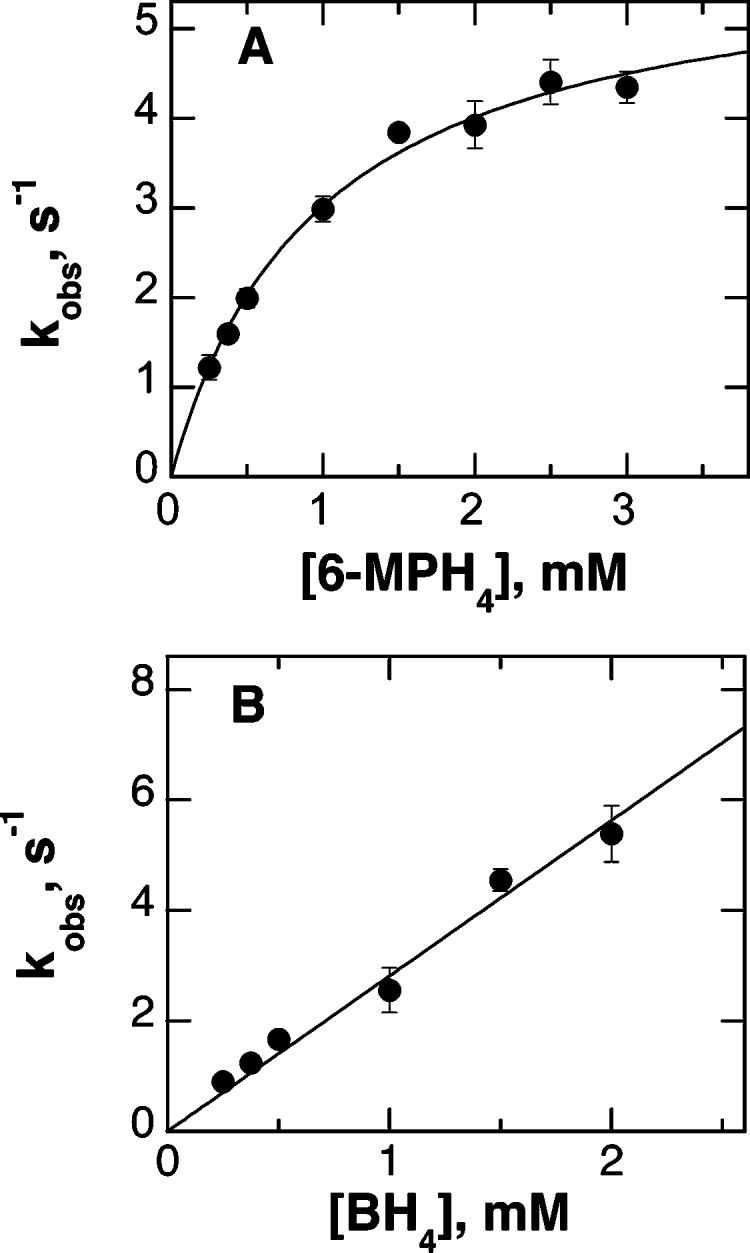
Kinetics of reduction of TyrH by tetrahydropterins: (A) reduction by 6-MPH4; (B) reduction by BH4. The line in (A) is from a fit to eq 2.
Table 1.
Kinetic Parameters for the Reduction of Wild-Type and Mutant TyrHa
| enzyme | reductant | redox potential (mV) |
k2b (mM−1 s−1) | klimc (s−1)b | Kdd (mM) |
|---|---|---|---|---|---|
| wild-type TyrH | 6-methyltetrahydropterin | 270e | 6.2 ± 0.1 | 5.9 ± 0.2 | 0.95 ± 0.10 |
| tetrahydrobiopterin | 270e | 2.8 ± 0.1 | |||
| 5-methyltetrahydrobiopterin | 400f | 0.11 ± 0.01 | |||
| ascorbate | 330g | 0.035 ± 0.001 | |||
| 1,4-benzoquinone | 99h | 0.16 ± 0.01 | |||
| glutathione | 900i | 0.00044 ± 0.00001j | |||
| E332A TyrH | 6-methyltetrahydropterin | 8.1 ± 0.8 | 9.5 ± 1.5 | 1.2 ± 0.3 | |
| S40E TyrH | 6-methyltetrahydropterin | 8.7 ± 0.6 | 7.6 ± 0.5 | 0.87 ± 0.12 | |
| tetrahydrobiopterin | 2.4 ± 0.1 |
Conditions: 100 mM Hepes, pH 7.5, 10% glycerol, 0.1 M KCl, 20 °C.
k2 is the second-order rate constant for reduction. Where the reaction exhibited simple second-order kinetics, k2 was determined from a linear fit of the data. Where the reaction exhibited saturation kinetics, k2 was determined from a fit to eq 2.
klim is the first-order rate constant for reduction from a fit of the data to eq 2.
Kd is the dissociation constant for the reductant determined from a fit of the data to eq 2.
From the one-electron redox potential for BH4 (36), assuming that the change from a dihydroxypropyl group to a methyl group at C6 does not have a significant effect.
Reference 36.
Reference 37.
Reference 38.
Reference 39.
The reduction of TyrH by glutathione was only characterized at a single glutathione concentration. The second-order rate constant for reduction of the wild-type TyrH by glutathione was determined by fitting the data to eq 1 and dividing the observed first-order rate constant by the concentration of glutathione (1 mM).
Wei et al. (19) studied the effect of replacing the BH4 in nitric oxide synthase with 5-methyltetrahydrobiopterin (5-MBH4). In that enzyme, the rate of reduction of the heme–dioxy intermediate was 3-fold greater than the rate with BH4. In addition, the lifetime of the resulting pterin-based radical intermediate was longer. On the basis of this precedent, 5-MBH4 was characterized as a reductant for TyrH. The results were qualitatively similar to those seen upon reacting ferric TyrH with BH4, but the second-order rate constant for enzyme reduction was decreased 24-fold (Table 1). 5-MBH4 is not capable of supporting either DOPA formation or tetrahydropterin oxidation by TyrH (results not shown).
Identification and Stoichiometry of the Pterin Product
Since tetrahydropterins are two-electron reductants, one molecule of 6-MPH4 should be able to reduce two iron atoms in TyrH. Indeed, this stoichiometry has previously been established under somewhat different conditions (2). An HPLC-based assay was used to determine the amounts of tetrahydropterin consumed and dihydropterin produced under the conditions used here for the kinetics. TyrH (50 μM) was mixed with 100 μM 6-MPH4, and the reaction was quenched after a few seconds with 1% trifluoroacetic acid. The amount of dihydropterin produced was then determined by HPLC, again using 1% trifluoroacetic acid as the mobile phase to prevent further tetrahydropterin oxidation. This yielded a stoichiometry of 0.5 mol of 6-methyldihydropterin produced per iron atom present.
There are two forms of dihydropterin, 7,8-dihydropterin (7,8-PH2) and quinonoid dihydropterin (20). The acidic mobile phase for the HPLC effectively prevented tetrahydropterin autoxidation, but at the expense of increasing the rearrangement of q-PH2 to 7,8-PH2, precluding identification of the immediate product of enzyme reduction. Dihydropteridine reductase, which requires q-PH2 as substrate (21), was used to determine if q-PH2 is produced upon reduction of TyrH. A solution of ferric TyrH, sheep dihydropteridine reductase, and NADH was made anaerobic. When 6-MPH4 was added to initiate the reaction, there was no significant NADH consumption, suggesting that q-PH2 is not the product of the anaerobic reduction of TyrH by 6-MPH4. In contrast, under aerobic conditions, all of the NADH was consumed within 100 s of 6-MPH4 addition, consistent with q-6-MPH2 as the product of autoxidation.
Rapid Freeze–Quench EPR
To confirm that the absorbance changes monitored in the stopped-flow experiments correspond to iron reduction, rapid freeze–quench EPR spectroscopy was used to follow the loss of the ferric iron signal. 6-MPH4 (1 mM final concentration) was mixed with ferric TyrH (100 μM final concentration) under the conditions used to determine the kinetics. Samples were rapidly quenched by freezing in isopentane at −130 °C. EPR spectra were then taken at 7 K to determine the amount of ferric iron remaining as a function of time after mixing. The decrease in the EPR signal was well described by a single exponential decay with a rate constant of 2.1 ± 0.8 s−1 (Figure 4). This value is within error of the value determined by stopped-flow absorbance spectroscopy under the same conditions (3.0 ± 0.1 s−1), confirming that the change in absorbance corresponds to iron reduction. After each EPR spectrum was taken, the sample was warmed to 100 K in order to determine if an EPR signal with radical characteristics could be detected. No radical signals were found in any of the samples. A similar analysis was carried out with 10 mM BH4, and again no signal consistent with a radical could be detected (results not shown). In addition, the experiment was repeated with 5-MBH4; no radical signals were seen.
Figure 4.
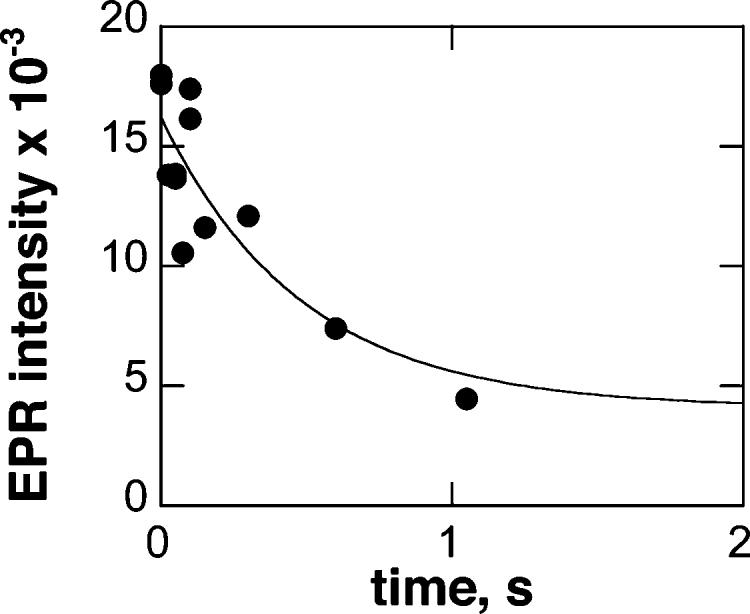
Rapid freeze–quench EPR time course of TyrH reduction. TyrH (100 μM) was reacted with 1 mM 6-MPH4 at 20 °C, and the reaction was quenched at −130 °C at the indicated times. Spectra were obtained at 7 K at 9.39 GHz with the following settings: microwave power, 1.0 mW; modulation amplitude, 20 G; receiver gain, 20000; sweep time, 167 s; time constant, 0.16 ms.
Reduction of TyrH by Alternate Reductants
The kinetic parameters for reduction of TyrH by several other biological reductants were also determined. When ferric TyrH was mixed anaerobically with ascorbate, a monophasic decrease was seen at 350 nm, similar to the results with tetrahydropterins (results not shown). The rate constant for this reaction varied directly with the concentration of ascorbate (Figure 5), consistent with an irreversible second-order reaction.2 The second-order rate constant for iron reduction by ascorbate was 80-fold lower than that for reduction by BH4 (Table 1). Glutathione would also reduce ferric TyrH, eliciting spectral changes similar to those seen with ascorbate and tetrahydropterins. However, the reaction was much slower even than that with ascorbate. The absorbance changes during the reduction of 100 μM TyrH by 1 mM glutathione consisted of a single exponential decrease in absorbance. From the first-order rate constant for the reaction at this concentration of glutathione, the second-order rate constant for reduction of the enzyme by glutathione could be calculated by assuming a simple second-order reaction. This rate constant was 6400-fold slower than that for BH4 (Table 1). Due to the slowness of the reaction, the kinetics were not investigated further. Finally, 1,4-benzoquinone was characterized as a reductant for TyrH. Photodiode-array-monitored reactions (300–700 nm) showed an increase in absorbance at 410 nm upon mixing with Fe(III)-TyrH, presumably due to absorbance by the quinone product. This wavelength was used to determine the effect of the concentration of 1,4-benzoquinone on the rate of reduction. The resulting single-wavelength traces could be fit to two exponential processes, with the first being dependent upon the concentration of 1,4-benzoquinone. The rate constant for the first phase varied directly with the concentration of the quinone, consistent with a second-order reaction 18-fold slower than reduction by BH4 (Table 1). The slower second phase was not investigated further.
Figure 5.
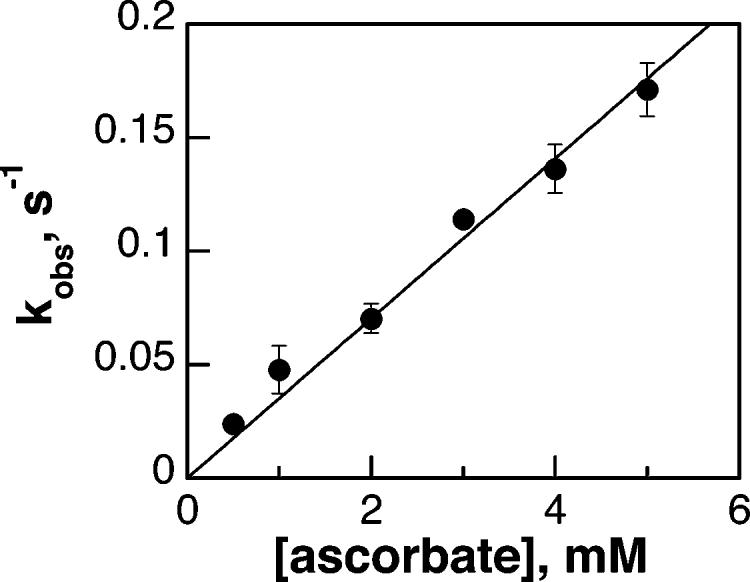
Kinetics of reduction of TyrH by ascorbate under anaerobic conditions. Fe(III)-TyrH (100 μM in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl) was mixed with varying concentrations of ascorbate dissolved in 2 mM HCl at 20 °C. The reaction was monitored at 350 nm, and the resulting traces were fit to eq 1 to obtain the pseudo-first-order rate constants.
Reduction of E332A TyrH by 6-MPH4
The binding sites for BH4 in TyrH and PheH have been identified by structure determination and mutagenesis. Glutamate 332 in TyrH, like the corresponding residue in PheH, glutamate 286, is the only amino acid side chain involved in binding the tetrahydropterin; all other interactions involve the peptide backbone (22). Mutation of glutamate 332 to alanine in TyrH results in an increase in the Km value for 6-MPH4 of 10-fold and an increase in the kcat/Km value of about 30-fold (13). To determine if reduction of the iron involves binding at this site, the kinetic parameters of enzyme reduction were determined using E332A TyrH. These kinetic parameters are similar to those for wild-type TyrH (Table 1).
Kinetics of Oxidation of TyrH
The differences in the near-UV absorbance spectrum between ferric and ferrous TyrH were also used to determine the kinetics of oxidation of the ferrous enzyme. This enzyme form was most readily prepared by addition of a stoichiometric amount of ferrous ammonium sulfate to the apoenzyme anaerobically. Figure 6 shows representative absorbance traces at 350 nm upon mixing ferrous TyrH with oxygenated buffer. The range of oxygen concentrations was limited by solubility, so that the reaction could not be carried out under pseudo-first-order conditions. Instead, the traces were analyzed by fitting to a second-order reaction directly. As shown in Figure 6, a reasonable fit of the data at multiple oxygen concentrations was obtained with a second-order rate constant of 210 M−1 s−1. In light of the role of dopamine in regulating TyrH (Scheme 1), the effect of dopamine on the oxidation kinetics was determined. At 120 μM oxygen, the presence of dopamine resulted in an increase in the rate constant for enzyme oxidation (results not shown). The dependence of the increase on the dopamine concentration could be fit to eq 2 to yield a maximal increase in the rate constant of 2-fold, a concentration of 260 ± 66 μM for a half-maximal increase, and a second-order rate constant for oxidation by dopamine at low dopamine concentrations of 39 ± 13 mM−1 s−1.
Figure 6.
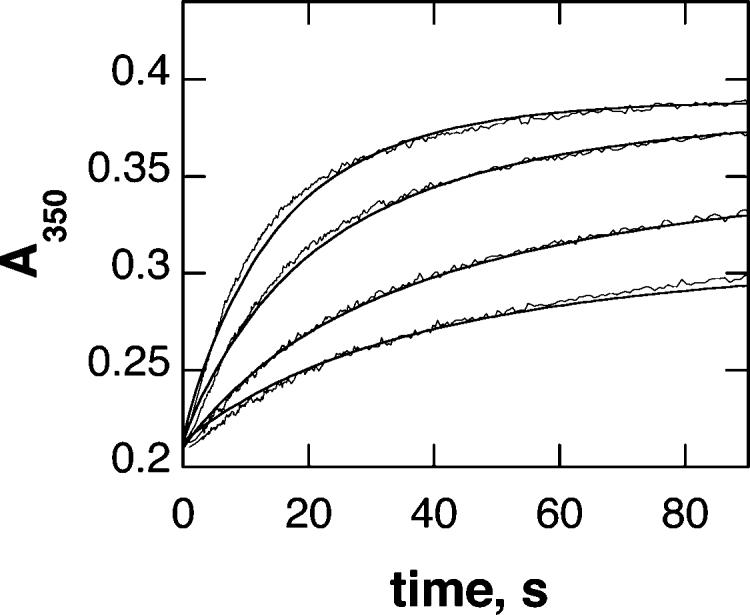
Time course of the reaction of ferrous TyrH with oxygen. TyrH (160 μM) was mixed with 60, 120, 240, or 360 μM oxygen in 200 mM Hepes, pH 7.5, 10% glycerol, and 0.1 M KCl at 20 °C. The lines are from a simulation of a second-order mechanism with a rate constant of 210 M−1 s−1.
To determine if hydrogen peroxide or superoxide was involved in the oxidation, the effects of catalase and superoxide dismutase on the oxidation kinetics were determined. No effect was seen if catalase was present. Addition of increasing amounts of superoxide dismutase decreased the rate constant for oxidation up to 40% (results not shown). This is consistent with some of the oxidation being due to superoxide produced in the initial reaction of molecular oxygen with the iron. The second-order kinetic pattern for the oxidation reaction in the absence of superoxide dismutase suggests that the reaction with superoxide is much more rapid than the initial reaction with O2.
S40E TyrH
Upon phosphorylation of TyrH at Ser40, the rate constant for dissociation of dopamine decreases by 3 orders of magnitude (9). Thus far, this is the only significant effect seen upon phosphorylation. The S40E mutation effectively mimics the effects of phosphorylation at Ser40 on dopamine binding (14). To determine if phosphorylation influences the kinetics of reduction or oxidation of the enzyme, the kinetic parameters for oxidation and reduction of S40E TyrH were determined. The kinetic behavior during reduction of S40E TyrH by tetrahydropterins was indistinguishable from that of the wild-type enzyme. These kinetic parameters are given in Table 1. Similarly, the reaction of ferrous S40E TyrH with oxygen resembled the reaction of the wild-type enzyme. The data could be fit to a second-order mechanism with a second-order rate of 190 M−1 s−1. The kinetic parameters for both oxidation and reduction of the S40E enzyme are similar enough to the values for the wild-type enzyme to suggest that phosphorylation does not alter the kinetics in either direction.
DISCUSSION
The need for the iron in TyrH and the other pterin-dependent amino acid hydroxylases to be in the ferrous form for activity is well established (4, 10, 11, 23). Both TyrH and PheH as isolated contain ferric iron (24-26), and in both cases the iron can be reduced by tetrahydropterins and other reductants (2, 26), with a stoichiometry of 0.5 tetrahydropterin per iron (2, 10). It does not appear that the kinetics of oxidation of the reduced enzymes by molecular oxygen have been analyzed previously, although oxidation of PheH by peroxides has been characterized (27). The results presented here provide insight into the mechanisms of reduction and oxidation of the iron atom in TyrH and the specificity of the reductive step.
With all of the reductants studied here the kinetics of reduction at a single concentration of reductant can be described as a single first-order process with no indication of intermediates. In the case of 6-MPH4, the corresponding rate constant reaches a limiting value at high concentrations of the tetrahydropterin. Similar saturation kinetics may also occur with ascorbate and BH4, but in both cases the concentrations required are well beyond those found physiologically, and even with 6-MPH4 the Kd is in the millimolar range. Thus, the kinetics of reduction of TyrH by the various reductants are best compared using the second-order rate constant k2 and considering this as a simple second-order reaction. Tetrahydropterins are clearly the best reductants based on this criterion, with 6-MPH4 somewhat more effective than BH4 (Table 1). Shiman et al. (12) previously reported that 6-MPH4 is similarly about 3-fold more effective than BH4 in reductively activating PheH. The basis for this preference is not clear, since the replacement of the dihydroxypropyl side chain of BH4 with a methyl moiety is unlikely to significantly alter the redox potential of the tetrahydropterin. The difference in rate constants may simply reflect the increased size of BH4 compared to 6-MPH4.
While the redox potential of TyrH has not been reported, the Em value of human PheH is 207 mV (28), and the identical arrangements of the iron ligands in the two enzymes (5) suggest that they will have similar redox potentials. The one-electron redox potential of the reductants in Table 1 spans this value. Unsurprisingly, for the compounds with redox potentials greater than that of BH4, the rate constant for reduction decreases with increasing redox potential. However, 1,4-benzoquinone, with a significantly lower redox potential than that of BH4, is also a slower reductant. This suggests that there is some specificity for the reductant. This specificity and the apparent saturation kinetics seen at high concentrations of 6-MPH4 raise the possibility that there is a binding site for the reductant. The structures of TyrH and PheH with bound pterin have been described (29, 30). The tetrahydropterin binds close to the iron, with its C4a carbon 5.9 Å from the iron. While there are several hydrogen bonds between the tetrahydropterin and the peptide backbone, the only interaction with the side chain of an amino acid residue is with a glutamyl residue, Glu322 in TyrH. The lack of an effect of the E322A mutation on the kinetic parameters for reduction of TyrH by 6-MPH4 (Table 1) indicates that reduction of the metal does not occur predominantly from tetrahydropterin bound in the same fashion as is utilized for catalysis. A minimal model consistent with the kinetic data is that the reductive step is adequately described as a simple collisional event.
Reduction of the iron atom from ferric to ferrous is a one-electron reduction, so that a pterin radical must be involved, at least transiently. There are two possible mechanisms for the decay of such a trihydropterin radical, dismutation to form a tetrahydropterin and dihydropterin or reaction with another ferric ion (Scheme 2). No pterin radical could be detected when the reduction was monitored by freeze–quench EPR spectroscopy. The conditions for these experiments were such that an accumulation of a radical species to 10% of the enzyme concentration (i.e., 10 μM) would have been detectable. This suggests that if a radical species is formed, it decays very rapidly. To evaluate the likely fate of any trihydropterin radical, the program Ksim (17) was used to simulate the reduction using the mechanism of Scheme 2. Initial concentrations of 10 mM BH4 and 100 μM TyrH were used. The published rate constant for the dismutation of the neutral trihydropterin radical at pH 7.4 of 93 mM−1 s−1 (31) was used for kd, and the measured k2 value of 2.8 mM−1 s−1 was used for kr. With these rate constants, if dismutation were the predominant pathway for the breakdown of the pterin radical, it would accumulate to ∼55 μM, well above the expected limit of detection. The value of kr2 had to be at least 5000 mM−1 s−1 to prevent accumulation of the trihydropterin radical to more than 5% of the enzyme concentration. This suggests that the predominant pathway for decay of the trihydropterin radical is via reduction of another iron atom to form the 7,8-dihydropterin.
Scheme 2.

The kinetic mechanism for oxidation of ferrous TyrH by molecular oxygen is well described as a simple second-order reaction. The oxidation of the ferric ion would produce superoxide as an initial product, and the superoxide would be expected to be able to oxidize another iron atom. The decrease in the rate constant for oxidation of ferrous TyrH in the presence of superoxide dismutase suggests that superoxide is responsible for a significant fraction of the oxidation. Since the kinetics are adequately described by a second-order mechanism, the reaction between superoxide and ferrous TyrH must be substantially more rapid than the reaction of molecular oxygen, consistent with the radical nature of superoxide.
The kinetics of reduction and oxidation of the active site iron in TyrH must be placed in a physiological context to evaluate the biological relevance of these reactions. The physiological concentration of BH4 is 10–100 μM (32). At these concentrations, a second-order rate constant for reduction of TyrH of 2.8 mM−1 s−1 would result in an in vivo half-life for the reduction of TyrH by BH4 of 3–25 s. This is consistent with BH4 being a physiological reductant. The magnitude of the rate constant for oxidation is also sufficient for oxidation of TyrH by O2 to be physiologically relevant. At a typical intracellular oxygen concentration of 20 μM in the brain (33), the oxidation of TyrH would be competitive with turnover, which occurs at about 1 s−1 (34). Dopamine has a stimulating effect on the rate constant for oxidation, suggesting that a direct oxidation of TyrH by dopamine is also possible. The average intracellular concentration of dopamine is 5–10 μM (35), at which the rate constant for oxidation of TyrH by dopamine would be about 0.3 s−1. However, most of this dopamine is present in secretory vesicles and hence inaccessible to TyrH, so that the concentration in the cytosol must be 1–2 orders of magnitude lower. This suggests that direct oxidation by dopamine, while possible, is less significant than oxidation by O2.
Mutagenesis of Ser40 of TyrH to glutamate has previously been shown to mimic the dramatic effect of phosphorylation of that residue on dopamine binding (14). The lack of an effect of this mutation on the kinetics of reduction and oxidation suggests that these steps are not regulated by phosphorylation of this residue. The kinetic parameters for oxidation and reduction reported here do establish that the interconversion of the ferrous and ferric enzyme can be sufficiently rapid in the cell for the regulatory model of Scheme 1 to be effective in regulating the enzyme. The slowest step by far in the interconversion of ferrous TyrH and the ferric enzyme–dopamine complex is the release of dopamine from the ferric enzyme (9), the step which is dramatically affected by phosphorylation.
Footnotes
This work was funded in part by NIH Grants R01 GM47291 (to P.F.F.), T32 GM08523 (to P.A.F.), and R01 GM39451 (to S.W.R.).
Abbreviations: TyrH, tyrosine hydroxylase; PheH, phenylalanine hydroxylase; DOPA, dihydroxyphenylalanine; BH4, tetrahydrobiopterin; 6-MPH4, 6-methyltetrahydropterin; 5-MBH4, 5-methyltetrahydrobiopterin; q-BH2, quinonoid dihydrobiopterin; 7,8-PH2, 7,8-dihydropterin.
With both tetrahydrobiopterin and ascorbate as reductants, the data could be fit to eq 2 with a slight improvement in the statistics of the fit. In the case of BH4, this yielded klim and Kd values of 35 ± 30 s−1 and 10.8 ± 10.4 mM, respectively. With ascorbate, the klim and Kd values were 0.78 ± 0.74 s−1 and 18 ± 10 mM, respectively. For both reductants, the alternate analysis had no effect on the value of the second-order rate constant for reduction at low concentrations of reductant, k2. Because of the large errors in the estimates using eq 2 and the very high Kd values, the simpler model of an irreversible second-order reaction was deemed sufficient to describe the reduction under physiological conditions.
REFERENCES
- 1.Fitzpatrick PF. The aromatic amino acid hydroxylases. In: Purich DL, editor. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley & Sons; New York: 2000. pp. 235–294. [DOI] [PubMed] [Google Scholar]
- 2.Ramsey AJ, Hillas PJ, Fitzpatrick PF. Characterization of the active site iron in tyrosine hydroxylase: Redox states of the iron. J. Biol. Chem. 1996;271:24395–24400. doi: 10.1074/jbc.271.40.24395. [DOI] [PubMed] [Google Scholar]
- 3.Sura G, Daubner SC, Fitzpatrick PF. Effects of phosphorylation by protein kinase A on binding of catecholamines to the human tyrosine hydroxylase isoforms. J. Neurochem. 2004;90:970–978. doi: 10.1111/j.1471-4159.2004.02566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fitzpatrick PF. The metal requirement of rat tyrosine hydroxylase. Biochem. Biophys. Res. Commun. 1989;161:211–215. doi: 10.1016/0006-291x(89)91582-9. [DOI] [PubMed] [Google Scholar]
- 5.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: specificity and thermodynamics. Biochemistry. 2000;39:773–778. doi: 10.1021/bi991901r. [DOI] [PubMed] [Google Scholar]
- 7.Andersson KK, Cox DD, Que L, Jr., Flatmark T, Haavik J. Resonance Raman studies on the blue-green-colored bovine adrenal tyrosine 3-monooxygenase (tyrosine hydroxylase). Evidence that the feedback inhibitors adrenaline and noradrenaline are coordinated to iron. J. Biol. Chem. 1988;263:18621–18626. [PubMed] [Google Scholar]
- 8.Daubner SC, Lauriano C, Haycock JW, Fitzpatrick PF. Site-directed mutagenesis of serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J. Biol. Chem. 1992;267:12639–12646. [PubMed] [Google Scholar]
- 9.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 10.Wallick DE, Bloom LM, Gaffney BJ, Benkovic SJ. Reductive activation of phenylalanine hydroxylase and its effect on the redox state of the non-heme iron. Biochemistry. 1984;23:1295–1302. doi: 10.1021/bi00301a043. [DOI] [PubMed] [Google Scholar]
- 11.Marota JJA, Shiman R. Stoichiometric reduction of phenylalanine hydroxylase by its cofactor: A requirement for enzymatic activity. Biochemistry. 1984;23:1303–1311. doi: 10.1021/bi00301a044. [DOI] [PubMed] [Google Scholar]
- 12.Shiman R, Gray DW, Hill MA. Regulation of rat liver phenylalanine hydroxylase. I. Kinetic properties of the enzyme's iron and enzyme reduction site. J. Biol. Chem. 1994;269:24637–24646. [PubMed] [Google Scholar]
- 13.Daubner SC, Fitzpatrick PF. Site-directed mutants of charged residues in the active site of tyrosine hydroxylase. Biochemistry. 1999;38:4448–4454. doi: 10.1021/bi983012u. [DOI] [PubMed] [Google Scholar]
- 14.McCulloch RI, Daubner SC, Fitzpatrick PF. Effects of substitution at serine 40 of tyrosine hydroxylase on catecholamine binding. Biochemistry. 2001;40:7273–7278. doi: 10.1021/bi010546d. [DOI] [PubMed] [Google Scholar]
- 15.Fitzpatrick P, Ralph EC, Ellis HR, Willmon OJ, Daubner SC. Characterization of metal ligand mutants of tyrosine hydroxylase: Insights into the plasticity of a 2-histi-dine-1-carboxylate triad. Biochemistry. 2003;42:2081–2088. doi: 10.1021/bi0271493. [DOI] [PubMed] [Google Scholar]
- 16.Ballou DP. Freeze-quench and chemical-quench techniques. Methods Enzymol. 1978;54:85–93. doi: 10.1016/s0076-6879(78)54010-x. [DOI] [PubMed] [Google Scholar]
- 17.Barshop BA, Wrenn RF, Frieden C. Analysis of numerical methods for computer simulation of kinetic processes: Development of KINSIM-a flexible, portable system. Anal. Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 18.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 19.Wei C, Wang Z, Arvai AS, Hemann C, Hille R, Getzoff ED, Stuehr DJ. Structure of tetrahydrobiopterin tunes its electron transfer to the heme-dioxy intermediate in nitric oxide synthase, Biochemistry. 2003;42:1969–1977. doi: 10.1021/bi026898h. [DOI] [PubMed] [Google Scholar]
- 20.Pfleiderer W. Chemistry of naturally occurring pterins. In: Blakley RL, Benkovic SJ, editors. Folates and Pterins. II. John Wiley & Sons; New York: 1985. pp. 43–114. [Google Scholar]
- 21.Shiman R. Phenylalanine hydroxylase and dihydropterin reductase. In: Blakley RL, Benkovic SJ, editors. Folates and Pterins. II. John Wiley & Sons; New York: 1985. pp. 179–249. [Google Scholar]
- 22.Andersen OA, Stokka AJ, Flatmark T, Hough E. 2.0 Å resolution crystal structures of the ternary complexes of human phenylalanine hydroxylase catalytic domain with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine or l-norleucine: Substrate specificity and molecular motions related to substrate binding. J. Mol. Biol. 2003;333:747–757. doi: 10.1016/j.jmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Moran GR, Daubner SC, Fitzpatrick PF. Expression and characterization of the catalytic core of tryptophan hydroxylase. J. Biol. Chem. 1998;273:12259–12266. doi: 10.1074/jbc.273.20.12259. [DOI] [PubMed] [Google Scholar]
- 24.Haavik J, Andersson KK, Petersson L, Flatmark T. Soluble tyrosine hydroxylase (tyrosine 3-monooxygenase) from bovine adrenal medulla: Large-scale purification and physicochemical properties. Biochim. Biophys. Acta. 1988;953:142–156. doi: 10.1016/0167-4838(88)90019-2. [DOI] [PubMed] [Google Scholar]
- 25.Gottschall DW, Dietrich RF, Benkovic SJ. Phenylalanine hydroxylase. Correlation of the iron content with activity and the preparation and reconstitution of the apoenzyme. J. Biol. Chem. 1982;257:845–849. [PubMed] [Google Scholar]
- 26.Fisher DB, Kirkwood R, Kaufman S. Rat liver phenylalanine hydroxylase, an iron enzyme. J. Biol. Chem. 1972;247:5161–5167. [PubMed] [Google Scholar]
- 27.Hill MA, Marota JJA, Shiman R. Reaction of rat liver phenylalanine hydroxylase with fatty acid hydroperoxides. Characterization and mechanism. J. Biol. Chem. 1988;263:5646–5655. [PubMed] [Google Scholar]
- 28.Hagedoorn P-L, Schmidt PP, Andersson KK, Hagen WR, Flatmark T, Martinez A. The effect of the substrate, dihydrobiopterin and dopamine on the EPR spectroscopic properties and the midpoint potential of the catalytic iron in recombinant human phenylalanine hydroxylase. J. Biol. Chem. 2001;276:22850–22856. doi: 10.1074/jbc.M009458200. [DOI] [PubMed] [Google Scholar]
- 29.Goodwill KE, Sabatier C, Stevens RC. Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 Å resolution: self-hydroxylation of phe300 and the pterin-binding site. Biochemistry. 1998;37:13437–13445. doi: 10.1021/bi981462g. [DOI] [PubMed] [Google Scholar]
- 30.Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J. Mol. Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 31.Patel KB, Stratford MRL, Wardman P, Everett SA. Oxidation of tetrahydrobiopterin by biological radicals and scavenging of the trihydrobiopterin radical by ascorbate. Free Radical Biol. Med. 2002;32:203–211. doi: 10.1016/s0891-5849(01)00777-8. [DOI] [PubMed] [Google Scholar]
- 32.Levine RA, Miller LP, Lovenberg W. Tetrahydrobiopterin in striatum: localization in dopamine nerve terminals and role in catecholamine synthesis. Science. 1981;214:919–921. doi: 10.1126/science.6117945. [DOI] [PubMed] [Google Scholar]
- 33.Rolett EL, Azzawi A, Liu KJ, Yongbi MN, Swartz HM, Dunn JF. Critical oxygen tension in rat brain: a combined 31P NMR and EPR oximetry study. Am. J. Physiol. 2000;279:9–16. doi: 10.1152/ajpregu.2000.279.1.R9. [DOI] [PubMed] [Google Scholar]
- 34.Fitzpatrick PF, Chlumsky LJ, Daubner SC, O'Malley KL. Expression of rat tyrosine hydroxylase in insect tissue culture cells and purification and characterization of the cloned enzyme. J. Biol. Chem. 1990;265:2042–2047. [PubMed] [Google Scholar]
- 35.Buu NT, Duhaime J, Kuchel O. Effects of l-DOPA on the concentrations of free and sulfoconjugated catecholamines in plasma, cerebrospinal fluid, urine, and central and peripheral nervous system tissues of the rat. J. Neurochem. 1985;44:787–792. doi: 10.1111/j.1471-4159.1985.tb12884.x. [DOI] [PubMed] [Google Scholar]
- 36.Gorren ACF, Kungl AJ, Schmidt K, Werner ER, Mayer B. Electrochemistry of pterin cofactors and inhibitors of nitric oxide synthase. Nitric Oxide. 2001;5:176–186. doi: 10.1006/niox.2001.0332. [DOI] [PubMed] [Google Scholar]
- 37.Buettner GR, Oberley LW. Considerations in the spin trapping of superoxide and hydroxyl radical in aqueous systems using 5,5-dimethyl-1-pyrroline-1-oxide. Biochem. Biophys. Res. Commun. 1978;83:69–74. doi: 10.1016/0006-291x(78)90398-4. [DOI] [PubMed] [Google Scholar]
- 38.Ilan YA, Czapski G, Meisel D. The one-electron transfer redox potentials of free radicals. I. The oxygen/superoxide system. Biochim. Biophys. Acta. 1976;430:209–224. doi: 10.1016/0005-2728(76)90080-3. [DOI] [PubMed] [Google Scholar]
- 39.Koppenol WH. A thermodynamic appraisal of the radical sink hypothesis. Free Radical Biol. Med. 1993;14:91–94. doi: 10.1016/0891-5849(93)90513-t. [DOI] [PubMed] [Google Scholar]