

Abstract
The role of a polypeptide loop in tyrosine hydroxylase (TyrH) whose homolog in phenylalanine hydroxylase (PheH) takes on a different conformation when substrates are bound has been studied using site-directed mutagenesis. The loop spans positions 177 to 191; alanine was introduced into those positions, introducing one alanine substitution per TyrH variant. Mutagenesis of residues in the center of the loop resulted in alterations in the KM values for substrates, the Vmax value for dihydroxyphenylalanine (DOPA) synthesis, and the coupling of tetrahydropterin oxidation to tyrosine hydroxylation. The variant with the most altered KM value for 6-methyltetrahydropterin was TyrH F184A. The variants with the most affected Ktyr values were those with substitutions in the center of the loop, TyrH K183A, F184A, D185A, P186A and D187A. These five variants also had the most reduced Vmax values for DOPA synthesis. Alanine substitution in positions 182–186 resulted in lowered ratios of tyrosine hydroxylation to tetrahydropterin oxidation. TyrH F184Y and PheH Y138F, variants with the residue at the center of the loop substituted with the residue present at the homologous position in the other hydroxylase, were also studied. The V/Ktyr to V/Kphe ratios for these variants were altered significantly, but the results did not suggest that F184 of TyrH or Y138 of PheH plays a dominant role in determining amino acid substrate specificity.
Keywords: tyrosine hydroxylase, conformational change, iron-oxygen chemistry, substrate specificity, alanine scanning
Introduction
Tyrosine hydroxylase (TyrH, E. C. 1.14.16.2) is a non-heme iron monooxygenase that catalyzes the hydroxylation of tyrosine to dihydroxyphenylalanine (DOPA), the rate-determining step of catecholamine biosynthesis. TyrH requires ferrous iron and tetrahydrobiopterin (BH4) to perform the reaction. The enzyme belongs to the family of aromatic amino acid hydroxylases, which also contains phenylalanine hydroxylase (PheH, EC 1.14.16.1).1 The reactions catalyzed by these two enzymes are shown in Scheme 1. The eukaryotic enzymes have regulatory (R) domains at their amino termini, coiled coils at their carboxyl termini, and catalytic (C) domains of about 330 residues at their cores.2,3 The R domains vary in length, with ∼150 residues in TyrH, and ∼115 in PheH, and are not homologous,4-7 while the sequences of the homologous C domains of rat TyrH and rat PheH are ∼60% identical. The C domains contain all of the residues required for catalysis.8-12
Scheme 1.

Reactions catalyzed by tyrosine hydroxylase and phenylalanine hydroxylase.
In light of the structural similarities, the catalytic mechanisms of the aromatic amino acid hydroxylases have generally been assumed to be the same. Mechanistic analyses of TyrH have been the most detailed. Studies with substrate analogs and isotopically labeled substrates and solvent have led to the mechanism shown in Scheme 2.13 The tetrahydropterin binds first, followed by oxygen, and then tyrosine. After the ternary complex is formed, irreversible chemistry occurs, giving the hydroxylating intermediate, X, in the rate-limiting step of the reaction.14 The subsequent hydroxylation of the amino acid substrate is followed by rapid product release. The hydroxylating intermediate has not been trapped for direct structural analysis, but existing data are consistent with an Fe(IV)O species.15 For TyrH, no reaction has been detected between Fe(II), oxygen, and tetrahydropterin in the absence of tyrosine. Based on this, a conformational change has been suggested to occur after amino acid binding.16
Scheme 2.

Kinetic mechanism of rat tyrosine hydroxylase.
Several structures have been determined of the C domains of TyrH17 and PheH.18 The first crystal structures showed that the arrangements of the residues that bind iron and pterin are essentially identical.19 Most of the subsequent structures have been of PheH in complex with substrates or inhibitors. The only structures available in which the iron is in the catalytically active ferrous form are of the PheH catalytic domain. One of these has both tetrahydropterin and an amino acid bound and is the only available structure containing an amino acid substrate.20 When this structure is compared to that of ferrous PheH with only tetrahydropterin bound, a polypeptide loop over the active site is seen to have very different conformations in the two structures. Figure 1 shows the position of the loop in the presence and absence of amino acid.20 The side-chain of Y138, at the middle of the loop, moves 20 Å, from an exposed position on the surface of the protein to a buried location in the active site, where it packs against the amino acid binding site. Residues 128–148 of PheH correspond to residues 174–194 of TyrH, and the residue at the position homologous to PheH Y138 is TyrH F184 (Scheme 3). The two available structures of TyrH are of the C domain of the ferric enzyme in the presence and absence of dihydropterin; there is no structure available with an amino acid bound. In the structure of the free enzyme, no electron density is seen for residues 178–188,17 while the structure of the dihydrobiopterin complex lacks residues 183–185,21 suggesting that this region of TyrH is also highly mobile. The movement of this flexible loop may constitute the conformational change that occurs after tyrosine binds, bringing together the iron and the substrates and allowing oxygen activation to occur.
Figure 1.
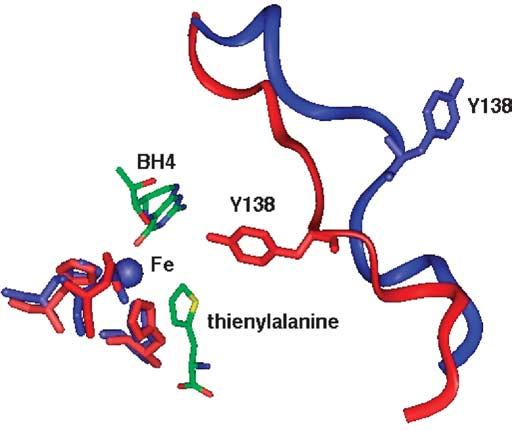
Different conformations of the 131–145 loop of tetrahydrobiopterin-bound PheH in the presence and absence of thienylalanine. The blue loop is from the binary complex (PDB ID 1DMW) and the red loop is from the ternary complex (PDB ID 1KW0). The active site iron is shown as a ball and is surrounded by its ligands H285, H290 and E376. The substrates tetrahydrobiopterin and thienylalanine are indicated.
Scheme 3.

Alignment of the amino acid sequences of PheH, line 1, and TyrH, line 2, in the region of the putative flexible loop. Identical amino acids are in bold, and similar ones are outlined.
Previous studies of the substrate specificities of TyrH and PheH identified the homologous residues V379 in PheH and D425 in TyrH as critical for the amino acid substrate specificity.22 The single mutation of D425 in TyrH to valine, the residue found at the corresponding position in PheH, changes the substrate preference in favor of phenylalanine over tyrosine by about 50,000-fold, such that TyrH D425V is almost completely unable to hydroxylate tyrosine but is very effective in the hydroxylation of phenylalanine. In contrast, the homologous variant PheH V379D shows only a modest decrease in the preference for phenylalanine as a substrate. Clearly, an aspartate at this critical position is required for efficient hydroxylation of tyrosine, but other residues are also critical for the substrate specificity of PheH. F184 and Y138, in the centers of the corresponding loops of TyrH and PheH, differ when comparing the two enzymes but are conserved across species for each hydroxylase. This suggests that the identity of this residue may be a determinant of the amino acid substrate specificity of these two enzymes.
Here, we describe the results of studies to determine whether this polypeptide loop is important for substrate binding and catalysis in TyrH. Two strategies were employed. The first was to replace each residue in the loop of TyrH with an alanine residue. The second was to characterize the amino acid substrate specificity of TyrH F184Y and PheH Y138F in order to determine whether this residue plays a role in the substrate specificity of the two hydroxylases.
Results
Alanine scanning mutagenesis of the flexible loop of TyrH
Residues 177–191 of TyrH correspond to the loop over the active site in PheH, which has different positions in the presence and absence of an amino acid substrate. In order to probe the importance of this putative flexible loop in TyrH, each residue from C177 to D191 was substituted in turn with alanine and the resulting proteins expressed in Escherichia coli. Each variant could be purified to homogeneity without complications. The KM values for tyrosine, phenylalanine, and 6-MePH4, as well as the Vmax values for DOPA formation were determined for each variant. These parameters are summarized in Table 1.
Table 1.
Steady-state kinetic parameters for variants with alanine substitutions in the flexible loop of tyrosine hydroxylase
| Enzyme | K6MePH4a (μM) | Ktyra (μM) | Vmaxa (min−1) | Kpheb (μM) | DOPA formation/ 6-MePH4 oxidation |
|---|---|---|---|---|---|
| Wild-type TyrH | 37±7 | 42±9 | 250±16 | 100±15 | 1.0±0.1 |
| C177A | 51±3 | 92±13 | 247±12 | 538±103 | 0.80±0.07 |
| H178A | 33± 2 | 86±4 | 222±4 | 188±20 | 0.83±0.02 |
| H179A | 30±4 | 57±8 | 148±6 | 385±129 | 0.51±0.003 |
| L180A | 43±3 | 175±3 | 96±8 | 636±89 | 0.27±0.01 |
| V181A | 60±6 | 20±3 | 57±2 | 65±10 | 0.23±0.05 |
| T182A | 50±10 | 24±4 | 113±7 | 85±10 | 0.48±0.01 |
| K183A | 47±3 | 39±4 | 38±1 | 117±23 | 0.35±0.01 |
| F184A | 21±3 | 11±4 | 12±1 | 51±11 | 0.16±0.01 |
| D185A | 61±4 | 34±11 | 19±2 | 51±10 | 0.38±0.01 |
| P186A | 57±5 | 11±1 | 31±1 | 21±5 | 0.14±0.01 |
| D187A | 43±5 | 18±4 | 62±5 | 80±29 | 0.45±0.015 |
| L188A | 41±4 | 22±4 | 107±23 | 40±21 | 0.52±0.01 |
| D189A | 33±3 | 47±20 | 48±11 | 140±17 | 0.43±0.01 |
| L190A | 66±6 | 65±7 | 179±10 | 262±32 | 1.08±0.03 |
| D191A | 60±7 | 35±7 | 352±30 | 85±39 | 0.88±0.03 |
Determined from the rate of DOPA formation in 50 mM Hepes (pH 7.0), 400 μM 6-MePH4, 100 μg/ml catalase, 1 mM dithiothreitol, 10 μM ferrous ammonium sulfate at 30 °C. For the measurement of the K6MePH4 value, the concentration of 6-MePH4 was varied from 8–400 μM at a tyrosine concentration of 100 μM; for the measurement of the Ktyr value, the concentration of tyrosine was varied from 5–1200 μM at a 6-MePH4 concentration of 400 μM.
Determined from rates of 6-MePH4 oxidation in the presence of 5–2000 μM phenylalanine in 50 mM Hepes (pH 7.0), 200 μM 6-MePH4,60 μg/ml catalase, 200 μM NADH, 10 μM ferrous ammonium sulfate, 0.05 unit/ml sheep dihydropteridine reductase at 30 °C.
The parameter affected least by substitution of alanine in the loop is the KM value for 6-methyltetrahydropterin, the first substrate to bind during turnover (Figure 2(a)).13 All of the K6-MePH4 values are comparable to that for wild-type TyrH. The residue showing the largest change is TyrH F184A; even in this case there is only a twofold decrease.
Figure 2.
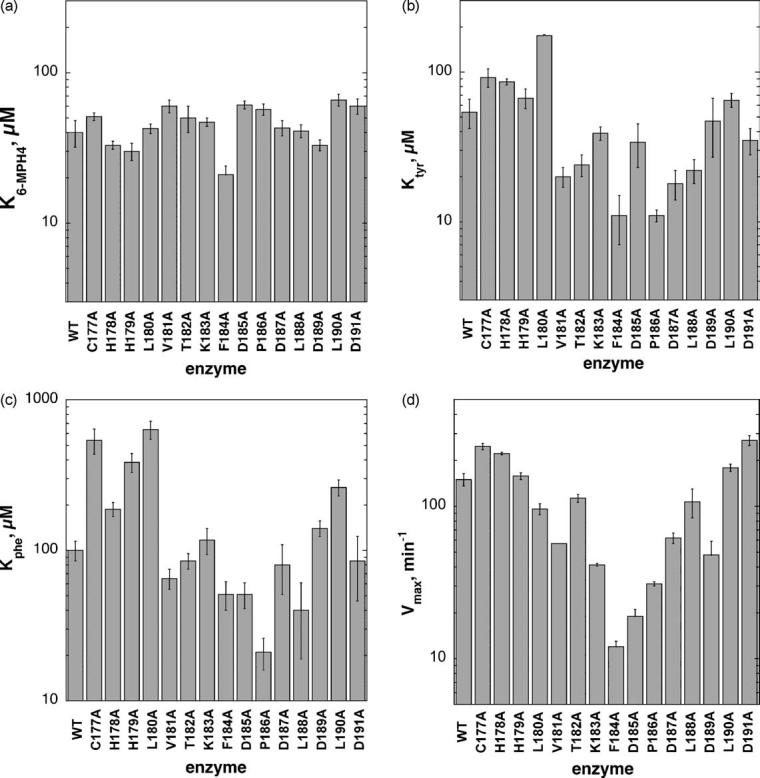
Steady-state kinetic parameters of alanine-scanning variants of TyrH: (a) KM values for 6-methyltetrahydropterin, (b) KM values for tyrosine, (c) KM values for phenylalanine, (d) Vmax values for DOPA synthesis.
The Ktyr values show a much larger range of effects of the mutations (Figure 2(b)). They vary from a high of 175 μM for TyrH L180A to a low of 11 μM for TyrH F184A and P186A, compared to a Ktyr value for wild-type TyrH of 42 μM. The variants with alanine substituted in positions 177–180 have higher or unchanged Ktyr values, and the variants with alanine from positions 181–188 have lower Ktyr values. Phenylalanine is also a good substrate for TyrH.22,23 The effects of alanine substitution on the Kphe values generally mirror the effects on the Ktyr values (Figure 2(c)). Once again TyrH F184A has the lowest value, alterations at positions 177–180 result in higher or unchanged Kphe values, and alterations at positions 181–188 result in lower Kphe values. However, while TyrH C177A and H179A have very altered Kphe values relative to wild-type TyrH, their Ktyr values are less affected.
The effects of the mutations on Vmax values for DOPA formation are shown in Figure 2(d). There is a clear relationship between the Vmax value and the position in the loop of the mutation. The variants with alanine in positions 181–188 (but particularly 183–186) have drastically lowered Vmax values. Variants at the outer edges of the loop have Vmax values comparable to wild-type TyrH. The catalytic mechanism of the aromatic amino acid hydroxylases can be divided into two partial reactions, the formation of the hydroxylating intermediate in a reaction that does not involve the amino acid substrate and the subsequent addition of oxygen to the amino acid by the hydroxylating intermediate.16 These two reactions are tightly coupled in the wild-type enzyme, so that 1 mol of DOPA is produced per mol of tetrahydropterin oxidized. A decrease in the Vmax value for DOPA formation could be due to a decrease in the rate of overall turnover or an increase in the unproductive breakdown of the hydroxylating intermediate. In order to determine whether only tyrosine hydroxylation or both hydroxylation and pterin oxidation were depressed in the variants with low Vmax values, the relative amounts of both products were measured for each. These data appear in Table 1 and Figure 3, and show that the mutations indeed alter the efficiency of coupling of oxygen activation to amino acid hydroxylation; again a mutation in positions 180–189 is more detrimental than a mutation at the ends of the loop.
Figure 3.
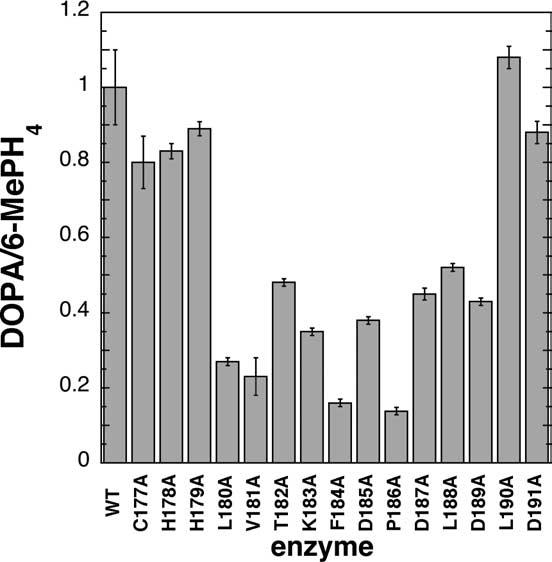
Coupling of tyrosine hydroxylation to tetrahydropterin oxidation by the TyrH variants with alanine substituted for the residues in the flexible loop. Coupling was measured by spectrophotometric determination of the amount (nmol) of 6-MePH4 oxidized followed by determination of the amount (nmol) of DOPA produced in the same reaction.
Contribution of F184 of TyrH and Y138 of PheH to substrate specificity
TyrH F184A displays the most altered kinetic parameters of any of the TyrH variants described here, demonstrating that this position is indeed important for binding and catalysis. The variants TyrH F184Y and PheH Y138F were characterized to determine if the identity of this residue plays a role in determining the substrate specificity of the two enzymes. The steady-state kinetic parameters for these two variants are given in Table 2.
Table 2.
Steady-state kinetic parameters for PheHY138F and TyrHF184Y
| Phenylalanine hydroxylation |
Tyrosine hydroxylation |
||||||
|---|---|---|---|---|---|---|---|
| Enzyme | Kphe (μM) |
Vmax (min−1) |
V/Kphe (min−1 μM−1) |
Ktyr (μM) |
Vmax (min−1) |
V/Ktyr (min−1 μM−1) |
(V/Ktyr)/(V/Kphe) |
| WT TyrH | 100±15a | 96±12a | 0.96±0.19a | 42±9 | 250±16 | 6.0±0.9 | 6.2±1.5 |
| TyrH F184Y | 39±7 | 130±17 | 3.4±0.5 | 51±12 | 122±16 | 2.4±0.3 | 0.71±0.14 |
| WT PheHa | 252±54 | 960±41 | 3.8±0.8 | nd | <0.1 | (3.6±0.3)×10−5 | (0.0095±0.0022)×10−3 |
| PheH Y138F | 365±74 | 680±40 | 1.9±0.37 | 1090±470 | 0.44±0.14 | (22±3)×10−5 | (0.15±0.067)×10−3 |
| PheH Y138F/V379D | 980±150 | 140±7 | 0.15±0.017 | 1650±640 | 1.8±0.67 | (117±27)×10−5 | (7.8±4.6)×10−3 |
| PheH V379Da | 191±27 | 38±2 | 0.2±0.03 | nd | <0.1 | (2.3±0.2)×10−5 | (0.12±.02)×10−3 |
nd, not determined.
From Daubner et al.22
Wild-type TyrH prefers tyrosine over phenylalanine as a substrate by about an order of magnitude based on the relative V/K values for the two amino acid substrates. In contrast, TyrH F184Y shows a slight preference for phenylalanine as a substrate. Overall, there is a decrease of about ninefold in the ratio of the V/Ktyr value for tyrosine to that for phenylalanine. This corresponds to a lower KM value for phenylalanine and a lower Vmax value for DOPA formation.
PheH has a much stronger preference for phenylalanine as a substrate than does TyrH for its native substrate. Indeed, for wild-type PheH, the low solubility of tyrosine and the high Ktyr value make it impossible to determine Ktyr or Vmax values; instead, only a V/Ktyr value can be determined.22 In contrast, the variant PheH Y138F has a Ktyr value in the measurable range, around 1 mM. Since a lower limit for the Ktyr value for the wild-type enzyme can be estimated as at least 10 mM, the change due to substitution of Y138 with a tyrosine residue is at least ninefold. The Vmax value for tyrosine formation by PheH Y138F is still quite low compared to wild-type TyrH, but it is measurable. Replacing Y138 of PheH with phenylalanine thus has an effect on the relative preference for tyrosine versus phenylalanine comparable to that seen upon the reverse mutation in TyrH, an increase of about tenfold in the ratio of the V/Ktyr value for tyrosine to that for phenylalanine.
Since V379 was previously shown to contribute to the amino acid specificity of PheH,22 the substrate specificity of the double variant PheH Y138F/V379D was characterized to determine whether the two mutations would have a synergistic effect. PheH Y138F/V379D is somewhat better than PheH Y138F at hydroxylation of tyrosine. The Ktyr value is not significantly decreased by incorporation of the second mutation. The Vmax and V/Ktyr values both increase about fivefold in the double variant compared to the effect of the single mutation. PheH Y138F/V379D is about 32-fold better at tyrosine hydroxylation than the wild-type enzyme when the V/Ktyr value is used as a criterion. The double mutant shows a larger loss in its ability to hydroxylate phenylalanine, a 13-fold decrease in the V/Kphe value compared to the PheH Y138F enzyme. These changes are the result of both altered Vmax and Kphe values. As a consequence of the changes in the V/K values for both substrates, PheH Y138F/V379D has a decrease in its preference for phenylalanine over tyrosine of about 52 when compared to either single mutant and about 800-fold when compared to the wild-type enzyme. The double variant is still not a good tyrosine hydroxylase; its V/Ktyr value is almost 8000-fold lower than that for wild-type TyrH and it still prefers phenylalanine as a substrate by over 100-fold. The relative specificities of all these mutant proteins are shown in Figure 4.
Figure 4.
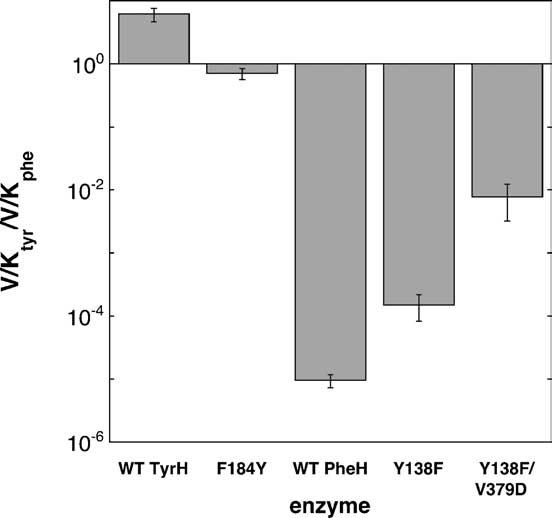
Amino acid substrate specificity of variants of TyrH and PheH at the central position of the flexible loop.
Combination of the V379D mutation with mutation of H264 to glutamine results in the PheH variant with the highest V/Ktyr value reported to date.22 Consequently, we also determined the substrate specificity of the triple variant PheH Y138F/H264Q/V379D; however, this enzyme was no more able to hydroxylate tyrosine than PheH Y138F/V379D (results not shown).
Discussion
The present results firmly support the region of TyrH from residues 179–189 as critical for the optimal functioning of TyrH. Replacement with alanine of residues in this loop resulted in sweeping alterations in steady-state kinetic parameters. TyrH variants with alanine substituted for residues K183 to P186 had the greatest alteration of Ktyr values and Vmax values, consistent with the central region of the loop being most critical for binding and catalysis. Furthermore, mutation of any residue from L180 to D189 to alanine resulted in smaller amounts of tyrosine hydroxylation relative to tetrahydropterin oxidation.
These results contribute further details to our understanding of the molecular events that occur during the catalytic reactions of TyrH and the two other aromatic amino acid hydroxylases. A great deal is known about the active site of TyrH, based on the structures of TyrH and PheH and the effects of mutagenesis of active site residues. The active site of TyrH is a cleft in the enzyme surface about 17 Å deep and 15 Å wide. The iron lies about 10 Å into the cleft,17 bound by two histidine residues (H331 and H336, TyrH numbering) and one glutamate (E376).16 The tetrahydropterin binds on one side of the iron. Site-directed mutagenesis of TyrH has established that the side-chains of F300 and E3328,12,24 are involved in tetrahydropterin binding; the crystal structures of both PheH25 and TyrH21 confirm the interactions of these residues with the tetrahydropterin. Site-directed mutagenesis has also established that the side-chains of R316 and D328 are involved in tyrosine binding; in the PheH structure, R270 (the residue corresponding to TyrH R316) interacts directly with the amino acid carboxylate while the corresponding aspartate orients the side-chain of the arginine residue.
The steady-state kinetic mechanism of TyrH is ordered, as shown in Scheme 2, with the tetrahydropterin binding first. For both the amino acid and the tetrahydropterin, the KM value is close to or equal to the Kd value,16,26 allowing the steady-state kinetic parameters to be used as measures of binding. Mutation of residues 177–191 to alanine has little effect on the KM value for 6-MePH4, with the exception of a modest change for the F184A enzyme. This is consistent with a lack of direct interaction of side-chains of loop residues with this substrate. In PheH, the 128–148 loop has the same position in the presence and absence of bound pterin,25 and the visible residues in the loop in TyrH can be overlaid upon those in PheH, both in the presence and absence of bound pterin.
Mutagenesis of loop residues to alanine results in much larger changes in the KM value for the amino acid substrate, whether tyrosine or phenylalanine is used. This suggests that the side-chains of residues in the loop are important for proper formation of the amino acid substrate binding site. Mutagenesis of residues 177–180 results in increases in the KMvalues of up to eightfold. In contrast, mutagenesis of residues 181–188 results in decreases in the KM values, suggesting that binding of the amino acid substrate is actually tighter in these mutant enzymes. These effects can be accounted for by a model in which side-chains of loop residues contribute to the structure of the amino acid binding site and binding of the amino acid to the wild-type enzyme involves some unfavorable interactions. One reasonable role for such unfavorable interactions would be to tightly restrict the amino side-chain in an orientation highly favorable to oxygen addition. Removal of a side-chain from a residue between 181 and 188 would relax the binding site for the amino acid, relieving some of the unfavorable interactions and increasing the amino acid affinity. However, this would result in the amino acid being less than optimally placed for hydroxylation, decreasing the rate of hydroxylation. Such a model is consistent with the result that the enzymes with the lowest Vmax values also have the lowest KM values for the amino acid substrate.
The decreased efficiency with which the reducing equivalents of the tetrahydropterin are coupled to amino acid hydroxylation can be explained by a slower hydroxylation rate combined with disruption of the loop by the mutations. For most of the mutant enzymes, the unproductive oxidation of tetrahydropterin completely accounts for the decrease in the Vmax value. We had previously proposed that the one role of the flexible loop is to exclude water from the active site, where it could react with the hydroxylating intermediate.15 Crystal structures of PheH show there to be fewer water molecules in the active site of the ternary complex than in the uncomplexed protein. Solvent isotope effects with TyrH E326A, which also consumes a large excess of tetrahydropterin unproductively, suggest that the E326A mutation disrupts an interaction with the loop and allows access of water to the iron during catalysis, discharging the hydroxylating intermediate before hydroxylation can occur.15 The data presented here support the idea that the role of the flexible loop is to create a smaller hydrophobic space, which prevents water from reacting with the hydroxylating intermediate. Increased access of solvent water would lead to more opportunity for water to discharge the hydroxylating intermediate before it reacted with the amino acid substrate. Such a model does not completely account for the effects of mutating residues 183–185. For these enzymes, the rate constant for the reaction of the hydroxylating intermediate with the amino acid substrate must also be decreased several-fold, presumably due to its less than optimal orientation in the active site.
The substrate specificity of the aromatic amino acid hydroxylases is determined solely within the catalytic domains.6 Several residues have been identified as contributing to the amino acid substrate specificity of these enzymes.22,27,28 The most dramatic changes in specificity to date occur upon mutating TyrH Q310 and D425, and the corresponding residues in PheH, H264 and V379D. TyrH D425V has an 8000-fold preference for phenylalanine over tyrosine, and is thus a clear example of reversed substrate specificity upon the mutagenesis of a single residue. In contrast, PheH H264Q/V379D has a 3000-fold decrease in its preference for phenylalanine, but still greatly prefers phenylalanine over tyrosine as a substrate.22 The inability to reverse the substrate specificity of PheH by single mutations, in contrast to the results with TyrH, suggests that other as yet unidentified residues in PheH are important in determining its substrate specificity. Because the central residue of the loop in PheH, Y138, has very different conformations in crystals depending on whether the amino acid substrate is present, we focused on this residue and its homolog in TyrH, F184. Mutation of F184 of TyrH to tyrosine and of Y138 of PheH to phenylalanine did result in changes in the relative preferences of the two enzymes for tyrosine versus phenylalanine, but the effects are better described as a loss of specificity rather than a reverse of specificity.
The effects of mutating TyrH F184 to tyrosine are quite modest, and much less than the effects of mutating D425, Q310, or H323. In the present case, the mutant enzyme shows little preference for tyrosine versus phenylalanine, while mutation of the other three residues reverses the substrate specificity of TyrH.22 The analogous mutation of Y138 in PheH to phenylalanine has little effect on the kinetic parameters for phenylalanine as substrate, but does yield an enzyme with clearly detectable activity at tyrosine hydroxylation. The V/Ktyr value for this enzyme is the highest of any single mutant of PheH to date, although it is still quite low. It is possible that Y138 interferes with tyrosine binding but not phenylalanine binding because the active site pocket does not have space for both hydroxyl groups. The unchanged Kphe value for PheH Y138F with respect to wild-type PheH is consistent with this. The substrate specificity of PheH Y138F is very close to that of the V379D enzyme, consistent with the proximity of these residues in the active site. Incorporation of the latter mutation does alter the specificity in a synergistic fashion, further increasing the V/Ktyr value and decreasing the V/Kphe value. Still, the variant phenylalanine hydroxylases bear little resemblance to native tyrosine hydroxylase. Clearly there are other residues that restrict the formation of DOPA from tyrosine.
In summary, we have identified a polypeptide loop in TyrH, and especially one residue in the very center of the loop, F184, which optimizes the coupling of tetrahydropterin oxidation to DOPA synthesis from tyrosine. Although this position is filled by Y138 in PheH, the identity of this residue is not a dominant factor in the different substrate specificities of the two hydroxylases. The movement of this flexible loop, which encompasses positions 179–189 in TyrH, may be involved in the rate-limiting step of the TyrH reaction by bringing the substrates and the iron into the close proximity required for formation of the hydroxylating intermediate. The loop also protects the intermediate from solvent.
Experimental Procedures
Materials
Custom oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Restriction endonucleases were from New England Biolabs Inc. (Beverly, MA). Pfu DNA polymerase was obtained from Stratagene USA (La Jolla, CA). DNA sequencing was performed using the BigDye kit of ABI (Foster City, CA). Plasmids were purified using kits from Qiagen Inc. (Valencia, CA). (6R)-BH4 and 6-methyltetrahydropterin (6-MePH4) were purchased from B Schircks Laboratories (Jona, Switzerland). Leupeptin and pepstatin were obtained from Peptides International (Louisville, KY). Catalase was obtained from Roche (Gaithersburg, MD). Distilled glycerol was from Invitrogen (Carlsbad, CA). Escherichia coli strain Over-Expresse™ C41(DE3) from Avidis (Saint Beauzire, France) was used for expression of TyrH. E. coli strain BL21(DE3) was used for PheH expression (Novagen, WI), and OmniMaxe™ was used for DNA preparations and cloning (Invitrogen). l-Tyrosine, l-DOPA, l-phenylalanine and dihydropteridine reductase were from Sigma Corp. (St Louis, MO). Heparin-Sepharose CL-6B and phenyl-Sepharose Fast Flow were purchased from Amersham-Pharmacia Biotech Inc, now GE Healthcare (Piscataway, NJ).
Expression and purification of recombinant proteins
Plasmids for expression of TyrH and PheH have been described.6 All the mutagenesis reactions used the QuikChange site-directed mutagenesis method (Strata-gene). For each construct, the entire coding region was sequenced to ensure that no other mutations were present. For expression of tyrosine hydroxylases, competent E. coli C41(DE3)29 were transformed with the plasmids; for phenylalanine hydroxylases, BL21(DE3)30 was used. In all cases, expression and enzyme purification were carried out as described for the wild-type enzymes.6 The concentrations of TyrH were determined using a value of of 1.04 and a mass of 56,000 Da.31 Concentrations of PheH were determined using of 1.0 and a mass of 52,500 Da.32 Enzyme purity was assessed by denaturing polyacrylamide gel electrophoresis.33
Enzyme assays
TyrH activity was determined using a colorimetric assay that measures the amount of DOPA produced.6 Standard conditions were 0.1 μM TyrH, 100 μM tyrosine, 400 μM 6-MePH4, 100 μg/ml of catalase, 10 μM ferrous ammonium sulfate, 1 mM dithiothreitol, 50 mM Hepes (pH 7.0), 30 °C. Assays were carried out for 2 min. For K6-MePH4 determinations the concentration of 6-MePH4 was 40–400 μM, and for Ktyr determinations the concentration of tyrosine was 5–1200 μM.
To measure tyrosine hydroxylation by PheH, a radio-metric assay was used to measure the amount of tritium released from 3,4-[3H]tyrosine as described.22 The ingredients for the assay were the same as for the colorimetric assay with the substitution of 1 mM β-mercaptoethanol for dithiothreitol.
Rates of tetrahydropterin oxidation were determined using a coupled assay with dihydropteridine reductase, monitoring the decrease in absorbance at 340 nm due to NADH oxidation.34 The assays contained 5–2000 μM phenylalanine in addition to 200 μM 6-MePH4, 80mM Hepes (pH 7.0), 60 μg/ml of catalase, 200 μM NADH, and 0.05 unit/ml of sheep dihydropterin reductase at 30 °C.
Tyrosine formation from phenylalanine was measured by monitoring absorbance changes at 275 nm due to the production of tyrosine.35 The assays contained 80 mM Hepes (pH 7.0), 5–3000 μM phenylalanine, 200 μM 6-MePH4, 100 μg/ml of catalase, 1 mM dithiothreitol, 10 μM ferrous ammonium sulfate at 30 °C.
Steady-state kinetic data were fit directly to the Michaelis-Menten equation (equation (1)). In some cases the data were fit to an equation that takes into account inhibition by high levels of tyrosine (equation (2)):36
| (1) |
| (2) |
To determine ratios of tetrahydropterin oxidation to tyrosine hydroxylation, the coupled assay with dihydropteridine reductase was performed, and 10 ml of 50% HCl was added to quench the reaction after 90 s. The amount of DOPA in the quenched reaction was determined using the colorimetric assay.
Acknowledgements
This work was supported, in part, by NIH grant GM47291.
Abbreviations used
- TyrH
tyrosine hydroxylase
- PheH
phenylalanine hydroxylase
- DTPA
diethylenetriaminepentaacetate
- 6-MePH4
6-methyltetrahydropterin
- BH4
tetrahydrobiopterin
- DOPA
dihydroxyphenylalanine
- Ktyr
KM value for tyrosine
- Kphe
KM value for phenylalanine
- K6-MePH4
KM value for 6-methyltetrahydropterin
Footnotes
Publisher's Disclaimer: This article was originally published in a journal published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues that you know, and providing a copy to your institution's administrator.
References
- 1.Fitzpatrick PF. Thetetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, Vrana KE. Leucine zippers and coiled-coils in the aromatic amino acid hydroxylases. Neurochem. Int. 1991;18:27–31. doi: 10.1016/0197-0186(91)90031-8. [DOI] [PubMed] [Google Scholar]
- 3.Walker SJ, Liu X, Roskoski R, Vrana KE. Catalytic core of rat tyrosine hydroxylase: terminal deletion analysis of bacterially expressed enzyme. Biochim. Biophys. Acta. 1994;1206:113–119. doi: 10.1016/0167-4838(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 4.Ledley FD, DiLella AG, Kwok SCM, Woo SLC. Homology between phenylalanine and tyrosine hydroxylases reveals common structural and functional domains. Biochemistry. 1985;24:3389–3394. doi: 10.1021/bi00335a001. [DOI] [PubMed] [Google Scholar]
- 5.Grenett HE, Ledley FD, Reed LL, Woo SLC. Full-length cDNA for rabbit tryptophan hydroxylase: functional domains and evolution of aromatic amino acid hydroxylases. Proc. Natl Acad. Sci. USA. 1987;84:5530–5534. doi: 10.1073/pnas.84.16.5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daubner SC, Hillas PJ, Fitzpatrick PF. Characterization of chimeric pterin dependent hydroxylases: contributions of the regulatory domains of tyrosine and phenylalanine hydroxylase to substrate specificity. Biochemistry. 1997;36:11574–11582. doi: 10.1021/bi9711137. [DOI] [PubMed] [Google Scholar]
- 7.Abate C, Smith JA, Joh TH. Characterization of the catalytic domain of bovine adrenal tyrosine hydroxylase. Biochem. Biophys. Res. Commun. 1988;151:1446–1453. doi: 10.1016/s0006-291x(88)80524-2. [DOI] [PubMed] [Google Scholar]
- 8.Daubner SC, Fitzpatrick PF. Site-directed mutants of charged residues in the active site of tyrosine hydroxylase. Biochemistry. 1999;38:4448–4454. doi: 10.1021/bi983012u. [DOI] [PubMed] [Google Scholar]
- 9.Ramsey AJ, Daubner SC, Ehrlich JI, Fitzpatrick PF. Identification of iron ligands in tyrosine hydroxylase by mutagenesis of conserved histidinyl residues. Protein Sci. 1995;4:2082–2086. doi: 10.1002/pro.5560041013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balasubramanian S, Carr RT, Bender CJ, Peisach J, Benkovic SJ. Identification of metal ligands in Cu(II)-inhibited Chromobacterium violaceum phenylalanine hydroxylase by electron spin echo envelope modulation analysis of histidine to serine mutations. Biochemistry. 1994;33:8532–8537. doi: 10.1021/bi00194a019. [DOI] [PubMed] [Google Scholar]
- 11.Teigen K, Frøystein NÅ, Martinez A. The structural basis of the recognition of phenylalanine and pterin cofactors by phenylalanine hydroxylase: implications for the catalytic mechanism. J. Mol. Biol. 1999;294:807–823. doi: 10.1006/jmbi.1999.3288. [DOI] [PubMed] [Google Scholar]
- 12.Dickson PW, Jennings IG, Cotton RGH. Delineation of the catalytic core of phenylalanine hydroxylase and identification of glutamate 286 as a critical residue for pterin function. J. Biol. Chem. 1994;269:20369–20375. [PubMed] [Google Scholar]
- 13.Fitzpatrick PF. The steady state kinetic mechanism of rat tyrosine hydroxylase. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 14.Fitzpatrick PF. Studies of the rate-limiting step in the tyrosine hydroxylase reaction: alternate substrates, solvent isotope effects, and transition state analogs. Biochemistry. 1991;30:6386–6391. doi: 10.1021/bi00240a006. [DOI] [PubMed] [Google Scholar]
- 15.Frantom PA, Fitzpatrick PF. Uncoupled forms of tyrosine hydroxylase unmask kinetic isotope effects on chemical steps. J. Am. Chem. Soc. 2003;125:16190–16191. doi: 10.1021/ja0383165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited diseases. Nature Struct. Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 18.Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC. Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nature Struct. Biol. 1997;4:995–1000. doi: 10.1038/nsb1297-995. [DOI] [PubMed] [Google Scholar]
- 19.Loeb KE, Westre TE, Kappock TJ, Mitic N, Glasfeld E, Caradonna JP, et al. Spectroscopic characterization of the catalytically competent ferrous site of the resting, activated, and substrate-bound forms of phenylalanine hydroxylase. J. Am. Chem. Soc. 1997;119:1901–1915. [Google Scholar]
- 20.Andersen OA, Flatmark T, Hough E. Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine, and its implications for the mechanism of catalysis and substrate activation. J. Mol. Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
- 21.Goodwill KE, Sabatier C, Stevens RC. Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 Å resolution: self-hydroxylation of phe300 and the pterin-binding site. Biochemistry. 1998;37:13437–13445. doi: 10.1021/bi981462g. [DOI] [PubMed] [Google Scholar]
- 22.Daubner SC, Melendez J, Fitzpatrick PF. Reversing the substrate specificities of phenylalanine and tyrosine hydroxylase: aspartate 425 of tyrosine hydroxylase is essential for l-DOPA formation. Biochemistry. 2000;39:9652–9661. doi: 10.1021/bi000493k. [DOI] [PubMed] [Google Scholar]
- 23.Fitzpatrick PF. Kinetic isotope effects on hydroxylation of ring-deuterated phenylalanines by tyrosine hydroxylase provide evidence against partitioning of an arene oxide intermediate. J. Am. Chem. Soc. 1994;116:1133–1134. [Google Scholar]
- 24.Ellis HR, Daubner SC, McCulloch RI, Fitzpatrick PF. Phenylalanine residues in the active-site of tyrosine hydroxylase: mutagenesis of Phe300 and Phe309 to alanine and metal ion-catalyzed hydroxylation of Phe300. Biochemistry. 1999;38:10909–10914. doi: 10.1021/bi991160u. [DOI] [PubMed] [Google Scholar]
- 25.Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J. Mol. Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 26.Francisco WA, Tian G, Fitzpatrick PF, Klinman JP. Oxygen-18 kinetic isotope effect studies of the tyrosine hydroxylase reaction: evidence of rate limiting oxygen activation. J. Am. Chem. Soc. 1998;120:4057–4062. [Google Scholar]
- 27.Daubner SC, Fitzpatrick PF. Mutation to phenylalanine of tyrosine 371 in tyrosine hydroxylase increases the affinity for phenylalanine. Biochemistry. 1998;37:16440–16444. doi: 10.1021/bi981648f. [DOI] [PubMed] [Google Scholar]
- 28.McKinney J, Teigen K, Frøystein NA, Salaün C, Knappskog PM, Haavik J, Martĺnez A. Conformation of the substrate and pterin cofactor bound to human tryptophan hydroxylase. Important role of Phe313 in substrate specificity. Biochemistry. 2001;40:15591–15601. doi: 10.1021/bi015722x. [DOI] [PubMed] [Google Scholar]
- 29.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 30.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 31.Andersson KK, Vassort C, Brennan BA, Que L, Jr, Haavik J, Flatmark T, et al. Purification and characterization of the blue-green rat phaeochromocytoma (PC12) tyrosine hydroxylase with a dopamine-Fe(III) complex reversal of the endogenous feedback inhibition by phosphorylation of serine-40. Biochem. J. 1992;284:687–695. doi: 10.1042/bj2840687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiman R, Gray DW. Substrate activation of phenylalanine hydroxylase. A kinetic characterizaion. J. Biol. Chem. 1980;255:4793–4800. [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Lazarus RA, Dietrich RF, Wallick DE, Benkovic SJ. On the mechanism of action of phenylalanine hydroxylase. Biochemistry. 1981;20:6834–6841. doi: 10.1021/bi00527a015. [DOI] [PubMed] [Google Scholar]
- 35.Shiman R, Jones SH, Gray DW. Mechanism of phenylalanine regulation of phenylalanine hydroxylase. J. Biol. Chem. 1990;265:11633–11642. [PubMed] [Google Scholar]
- 36.Royo M, Daubner SC, Fitzpatrick PF. Effects of mutations in tyrosine hydroxylase associated with progressive dystonia on the activity and stability of the protein. Proteins: Struct. Funct. Genet. 2005;58:14–21. doi: 10.1002/prot.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]