

Abstract
The blood coagulation protein factor XI (FXI) consists of a pair of disulfide-linked chains each containing four apple domains and a catalytic domain. The apple 4 domain (A4; F272-E362) mediates noncovalent homodimer formation even when the cysteine involved in an intersubunit disulfide is mutated to serine (C321S). To understand the role of noncovalent interactions stabilizing the FXI dimer, equilibrium unfolding of wild-type A4 and its C321S variant was monitored by circular dichroism, intrinsic tyrosine fluorescence and dynamic light scattering measurements as a function of guanidine hydrochloride concentration. Global analysis of the unimolecular unfolding transition of wild-type A4 revealed a partially unfolded equilibrium intermediate at low to moderate denaturant concentrations. The optically detected equilibrium of C321S A4 also fits best to a three-state model in which the native dimer unfolds via a monomeric intermediate state. Dimer dissociation is characterized by a dissociation constant, Kd, of ~90 nM (in terms of monomer), which is in agreement with the dissociation constant measured independently using fluorescence anisotropy. The results imply that FXI folding occurs via a monomeric equilibrium intermediate. This observation sheds light on the effect of certain naturally occurring mutations, such as F283L, which lead to intracellular accumulation of non-native forms of FXI. To investigate the structural and energetic consequences of the F283L mutation, which perturbs a cluster of aromatic side chains within the core of the A4 monomer, it was introduced into the dissociable dimer, C321S A4. NMR chemical shift analysis confirmed that the mutant can assume a native-like dimeric structure. However, equilibrium unfolding measurements show that the mutation causes a four-fold increase in the Kd for dissociation of the native dimer and a 1 kcal/mol stabilization of the monomer, resulting in a highly populated intermediate. Since the F283 side chain does not directly participate in the dimer interface, we propose that the F283L mutation leads to increased dimer dissociation by stabilizing a monomeric state with altered side chain packing that is unfavorable for homodimer formation.
Keywords: FXI, blood coagulation, protein folding, NMR, fluorescence, CD
Introduction
Factor XI (FXI) is a human blood coagulation zymogen that is converted into its proteolytically active form, factor XIa (FXIa), in response to blood vessel injury (reviewed in ref. 1). FXI circulates in the blood plasma at a concentration of ~30 nM as a 160 kDa homodimeric glycoprotein. The enzymatic activators of FXI (thrombin, factor XIIa or FXIa) cleave an internal R369-I370 bond in each monomer of FXI, yielding FXIa, which consists of a heavy chain of 369 residues and a light chain of 238 residues. The heavy chain comprises four 90 to 91-residue tandem repeat sequences termed apple domains (A1–A4) and the light chain represents a trypsin-like serine protease domain with active site residues H413, D464, and S557. The physiological substrate of FXIa is factor IX, which it activates by cleaving two scissile bonds at R145 and R180.2 The primary role of FXIa is recognized as part of a feedback loop, referred to as the consolidation phase of blood coagulation, resulting from activation of FXI by the small quantities of thrombin that are generated by the tissue factor pathway prior to its inhibition by tissue factor pathway inhibitor.3,4
The apple domains of FXI are 23–34% identical to one another at the sequence level, and 58% identical with their respective apple domains of the intrinsic pathway zymogen prekallikrein (PK), which circulates as a monomer. Each apple domain of FXI contains regions involved in interacting with other proteins and with the platelet surface.1 Papagrigoriou et al. recently reported a crystal structure of the dimeric FXI zymogen, which confirms that self-association of FXI is mediated by dimerization of A4 involving both noncovalent interactions and an intersubunit disulfide bond (Figure 1).5 FXI A4 directs reversible dimer formation even when the cysteine responsible for the covalent linkage between the monomers, Cys321, is changed to serine, resulting in a fully active protein (C321S A4) that exists as a dimer at concentrations as low as 80 nM (monomer concentration), suggesting that the dissociation constant (Kd) for reversible dimer dissociation is comparable to this value.6
Figure 1.
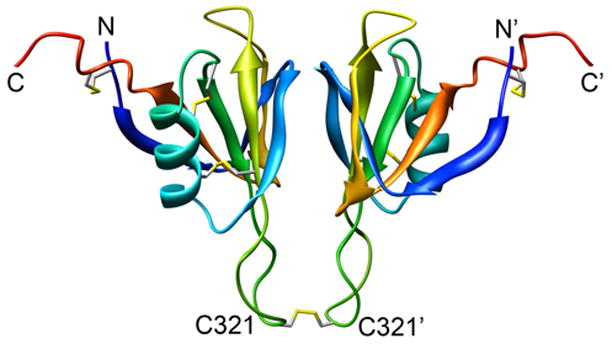
Ribbon diagram of the of the A4 dimer (residues F272 through E361 of FXI), based on the crystal structure of the FXI zymogen.5 Disulfide bonds, including the one linking the monomers via Cys321, are shown in stick representation.
FXI is the only homodimeric protein in the blood coagulation cascade, and the dimeric property of FXI is thought to be important for concerted cleavage of both scissile bonds in factor IX.7 The physiological relevance of FXI dimerization was examined using FXI and PK chimeric proteins in which the A4 domain of FXI was replaced by the monomeric PK A4 domain. The resulting monomeric chimera, FXI/PKA4, failed to activate factor IX in the presence of platelets, but was fully active in solution and in kaolin-triggered plasma clotting assays.8 This finding suggests that the dimeric structure of FXIa is required for normal factor IX activation in the presence of platelets.8
Several mutations in the population at large have been observed to lead to FXI deficiency, which gives rise to a bleeding disorder of variable, but usually mild severity.1 Some of these mutations, especially those in the FXI A4 domain, are thought to affect the folding and dimerization of FXI, due to impaired secretion into plasma from hepatocytes.9,10 The type III mutation, which results in a F283L amino acid change in A4 due to a single base substitution in exon 9 of the gene encoding FXI,11 is one of the more common mutations causing FXI deficiency. Along with the type II (E117stop) mutation, the F283L mutation accounts for more than 95% of the known mutations from FXI deficient patients.12 The FXI levels in the plasma of FXI deficient patients homozygous for this mutation are <10% of normal levels, while heterozygous patients have 67% of normal levels.11,13 Although the catalytic activity in plasma is <10%, purified FXIa F283L exhibits normal enzymatic activity,10 and pulse-chase and immunoprecipitation experiments showed increased levels of monomeric FXI within the cell,10,14 suggesting that in vivo, the F283L mutation induces retention of FXI within liver cells. Thus, elucidating the structural and thermodynamic basis for FXI homodimer formation is important for understanding its role in the consolidation phase of blood coagulation.
In order to study dimerization and unfolding of the FXI A4 domain, the cysteine (C321) responsible for covalent linkage with the adjacent monomer was mutated to serine (C321S), and a global analysis method was used to fit guanidine hydrochloride (GuHCl) dependent unfolding data of C321S A4 measured by circular dichroism (CD) and intrinsic fluorescence, and to determine the free energy of dissociation describing the transition from dimeric to monomeric states as well as the free energy of monomer unfolding. In addition, the type III mutation (F283L) was introduced into the C321S construct, and its overall secondary structure studied using 15N HSQC NMR spectra and unfolding behavior, relative to the C321S A4 protein. These data show that the F283L mutation in FXI does not cause major disruption of the structure, but perturbs the structure of the dimer interface enough to raise the Kd by four-fold, which has a major impact on protein maturation during FXI synthesis, helping to explain the lowered levels of expression of FXI F283L.
Results
Optically detected unfolding and dimer dissociation
In order to understand the role of the A4 domain in mediating dimerization of FXI, we used various spectroscopic and hydrodynamic methods to monitor the reversible denaturant-induced unfolding transition of the covalently linked A4 dimer and its dissociable variant, C321S (see Figure 1). To ensure complete equilibration, a series of samples at different denaturant concentrations were incubated overnight prior to spectroscopic analysis (see Methods). The CD signal at 225 nm was used to monitor changes in helical secondary structure content as a function of GuHCl. As shown in Figure 2A, wt A4 undergoes a gradual increase in ellipticity with increasing GuHCl concentration up to about 4 M GuHCl, followed by a sharp transition centered at ~4.8 M GuHCl, corresponding to a significant loss in helix content concomitant with the main structural unfolding transition. The linear portion above 5.5 M GuHCl is indicative of non-cooperative changes within the denatured population. In contrast, the changes in CD signal below 4 M GuHCl show some curvature and can be attributed to a conformational transition from native A4 dimer to a partially structured intermediate state. The intermediate possesses ~25% higher CD signal at 225 nm compared to the 0 M GuHCl sample.
Figure 2.
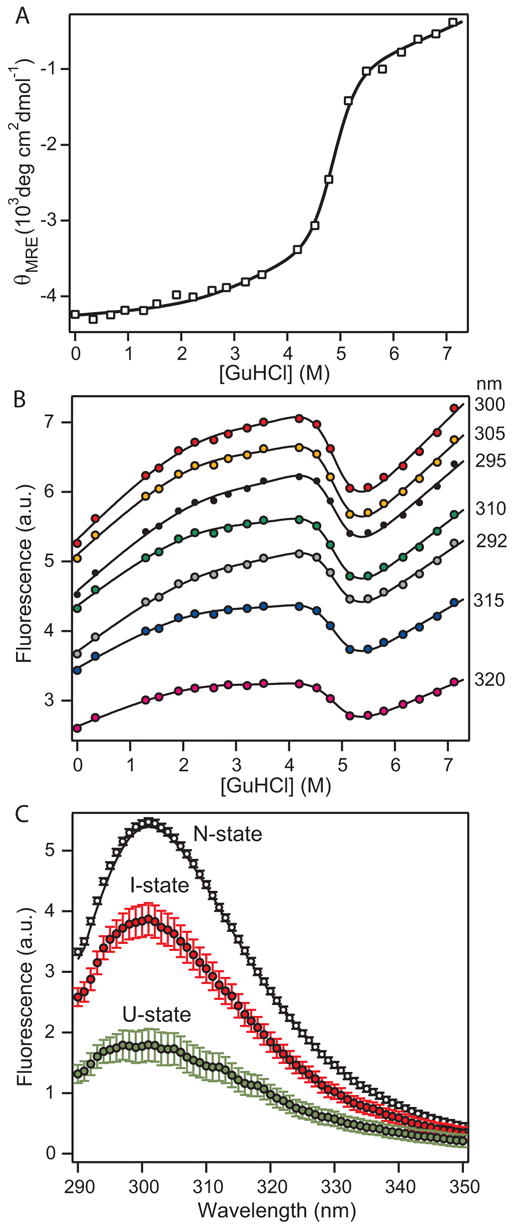
GuHCl-induced unfolding/dimer dissociation of wt A4 monitored by CD and tyrosine fluorescence. A. Molar mean-residue ellipticity at 222 nm (θMRE) vs. GuHCl concentration. B. Tyrosine fluorescence signal at representative emission wavelengths vs. GuHCl concentration. The lines represent a global fit of the combined fluorescence data (transition curves at 1 nm increments from 290 to 390 nm) to a unimolecular three-state model of protein unfolding (equation 1). The optimized global fit parameter (Cm and m-values for each transition; Table 1) also reproduce the CD-detected unfolding curve (line in panel A). C. Intrinsic tyrosine fluorescence spectra in the absence of denaturant for each of the three equilibrium states obtained by global fitting of the combined fluorescence data.
As a complementary probe to monitor unfolding we measured the changes in tyrosine fluorescence as a function of denaturant concentration (A4 contains four Tyr and no Trp residues). Following procedures developed previously,15,16 we recorded a series of fluorescence emission spectra at different GuHCl concentrations and used a global analysis procedure to fit the combined data to a unimolecular three-state mechanism. Figure 2B shows representative transition curves obtained by plotting the fluorescence signal at a given wavelength vs. GuHCl concentration. The analysis is complicated by the relatively small change in tyrosine fluorescence associated with the conformational transitions (~15% of the total signal) and the steep baselines at low and high GuHCl concentrations. Nevertheless, we were able to accurately reproduce the transition curves at each wavelength with a common set of optimized transition parameters (Table 1) obtained by global fitting of a three-state model to the combined data set (see Figure 2B for examples). The sharp decrease in fluorescence between 4 and 5 M GuHCl parallels the loss in helix content observed by CD (Figure 2A), indicating that disruption of both secondary and tertiary structure occurs during this cooperative unfolding transition. The bend in the fluorescence curves near 2 M GuHCl can be attributed to a decrease in fluorescence in going from the native state to an equilibrium intermediate; because of the pronounced solvent-dependent increase in tyrosine fluorescence in all three conformational states, this results in a plateau rather than a net decrease in the observed signal (Figure 2B). The fact that a single set of three-state equilibrium parameters (Table 1) accurately reproduces both the combined fluorescence and CD data (lines in Figures 2A and 2B) strongly supports our conclusion that a distinct structural transition occurs at denaturant concentrations below the main unfolding transition. A unimolecular three-state unfolding equilibrium with an ensemble of non-native states populated at intermediate denaturant concentrations (<4 M GuHCl) is the simplest model consistent with the data; a two-state fit would require the ad hoc assumption that the native state experiences complex (non-linear) solvent-induced changes in the CD and fluorescence signals.
Table 1.
Thermodynamic parameters describing unfolding of wt A4 and dimer dissociation coupled with unfolding for C321S and F283L/C321S variants1
| Cm1 (M) | m1 | ΔG1 | Kd (nM)3 | Cm2 (M) | m2 | ΔG2 | |
|---|---|---|---|---|---|---|---|
| wt A4 | 3.20 ± 0.06 | 0.80 ± 0.1 | 2.6 ± 0.3 | N.A. | 4.84 ± 0.01 | 2.57 ± 0.03 | 12.4 ± 0.2 |
| C321S A4 | (2.75)2 | 1.20 ± 0.06 | 9.5 ± 0.45 | 90 ± 50 | (4.37)4 | 0.60 ± 0.03 | 2.61 ± 0.17 |
| F283L/C321S A4 | (1.35)2 | 2.50 ± 0.006 | 8.71 ± 0.02 | 350 ± 20 | (4.30)4 | 0.82 ± 0.01 | 3.63 ± 0.04 |
The units for m and ΔG are kcal l mol−2 and kcal mol−1, respectively (1 kcal = 4.187 kJ).
Apparent midpoint of second-order transition (N2 ⇔ 2M) from Figures 4B and 9B.
Dimer dissociation constant, Kd = exp(−ΔG1/RT), where R=1.987 10−3 kcal mol−1 K−1 is the gas constant.
Midoint of the unimolecular transition (M⇔U), Cm2 = ΔG2/m2.
By plotting the fitted baseline values (intercepts at 0 M GuHCl) for each state vs. wavelength (Figure 2C), the global analysis also yields information on the intrinsic fluorescence emission spectra for the native (N), intermediate (I) and unfolded (U) states. The fitted spectrum of the N-state (black symbols) has a maximum at 301 nm and accurately reproduces the equilibrium spectrum recorded in the absence of denaturant (black line). Because of the steep positive slope of the unfolded baseline the fitted U-state spectrum (green symbols), which represents the unfolded state in the absence of denaturant, the emission maximum of the N-state is much lower. The fluorescence spectra of the I-state (red symbols) is similar in shape to that of the N-state, but 30% lower in intensity and slightly shifted to lower wavelengths. These spectral changes are consistent with a progressive increase in solvent exposure for some of the tyrosine side chains in the I- and U-states relative to the N-state, which gives rise to more efficient solvent-induced fluorescence quenching.
In order to investigate the role of the covalent disulfide linkage in stabilizing the A4 dimer, we studied the unfolding behavior of the C321S variant as a function of both GuHCl and protein concentration (Figure 3). Since unfolding of this non-covalent dimer results in a monomeric unfolded state, the transition curves also contain information on the equilibrium constant for dimer dissociation, Kd. Figure 3A compares the normalized CD-detected transition curves for the C321S A4 mutant at several protein concentrations. Compared to wt A4, the onset of unfolding as observed by CD begins at lower GuHCl concentrations, and the transitions are substantially broader, but the sloping native and unfolded-state baselines in wt A4 are also observed for C321S A4. With increasing protein concentration, the transition curves become steeper and the midpoint shifts to higher GuHCl concentrations. The fact that the denaturant-induced loss in (helical) secondary structure varies with protein concentrations indicates that both unfolding and dimer dissociation events contribute to the apparent transition curves. The observation that protein concentration primarily affects the data between 1 and 4 M GuHCl while the upper portion of the transition (>4M GuHCl) is less sensitive suggests the presence of two distinct, but overlapping transitions where only the former involves a dimer-monomer equilibrium.
Figure 3.
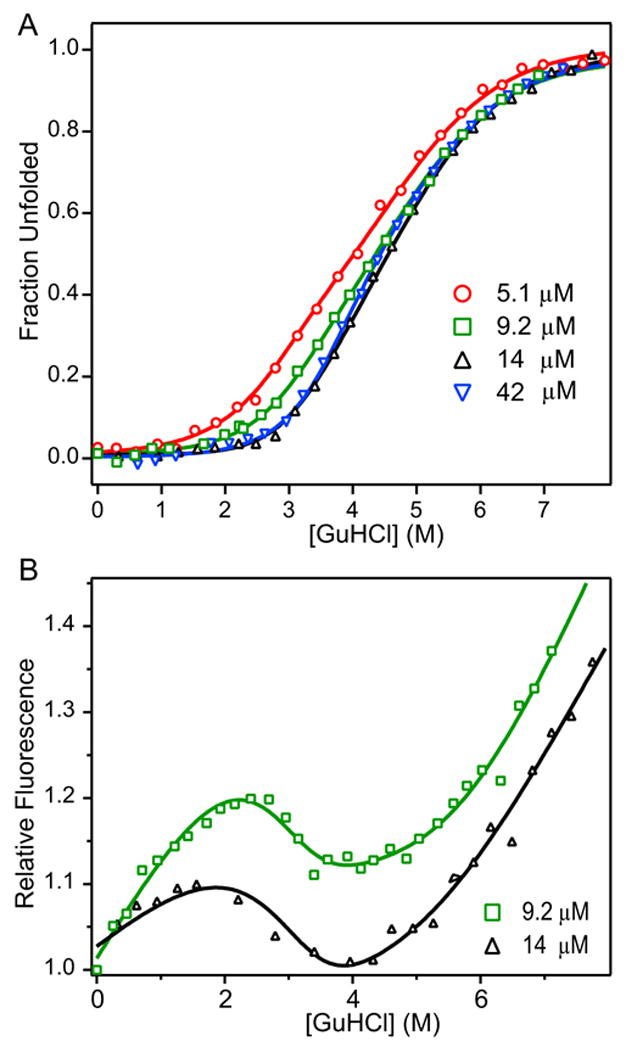
Normalized ellipticity (A) and relative fluorescence (B) of C321S A4 protein as a function of GuHCl globally fitted to three-state dimeric model with monomeric intermediate. Normalized molar residue ellipticity (A) and relative fluorescence (B) of C321S A4 at 5.1 μM (red circles), 9.2 μM (green squares), 14 μM (black triangles), and 42 μM (blue inverted triangles) vs. GuHCl concentration. Data were fit to a three-state dimeric model of protein unfolding with a partially structured monomeric intermediate (Scheme 3, equation 4).
We also used tyrosine fluorescence to monitor the GuHCl-induced conformational changes for the C321S mutant of A4 (Figure 3B). As in the case of wt A4 (Figure 2B), interpretation of these data is complicated by the steep slope of the baselines at low and high GuHCl concentrations and the more limited range in protein concentration that can be measured reliably. Nevertheless, the fluorescence data can be fitted adequately using the same set of equilibrium parameters derived from a unimolecular three-state fit of the CD data (see below).
For a protein like C321S A4, which exists as a stable dimer under native conditions and forms an unfolded monomeric state under denaturing conditions, one can consider three possible models (see, e.g., ref. 17). (Scheme 1) Native dimer (N2) is converted into unfolded monomers (U) through a cooperative mechanism without appearance of intermediates:
| (Scheme 1) |
(Scheme 2) unfolding of the native dimer precedes dissociation into monomers giving rise to a dimeric intermediate, M2:
| (Scheme 2) |
(Scheme 3) with increasing denaturant concentrations, the native dimer first dissociates into a monomeric intermediate (M) before undergoing complete unfolding:
| (Scheme 3) |
The CD and fluorescence data were globally fitted with equations representing the different models for dimer unfolding (see Methods, equations 5, 6, and 7). This combined fit was optimal when Scheme 3 was used (lines in Figure 3) whereas Schemes 1 and Scheme 2 were unable to reproduce the data (see below). The thermodynamic parameters thus obtained are listed in Table 1. The free energy for dimer dissociation in the absence of denaturant, ΔG1, is 9.5 ± 0.45 kcal mol−1, which corresponds to a dissociation constant Kd = 90 nM. The modest value of the free energy for unfolding of the monomeric intermediate, ΔG2 = 2.61 ± 0.17 kcal mol−1, indicates that the A4 domain is rather unstable in the absence of the stabilizing dimeric interactions.
Supplementary Figure 1 illustrates our unsuccessful attempt to fit the CD and fluorescence data using alternative equilibrium mechanisms. Neither Scheme 1 (solid lines) nor Scheme 2 (dashed lines) provide a satisfactory fit, judging from the χ2 parameters, which decreased from 0.307 for Scheme 1 to 0.0740 for Scheme 2 and 0.0394 for Scheme 3. In addition, the attempted global fitting of the CD and fluorescence data with Scheme 2 gave a very unrealistic value for the Kd of 50 μM. This value is inconsistent with gel filtration experiments, which show the C321S A4 protein to be completely dimeric at all loading concentrations >750 nM (data not shown). A key prediction of Scheme 3 is that the first transition (i.e., the one occurring at lower denaturant concentration) should become more pronounced at lower protein concentration where the N2 ⇔ 2M transition is shifted towards the monomeric intermediate. This effect is most apparent in Supplementary Figure 1A where the loss in native CD signal is more pronounced at the lower protein concentrations studied. This gives rise to an apparent decrease in the slope of the transition curves with decreasing protein concentration, as observed previously for another dimeric protein.17 In contrast, the slope of the transition curves predicted by the concerted unfolding/dimer dissociation mechanism (Scheme 1) is independent of protein concentration, and in the case of Scheme 2, any variation with protein concentration is expected to produce highly destabilizing conditions leading to dissociation of the dimeric intermediate.
Figure 4A shows the fraction of the disulfide-linked dimers of native (N), intermediate (I), and unfolded (U) species of wt A4 populated vs. GuHCl concentration, as predicted by the best fit of the CD and fluorescence-detected unfolding transitions (Figure 2; Table 1). The I-state fraction reaches a maximum of 75% at 4.3 M GuHCl and decays sharply as the fully unfolded state accumulates in a steep transition centered around 4.8 M GuHCl. Figure 4B shows similar plots for the dissociable C321S A4 variant at two protein concentrations (5 μM and 42 μM). The relative populations of the native dimer (N2), the monomeric intermediate (M) and unfolded (U) states were predicted on the basis of Scheme 3 (equation 4), using the fitting parameters in Table 1. As expected from mass-action considerations, the monomeric M-state accumulates more readily at lower protein concentration (5 μM) where it reaches a peak population of 70% at 3.0 M GuHCl. Conversely, higher protein concentrations favor the native dimer, causing a shift in the native population to higher denaturant concentrations.
Figure 4.
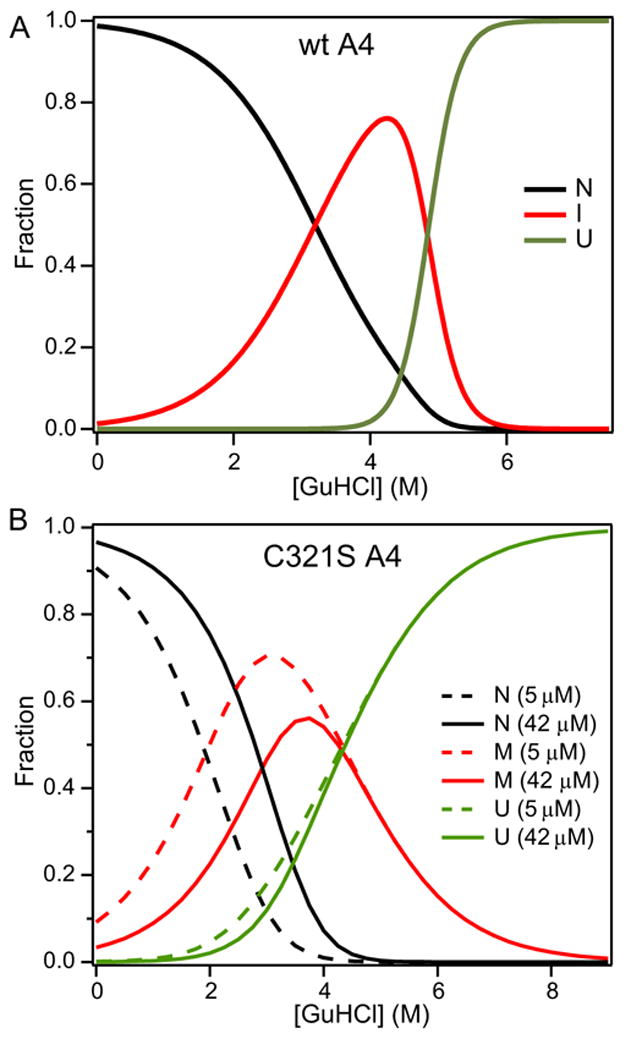
Relative populations of native (N, black lines), intermediate (I, red lines), and unfolded (U, green line) states of (A) wt A4 (20 μM) and (B) C321S A4 protein at 5.1 μM (dashed) and 42 μM (solid) as a function of GuHCl concentration. The populations for wt A4 (A) were calculated from a unimolecular three-state model (equation 1), using the parameters obtained by fitting the data in Figure 2. The population for the dissociable C321S mutant (B) were calculated from equation (4), which describes a three-state dimer-dissociation/unfolding transition with monmeric intermediate (Scheme 3), based on the fit of the CD and fluorescence data in Figure 2 (cf. Table 1).
Dynamic light scattering
To confirm the prediction that a monomeric, partially structured intermediate accumulates at moderate GuHCl concentrations during C321S A4 unfolding, we used dynamic light scattering (DLS) to measure the changes in hydrodynamic radius (Rh) across the unfolding transitions of wt A4 and the C321S variant (Figure 5). The translational diffusion coefficient measured by DLS can be related to Rh (using a globular protein model that assumes a spherical shape of the protein; see Methods), and thus to the association state of the protein. As the GuHCl concentration is raised from 0 to 4 M, the Rh for wt A4 (filled squares in Figure 5) increases from 2.4 to 2.7 nm. This slight expansion (~20% of the overall increase in Rh on unfolding) is consistent with the formation of a partially unfolded, but still relatively compact intermediate of the cross-linked A4 dimer. A major increase in hydrodynamic dimensions (from 2.7 to 4.5 nm) occurs between about 4.5 and 5.2 M GuHCl, confirming that this transition comprises the principal conformational change during unfolding of A4. As expected, the dimensions of the dissociable C321S A4 variant are the same as wt A4 in the absence of denaturant (Rh = 2.4 nm). However, in marked contrast to the wt protein, the Rh of C321S A4 decreases initially with increasing denaturant concentration, reaching a minimum value of ~1.9 nm at 3.5 M GuHCl. This observation strongly supports our conclusion that a monomeric, partially structured, intermediate accumulates under moderately denaturing conditions (Scheme 3). The subsequent increase in Rh coincides with the second conformational event in Scheme 3, i.e., unfolding of the monomeric intermediate, which is expected to be accompanied by an increase in Rh. Both the wt and C321S A4 DLS-detected unfolding transitions fit to their respective three-state unfolding models (Schemes 4 and 1, respectively), with the same fit parameters as those used for fitting the CD and fluorescence unfolding curves.
Figure 5.
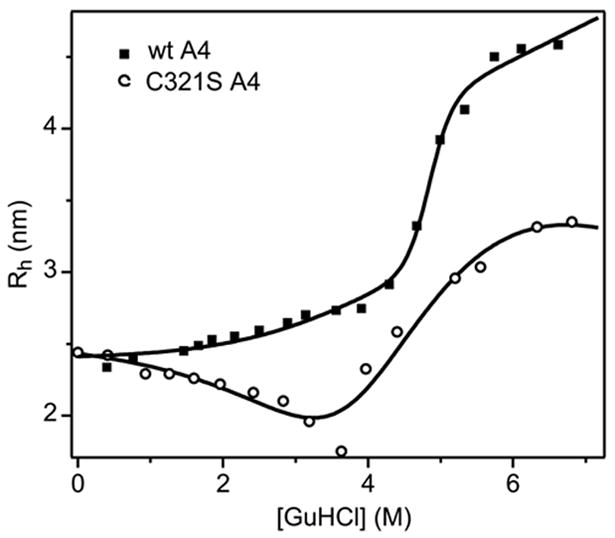
Hydrodynamic radius (Rh) versus GuHCl concentration for 75 μM wt A4 (filled squares) and 100 μM C321S A4 (open circles) measured by DLS. The wt A4 data were fitted on the basis of the unimolecular three-state unfolding model (equation 1), while the fit to the C321S A4 data represents a three-state dimeric unfolding model with monomeric intermediate (equation 4). Both fits were constrained using the parameters obtained from the analysis of the CD and fluorescence data (Table 1).
The Rh measured by DLS in the absence of denaturant is 2.34 nm for wt A4 and 2.44 nm for C321S A4. These sizes correspond to spherical volumes of 54 nm3 for wt A4 and 61 nm3 for C321S A4, which are consistent with the volume of an prolate spheroid (58.7 nm3) calculated from the overall dimensions of the A4 dimer in the crystal structure of FXI (dmax = 5.65 nm, dmin of 3.51 nm).5 The C321S mutant reaches a limiting Rh of ~3.4 nm at 6 M GuHCl, which is substantially smaller than that of the fully unfolded state of wt A4 (4.5 nm) consistent with the fact that the latter is a covalently linked dimer.
Characterization of C321S A4 by analytical ultracentrifugation
In order to unambiguously determine the oligomeric state of C321S A4, analytical ultracentrifugation sedimentation equilibrium (AUC SE) measurements were performed on C321S A4. Figure 6A shows raw absorbance data from equilibrium runs on a 201.4 μM sample of C321S A4 at two different centrifugal speeds (17,000 rpm, dash-dotted line and 25,000 rpm, solid line). Figure 6B shows a plot of the weight fraction of species vs. total monomer concentration derived from a global fit of a monomer-dimer-tetramer model of the raw data in Figure 6A. A global fit of a monomer-dimer-tetramer equilibrium (Figure 6B) converged on a unique set of fitting parameters, while a monomer-dimer equilibrium did not fully account for the data. The dimeric state of C321S A4 (solid line) is the predominant species over the concentration range from 32.9 μM to 212 μM, with a maximum of 7.2% of the protein present as tetramer or higher order aggregates at 212 μM (dashed line). Monomeric protein (dash-dotted line) decreases from ~17% at 32.9 μM to <5% at 212 μM.
Figure 6.
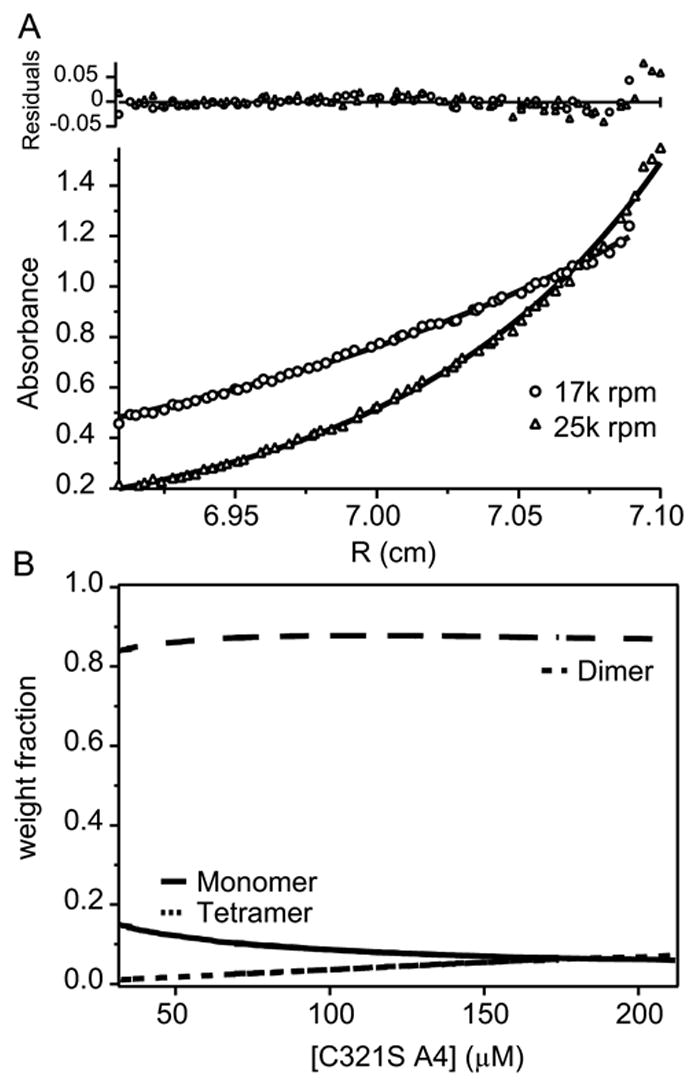
Analytical ultracentrifugation sedimentation equilibrium (AUC SE) analysis of C321S A4. (A) Global fit of absorbance at 280 nm vs. the radial distance from the center of the centrifuge rotor to the sample cell measured at 17,000 rpm (circles) and 25,000 rpm (squares). (B) The fit parameters from panel A were used to calculate the relative fractions of monomeric (solid line), dimeric (long dashes), or tetrameric (short dashes) species vs. the total monomeric protein concentration in μM.
Since the protein concentrations used for unfolding and fluorescence anisotropy analyses (see below) were less than or at the low end of this range, the AUC SE data show that the contribution of tetrameric and larger oligomers to the unfolding and dissociation data is negligible over the concentration ranges studied. In the highest C321S A4 protein concentration of 42 μM from unfolding experiments, the corresponding contribution from tetrameric protein is 1.4%, which is not expected to contribute to the observed unfolding transitions.
Fluorescence anisotropy
As an independent measure of the monomer-dimer equilibrium of C321S A4, fluorescence anisotropy experiments were performed on protein samples labeled with Alexa Fluor 488 (AF488). The labeled lysine residue was identified by recording MALDI-TOF mass spectra on trypsin-digested samples, which showed that AF488 was present only on K357 when the labeling ratio was less than or equal to 0.35. The protein concentration and labeling ratio were determined according to ref. 18.
Fluorescence anisotropy was measured as a function of protein concentration ranging from 7 nM to 870 nM (Figure 7). Upon dilution of the protein, the anisotropy decreases from ~0.11 to 0.02. A model of dimer-monomer dissociation (equation 8) qualitatively describes the observed trend, but does not exactly reproduce the data, which change over a narrower range of concentration than predicted for a monomer-dimer equilibrium (line in Figure 6).19,20,21 The anisotropy of the dimeric state (rD = 0.106 ± 0.004) is consistent with the anisotropy expected from the measured limiting polarization value (P0, calculated from the Perrin-Weber equation19). The P0 of 0.219 was measured at a protein concentration of 1 μM at 4º C in 56% sucrose, corresponding to a polarization value of 0.145 (anisotropy = 0.109), determined using the Perrin-Weber equation (ρ = 2.22 × 10−8 sec, τ = 4.10 × 10−9 sec). However, the limiting anisotropy for the monomeric state (rM) cannot be measured precisely due to sensitivity limitations, which leads to additional uncertainty in estimating Kd. Given the size of the C321S A4 monomer (MW = 10,057 Da), we expected a limiting anisotropy for the monomer, rM ~ 0.06. The fact that we measured substantially smaller values at low protein concentration may be due to an increase chromophore mobility in the monomeric state, which is known to lower the observed anisotropy value.22 Despite this uncertainty, fitting a dimer-monomer model to the anisotropy data of the dissociable A4 dimer (Figure 7) yields an apparent Kd of 50 ± 30 nM that is consistent with the value derived from the denaturant-induced equilibrium measurements (90 ± 50 nm; Table 1).
Figure 7.
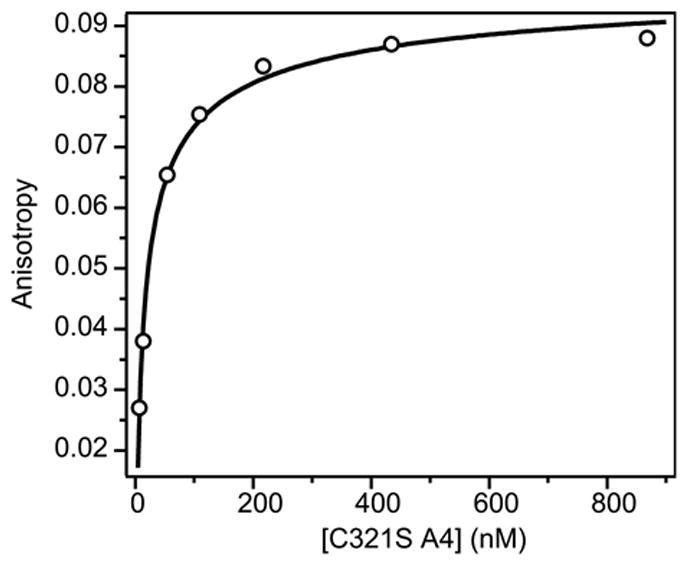
Dimer dissociation of fluorescence-labeled C321S A4 observed by fluorescence anisotropy. The fluorescence anisotropy of C321S A4-AF488 (circles) is plotted vs. total protein concentration in nM (per monomer). The line represents a fit of a dimer-monomer equilibrium model (equation 7).
Structural characterization and unfolding of F283L/C321S A4
Meijers et al. hypothesized that a Phe to Leu mutation at position 283 in FXI (a.k.a. type III mutation) causes a defect in folding and/or dimer formation of the protein, preventing secretion of FXI F283L by the kidney cell line used for these studies.10 In order to understand the effect of this important mutation in FXI deficient patients on the unfolding and dissociation properties of the non-covalent C321S A4 dimer, we introduced the F283L mutation into the C321S A4 construct. Optical, NMR and hydrodynamic methods were used to characterize the structure, folding equilibrium and oligomerization state of the mutant A4 domain.
As a sensitive assay of any structural differences between C321S and F283L/C321S A4, we recorded 1H-15N correlation NMR spectra (15N HSQC) for uniformly 15N labeled samples of both proteins under native conditions (50 mM sodium phosphate, pH 6.0, 100 mM sodium chloride). A comparison of the 15N HSQC spectra (Figure 8A) confirms that the mutant protein retains a well folded, globular structure, since the cross peaks are well dispersed across the typical range of 1H and 15N frequencies. However, the F283L mutation causes minor chemical shift changes for a significant fraction of the residues, and several peaks decrease in intensity, probably due to line broadening caused by changes in local mobility. Resonance assignments for F283L/C321S A4 were obtained based on comparison of 15N NOESY-HSQC and 15N TOCSY-HSQC data with an HSQC spectrum of C321S A4 under matching conditions. In Figure 8B, the normalized chemical shift changes (see Methods) associated with the mutation are plotted vs. residue number. Significant chemical shift changes (>0.1 ppm) are limited to four of the β-strands: β2, which includes the site of mutation, β4, β5 and β7. All of these segments contribute residues to a central cluster of hydrophobic side chains, including F283 and two other aromatic groups (F311 and Y351). Thus, the NMR chemical shift analysis (Figure 7B) reveals that the F283L mutation introduces local as well as medium range structural perturbations mediated by side chains within the core, but does not cause major disruption of the A4 structure.
Figure 8.
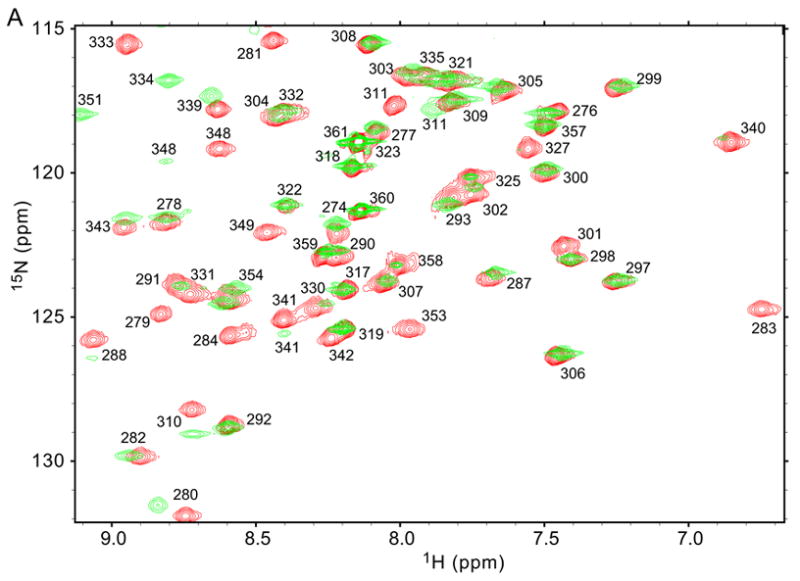
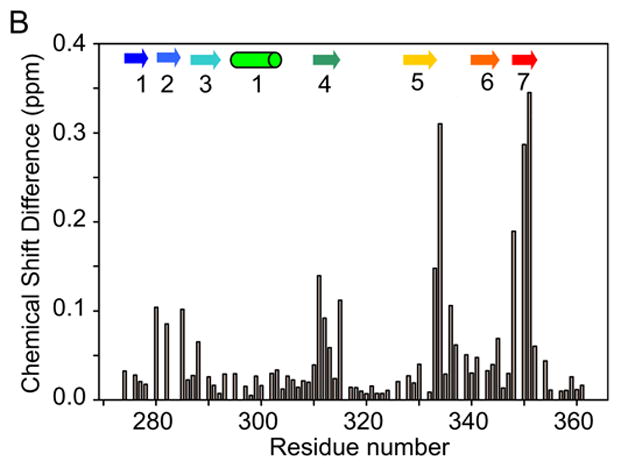
NMR chemical shift analysis of F283L/C321S A4 in comparison to C321S A4. (A) Comparison of the 15N HSQC spectrum of F283L/C321S (green) with that of C321S A4 (red). Peak assignments were obtained by standard heteronuclear NMR techniques (see Methods). (B) Graph of 15NH normalized chemical shift differences between F283L/C321S A4 and C321S A4, calculated according to equation (8). Secondary structure elements are indicated using cylinders for α-helices arrows for β-strands, based on the crystal structure of FXI.5
To determine the effect of the F283L mutation on structural stability and monomer-dimer equilibrium, we used the CD signal at 225 nm to follow the equilibrium unfolding transition of F283L/C321S A4 vs. GuHCl concentration (Figure 9A). As in the case of C321S A4 (Figure 3), unfolding measurements at pH 7.5 (in 50 mM sodium phosphate, 100 mM sodium chloride) were carried out at several protein concentrations (11.6, 25.5, and 84.8 μM). The data were globally fitted to a three-state dimer unfolding model with a monomeric intermediate (Scheme 3), which yielded an excellent simultaneous fit of all data (χ2 = 1.13 × 105). In contrast, the cooperative two-state (Scheme 1; χ2 = 1.57 × 105) and three-state dimeric unfolding model with dimeric intermediate (Scheme 2; χ2 = 1.35 × 105) did not reproduce the data as well. Compared to the C321S A4 parent protein (Figure 3A), the first phase in the GuHCl-induced signal change is shifted toward lower denaturant concentration, giving rise to a well-resolved two-step transition. The midpoint of the first (low-denaturant) transition varies with protein concentration, confirming that it represents a second-order dissociation step (N2 ⇔ 2M). The global fit yields a Kd of 350 ± 20 nM, which represents a 4-fold increase in dimer dissociation constant compared to C321S A4 (Table 1). The second step is independent of protein concentration, and can thus be assigned to a unimolecular conformational event, i.e., unfolding of the monomeric intermediate (M ⇔ U). Although the mutation has virtually no effect on the midpoint of the main unfolding transition, it causes a significant increase in m-value (Table 1), and the corresponding free energy in the absence of denaturant (ΔG2 = 3.63 ± 0.04 kcal mol− 1) is somewhat enhanced relative to the C321S A4 variant (ΔG2 = 2.61 ± 0.17 kcal mol− 1). Comparing the CD signal of F283L/C321S A4 with that of C321S A4 (Fig. 8A, open circles), we observe that there are striking differences in the shape of the unfolding transition for F283L/C321S A4 relative to C321S A4. Most notably, the plateau in the F283L/C321S A4 data between 1.75 and 2.5 M GuHCl, which corresponds to formation of a monomeric intermediate, is much less pronounced for C321S A4, indicating that the F283L mutation leads to increased accumulation of a monomeric intermediate state. Thus, the F283L mutation stabilizes the folded monomeric state at the expense of a weakened dimer interface.
Figure 9.
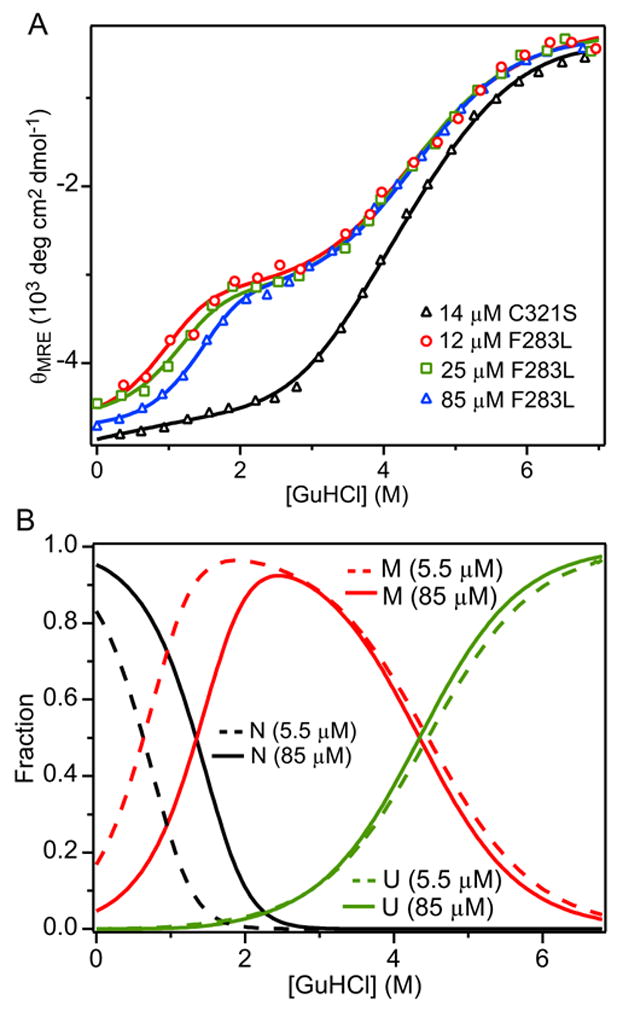
Unfolding/dimer-dissociation equilibrium of F283L/C321S A4. (A) CD signal at 225 nm vs.GuHCl concentration measured at protein concentrations 12 μM (red circles), 25 μM (green squares), and 85 μM (blue triangles) along with global fits of a three-state unfolding model with monomeric intermediate (lines). The unfolding transition for 5.1 μM C321S A4 (cf. Figure 2A) is shown for comparison (black triangles). (B) Relative populations of native (black lines), intermediate (red lines), and unfolded (green lines) states for 5.48 μM (dashed) and 84.8 μM (solid) F283L/C321S A4 protein vs. GuHCl concentration obtained using the global fitting parameters in Table 1.
DLS studies of F283L/C321S A4 unfolding (data not shown for brevity) gave results similar to that of C321S A4, and confirmed that the mutant also unfolds via a monomeric intermediate, as in Scheme 3 (N2 ⇔ 2M ⇔ 2U). In the absence of denaturant, the DLS measurements yield an Rh of 2.34 nm, which, upon addition of GuHCl, decreases to 2.0 nm (at 2.9 M GuHCl) before increasing again to a maximum of 3.4 nm at 6 M GuHCl. As in the case of C321S A4, the decrease in molecular dimensions observed at intermediate denaturant concentrations followed by renewed expansion at high denaturant concentrations is fully consistent with an equilibrium unfolding mechanism involving a monomeric intermediate (Scheme 3).
In Figure 9B we plot the relative populations of the N, M and U state for F283L/C321S A4 obtained by global fitting of the CD data vs. GuHCl concentration. In comparison to the C321S A4variant (Figure 4B), the N2 ⇔ 2M transition occurs at lower GuHCl concentration (due to the 4-fold increase in Kd) and the M ⇔ U transition is shifted to the right (due to the 1 kcal/mol stabilization of M relative to U). As a result, the M state population is increased, especially at low protein concentrations where the native dimer dissociates more readily. At GuHCl concentrations in the range of 1 to 2 M the M-state accounts for nearly 100% of the molecules.
Discussion
We have used several biophysical techniques, including CD, fluorescence, DLS and NMR, to characterize the equilibrium unfolding behavior of wt A4 and two dissociable variants containing C321S and C321S/F283L mutations, respectively. The wt A4 dimer (Figure 1) undergoes large-scale unfolding with a concomitant loss of helical secondary structure, solvent-exposure of aromatic side chains and increase in hydrodynamic radius in a sharp transition centered around 4.8 M GuHCl (Figure 2). This exceptionally high stability toward solvent denaturation is not surprising, given that the A4 dimer contains a total of seven disulfide bonds (three within each monomer and one between the subunits). However, further analysis using global fitting techniques reveals a second, subglobal, unfolding transition at moderate denaturant concentrations, which is accompanied by a 25% decrease in the CD signal due to α-helical secondary structure, tyrosine fluorescence changes consistent with increased solvent exposure and a ~20% increase in hydrodynamic radius (Figures 1 and 4, respectively). The combined unfolding data can be fitted on the basis of a unimolecular three-state mechanism consisting of two coupled unfolding transitions with free energy increments of 2.3 and 12.4 kcal mol− 1, respectively. While most globular proteins of small to moderate size exhibit two-state unfolding transitions, accumulation of equilibrium intermediates at moderate denaturant concentration is not without precedent.15,16,23 Although we have only limited structural information, the presence of a partially unfolded state may well be related to the fact that the disulfide bond connecting the subunits of the A4 dimer is located at the tip of an exposed pair of loops;5 disruption of the non-covalent interactions between monomers would result in a pair of domains connected through a flexible linker segment. This scenario is consistent with the observed increase in Rh, and the changes in optical parameters, which can be attributed to perturbations in the monomer structure upon disruption of the dimer interface.
With increasing denaturant concentration, the C321S variant, which lacks a covalent linkage, undergoes a series of transitions involving both unfolding and dissociation of the native dimer. By observing this second-order transition both as a function of denaturant and protein concentration, we can not only characterize the conformational equilibrium, but also determine the dissociation constant of the native dimer.17 Solvent-denaturation thus represents a valuable alternative to conventional methods, such as size-exclusion chromatography and fluorescence anisotropy, for measuring dissociation constants for homodimeric proteins and provides additional information on the energetically linked unfolding reactions. Our sedimentation equilibrium data (Figure 6) clearly show that in the absence of denaturant, the predominant form of C321S A4 is a dimer over the whole range of protein concentrations used in our biophysical experiments (~5 to 100 μM). At the other extreme of the unfolding transition (~6 M GuHCl), the protein appears to be fully dissociated into monomers, based on its reduced hydrodynamic radius compared to wt A4 (Figure 5). This leaves us with three possible folding/oligomerization mechanisms in which intermediate states are either absent (Scheme 1), dimeric (Scheme 2) or monomeric (Scheme 3), depending on the relative strengths of the intra- vs. inter-subunit interactions. Among these, a fully cooperative unfolding/dimer dissociation mechanism without intermediates (Scheme 1) can readily be ruled out, since it fails to explain the observed biphasic unfolding behavior and protein concentration-dependent changes in slope (Figures 3 and 9). The distinction between Schemes 2 and Scheme 3 is more subtle, since both mechanisms predict populated equilibrium intermediates. We initially favored Scheme 3, since it resulted in somewhat lower χ2 values in our global fitting of the optical data (Figure 3). The DLS measurements provided striking additional evidence in favor of Scheme 3, since this is the only mechanism that can account for the observed decrease in Rh at low to moderate denaturant concentrations, where the native dimer dissociates into folded monomers. In contrast, accumulation of a partially unfolded dimeric intermediate, according to Scheme 2, would give rise to an increase in Rh, similar to that observed for wt A4 (Figure 5).
On the basis of this model, global analysis of the GuHCl unfolding data allows a reliable determination of the dimer dissociation constant. The value obtained for C321S A4, Kd = 90 ± 50 nM, is consistent with the value of 50 ± 30 nM obtained independently using fluorescence anisotropy measurements vs. protein concentration (Figure 7). The Kd of full length dissociable FXI was recently reported to be ~70 nM from AUC SE studies of FXI G326C.24 Since this value is comparable with that measured in the present study for the isolated A4 dimer using GuHCl induced unfolding and fluorescence spectroscopy, FXI A4 must contain most, if not all of the dimer interface surface area between FXI monomers. This conclusion is consistent with the crystal structure of the full length FXI zymogen, which shows that all of the residues responsible for dimer formation reside within FXI A4.5
Qualitatively, if we compare the population plot for C321S A4 (Fig. 3B) with that of wt A4 (Fig. 3A), it is apparent that the C321 inter-subunit disulfide bond conveys a dramatic increase in both stability and cooperativity. For the dissociable variant, the fully unfolded form begins to appear around 1.5 M GuHCl and builds up over a wide range of denaturant concentrations extending above 6 M and is characterized by a relatively low m-value (0.6 kcal mol− 1 M− 1; Table 1). The wt protein, on the other hand, shows no detectable unfolded population at concentrations <4 M GuHCl, followed by a very sharp transition at concentrations >4 M GuHCl. The corresponding m-value (2.57 kcal mol− 1 M− 1) indicates a major increase in solvent-accessible surface area, as expected for a globular protein of the size of the A4 dimer.25 However, a more quantitative comparison of the equilibrium parameters is complicated by the accumulation of intermediate states and the fact that the unfolded states of the two proteins represent different oligomeric states. In the case of C321S A4, the dimeric interactions are lost in the first transition from native dimer to the monomeric intermediate, while in wt A4 inter-subunit contacts are at least partially preserved during the first unfolding transition. These additional interactions and buried surface area stabilize the intermediate for the covalently linked dimer and render its unfolding transition more cooperative relative to the dissociable variant. In Table 1, we express the denaturant-dependence of dimer dissociation for C321S A4 in terms of an m-value (m1 = 1.2 kcal mol-1 M− 1), which is related to the amount of surface area that is buried in the dimer and becomes exposed to the solvent upon dissociation. The m-value for the second transition, m2 = 0.6 kcal mol− 1 M− 1 reflects the exposure upon unfolding of residues buried within each monomer. Thus, the overall increase in solvation is proportional to mtot = m1 + 2×m2 = 2.4 kcal mol−1 M−1. In the case of wt A4, the m-value for complete unfolding is mtot = m1 + m2 = 3.4.kcal mol−1 M−1. This discrepancy cannot be readily attributed to the denatured state, since the cross-linked dimer is expected to retain more, not less, residual structure after unfolding. Therefore, the difference in total m-values must be attributed to structural differences of the folded states causing an increase in solvent-accessible surface area for C321S A4 relative to the wild type. A major contribution may come from the two disulfide-linked loops around C321, which are in close contact in the crystal structure of FXI,5 but were found to be more flexible in an NMR structure of the A4 dimer determined recently in our laboratory (Samuel, Cheng, Riley, Walsh and Roder, submitted for publication) Thus, the loops may undergo local unfolding upon disruption of the inter-subunit disulfide bond, giving rise to a 30% increase in solvent-accessible surface area.
Physiological concentrations of FXI in the blood plasma are ~62.5 nM (in terms of monomer), but the protein concentration of FXI during protein synthesis in the ER of liver cells is expected to be higher than secretion levels in the plasma. The contribution of the inter-monomer disulfide bond involving C321 toward stabilizing the folded dimer is expected to be primarily due to the loss of translational degrees of freedom of the cross-linked unfolded state compared to the C321S variant whose subunits can freely diffuse upon unfolding. However, a quantitative analysis of this effect is complicated by the fact that both wt A4 and C321S A4 exhibit multi-state unfolding transitions involving structurally distinct intermediates. The presence of an inter-subunit disulfide per se is probably not essential for function, since rabbit FXI, which lacks this covalent bond, functions normally.26 On the other hand, the formation of stable noncovalent interactions between monomers appears to be essential for secretion of fully active FXI. This is underscored by our observation that the F283L mutation stabilizes the monomer at the expense of the dimer (Figure 9), which is consistent with the previous findings that the mutation leads to elevated levels of retained monomeric protein inside cultured human kidney cells expressing FXI F283L relative to cells secreting wt FXI.10,14 These reports indicated that intermonomer disulfide bond formation occurred to a lesser extent in FXI F283L cells relative to wt FXI cells, strongly suggesting that the F283L mutation partially prevents formation of disulfide-linked homodimeric FXI protein in this cell culture model. While the in vivo secretory pathway of FXI in liver cells is not well understood and the intracellular localization of disulfide bond formation is not known, two separate reports from cell culture studies suggest that intermonomer disulfide bond formation is important for secretion of FXI at normal levels.10,14
The F283L mutation results in a four-fold increase in Kd relative to C321S A4 (350 nM vs. 90 nM), but does not inhibit formation of a native-like dimeric structure, as indicated by the close similarity between the 15N HSQC NMR spectra of the two proteins. The effect of this conservative amino acid change on dimer stability is surprising, since the side chain of F283 is not directly involved in the A4 dimer interface, although the adjacent L284 side chain participates in a hydrophobic inter-subunit contacts.5 As illustrated by Figure 10, F283 is part of a cluster of aromatic side chains, along with Y278, F311 and Y351, which make up the bulk of the hydrophobic core between the 5-stranded and the 2-stranded β-sheets of A4.5 A likely scenario for explaining the effect of the F283L mutation in shifting the equilibrium from the native dimer toward the monomeric intermediate is that the Phe to Leu amino acid change results in some side chain rearrangements within the core of A4 that are energetically favorable in terms of monomer stability, but cause unfavorable structural perturbations at the dimer interface. This scenario is consistent with the NMR chemical shift analysis shown in Figure 8, which showed significant chemical shift changes not only for residues at or near the site of mutation, but also residues in the C-terminal half of the protein located on the opposite side of the aromatic cluster (Figure 10).
Figure 10.
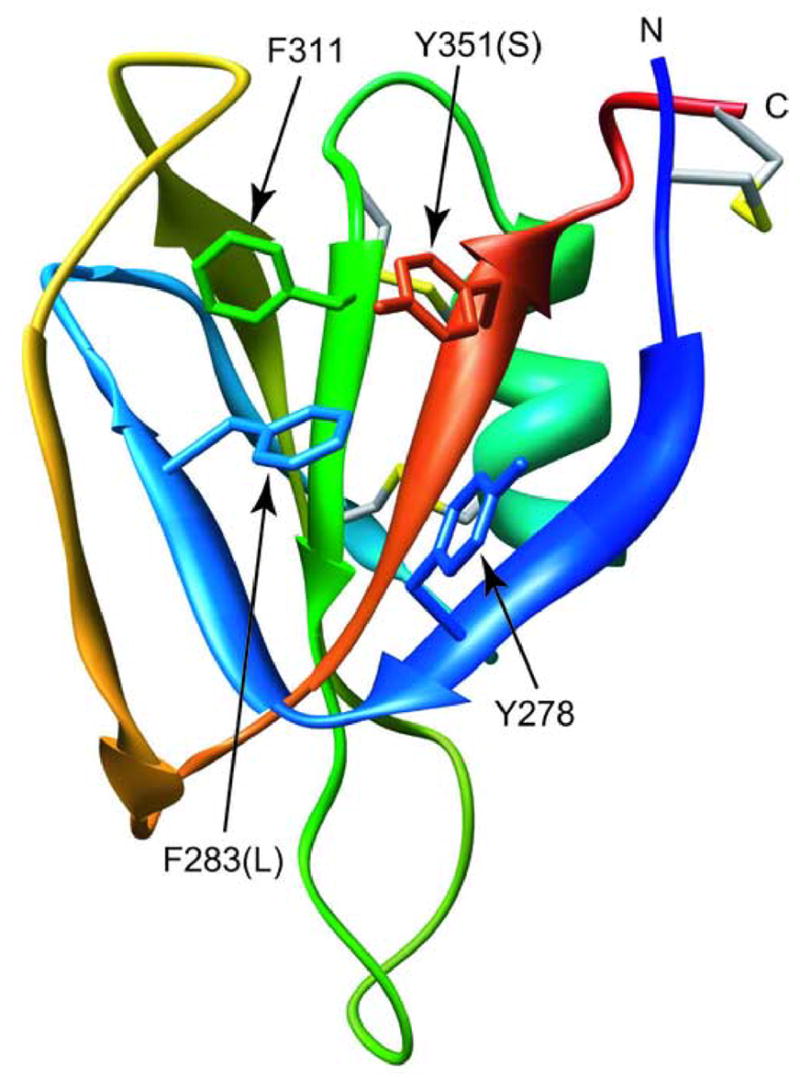
Ribbon diagram of a monomer of wt A4, based on the crystal structure of FXI.5 Side chains are shown for residues involved in an aromatic cluster, some of which are altered in FXI patient mutations (F283L, Y351S, as well as the adjacent G350A/E).
Meijers et al. hypothesized that the F283L mutation in FXI affects protein folding and interferes with efficient secretion of FXI F283L.10 The FXI F283L mutant shows much lower secretion levels compared to wild type (<10%) and the enzymatic activity in plasma is <10% of normal.11 When purified, however, FXIa F283L exhibits normal enzymatic activity in a specific clotting assay.10 A Phe at position 283 is highly conserved in FXI A4 domains from various species. A Phe is also found in the corresponding position of two other apple domains of human FXI, A1 and A3, but is replaced by a Met in A2. Since these homologous domains are monomeric, we can conclude that an aromatic or long aliphatic side chain is critical for the stability of the monomeric state of the apple domain. This is consistent with the fact that the F283 side chain is partially buried within a cluster of other aromatic side chains (Figure 10).
It is interesting that a second member of this aromatic cluster, Y351, is the site of another patient mutation (Y351S), and displays a similar phenotype as FXI F283L.27 In addition, two other patient mutations have been found to target G350, which precedes Y351 in the the C-terminal strand of the β-sheet (Figure 10). The G350E mutation resulted in a very low secretion,14 whereas the G350A mutant is secreted at ~44% of normal levels, but showed low catalytic activity.28 Thus, 4 patient mutations, from a total of 14 known point mutations within the A4 domain 29 affect residues participating in or adjacent to the aromatic cluster, indicating that this is a critical structural feature, both in terms of monomer stability and maintaining a favorable interface for homodimer formation.
In conclusion, our biophysical evidence that the folding/unfolding equilibrium of dissociable (C321S) variants of A4 feature a well populated monomeric intermediate state has implications not only for understanding the folding of this dimeric protein, but also provides a rationale for understanding functional effects of certain mutations in patients with clotting deficiencies. In particular, the phenotype of the F283L mutation, which results in retention of momomeric FXI in the cytosol of liver cells, is consistent with our observation that the mutation results in a structurally more stable monomeric intermediate while destabilizing the native dimeric form. We hypothesize that the Phe to Leu substitution perturbs the side chain packing within a central aromatic cluster that results in energetic stabilization of the monomer at the expense of interactions between the subunits of the dimer.
Materials and methods
Restriction enzymes EcoRI, and PstI, and T4 DNA ligase were from Invitrogen (Carlsbad, CA). 30% (37.5:1 (acryl:bis) acrylamide mixtures used for SDS-PAGE were purchased from National Diagnostics, (Atlanta, GA). Chemicals used in buffers and media were from Sigma-Aldrich Corp. (St. Louis, MO). Ultrapure GuHCl was from ICN Biochemicals (Irvine, CA). Pfu polymerase, QuikChange® Site-Directed Mutagenesis Kit, and competent cells were from Stratagene (La Jolla, CA). The Bacterial Protein Extraction Reagent (BPER®) and the GelCode® Blue gel stain reagent were from Pierce Chemical Co. (Rockford, IL). The Protean II mini-gel apparatus used was from Bio-Rad Laboratories (Hercules, CA). The AKTA FPLC system, Superose 12™ column, and HiLoad™ 16/60 Superdex™ 75 column was from Amersham Biosciences (Piscataway, NJ). The Alexa Fluor® 488 (AF488) labeling kit and NanoOrange® protein quantitation kit were from Invitrogen (Carlsbad, CA).
Protein purification and characterization
For E. coli bacterial expression and purification of wt A4 and the C321S and F283L/C321S variants, we followed the methods reported by Dorfman & Walsh30 with some modifications. Since the recombinant A4 protein is expressed in bacterial inclusion bodies, the 6xHis tag was removed with a PCR primer against the ribosomal binding site immediately upstream of the 5′ end of the FXI A4-6xHis tag fusion construct. The sequence of this primer was:5′-ATATGAATTCATTAAAGAGGAGAAATTAACTATGTTCTGCCA TTCTTCATTTTACCATGACACTGATTTTA-3′. The primer 3′PstI descibed in Dorfman & Walsh30 was the reverse primer, with the sequence 5′-AAAACTGCAG(TTA)CTCATTATCCATTTTACACAA. Purified PCR products were treated with EcoRI and PstI, ligations performed, and clones with the expected DNA sequence C321S A4 sequence without the 6xHis tag present were transformed into M15 [pREP4] bacterial cells. In addition, the F283L mutation was introduced into the C321S A4 construct using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). BPER® (Pierce Chemical Co. Rockford, IL) was used to isolate the inclusion body fraction from IPTG-induced cells. After purification of the inclusion bodies, solubilization was carried out by the method of Rudolph et al.,31 which called for resuspension of inclusion bodies in 6 M GuHCl, 20 mM HEPES pH 7.4, 100 mM NaCl, 100 mM DTT. After removal of the DTT using dialysis against 4 M urea, 100 mM HCl, the total protein concentration was determined using the NanoOrange® protein quantitation kit (Invitrogen, Carlsbad, CA) then diluted to 0.40 mg/ml for in vitro folding as described in Dorfman & Walsh.30 In vitro folding was performed using a thiol/disulfide exchange protocol involving cysteine as the reducing agent.32 Unfolded protein samples were dialyzed against 20 mM Tris-HCl pH 9.0, 100 mM NaCl in the presence of 2 M Urea and 2 mM cysteine. Six buffer changes followed, with respective urea/cysteine concentrations of 2 M/2 mM, 1 M/1 mM, 0.5 M/0.5 mM, 0.25 M/0.25 mM, 0.10 M/0.10 mM, and buffer only. Purification of the refolded A4 protein was performed by gel filtration chromatography using the AKTA FPLC system with a HiLoad™ 16/60 Superdex™ 75 column (GE Healthcare, Piscataway, NJ). For NMR studies, uniformly 15N-labeled samples of A4 were prepared in M9 minimal media using bacterial expression methods described previously.33
FPLC purified samples were analyzed by SDS-PAGE, using DTT to reduce intra and intermonomer disulfides. Samples were loaded onto a 15% acrylamide mini gel attached to a Protean II mini-gel apparatus (Bio-Rad Laboratories, Hercules, CA). Staining was performed using the GelCode® Blue gel stain reagent (Pierce Chemical Co. Rockford, IL).
In order to assess the purity of wt A4, C321S A4, and F283L/C321S A4 protein obtained from bacterial inclusion body fractions, SDS-PAGE was used to compare inclusion body fractions and FPLC-purified proteins in the absence or presence of 0.1 M DTT (data not shown for brevity). Comparison of C321S A4 inclusion body protein to FPLC purified C321S A4 showed that purification was completed to homogeneity. FPLC purified wt A4 shows the expected 20 kDa band on SDS-PAGE under non-reducing conditions due to the presence of the intermonomer disulfide bond at C321. Reduced wt A4 and both nonreduced and reduced C321S A4 samples show the same 10 kDa MW band on SDS-PAGE due to the absence of the intermonomer disulfide at C321. F283L/C321S A4 also shows the expected 10 kDa bands in the absence and presence of reducing agent. The C321S A4 and F283L/C321S A4 samples all show a 20 kDa band, representing < 5% of the intensity of the monomeric band in both the presence and absence of DTT, possibly due to strong noncovalent interactions between the monomers persisting in the presence of SDS. Molecular weights of the protein constructs were confirmed by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) measurements. Thus, these SDS-PAGE and MS data show that the expression and purification procedures for these proteins gave the expected results.
Unfolding equilibrium studies
For studies of the unfolding of the covalently linked wt A4 dimer, a unimolecular three-state model describing a transition from the native state through a partially structured intermediate to the unfolded state, was employed, as previously reported.16 The dependence of the measured signal (Y0) on GuHCl concentration is described by equation (1),
| (1) |
where Yn, Yi, and Yu are the intrinsic signals of protein in native, intermediate, and unfolded states, respectively, Fn is the fractional population of native protein (Fn = 1/(1+K1+K1K2)), and K1 and K2 are equilibrium constants for the transitions from native to intermediate state (K1) and from intermediate to unfolded state (K2). The equilibrium constants for each transition, Ki, are related to denaturant concentration, c, via the relationship Ki = exp(mi(c−Cmi)/RT), where Cmi and mi describe the midpoint and slope, respectively, for a given transition i. The corresponding free energy in the absence of denaturant is given by ΔGi(0) = mi × Cmi, assuming a linear dependence of the free energy of each transition on denaturant concentration, ΔGi(c) = ΔGi(0) − mi × c. For global analysis of the family of fluorescence curves vs. GuHCl concentration at different emission wavelengths (290–390 nm), we relied on procedures detailed in Latypov et al.16 The three-state equilibrium parameters (Cm1, m1, Cm2, m2) were treated as global fitting parameters. The intercepts and slopes of the N-state, I-state and U-state, respectively, were treated as local parameters. The slope for the I-state baseline was assumed to be the average of the N-state and U-state slopes.
To describe the unfolding transition of a native dimer, there are three possible equilibrium mechanisms, as outlined in Hobart et al.17 A cooperative two-state transition from the native dimer (N2) to the unfolded monomer (2U) without accumulation of an intermediate (Scheme 1) is described by equation (2), where, Y0 represents the CD or fluorescence signal at a given denaturant concentration, Yn andYu represent the signal in the native or unfolded states, respectively, and Fu is the fraction of unfolded protein.
| (2) |
Alternatively, two equations are given below that describe unfolding through a partially structured dimeric intermediate (Scheme 2, equation 3), or unfolding via a monomeric intermediate (Scheme 3, equation 4). In equations (3) and (4), Pt stands for the total protein concentration (in terms of monomer), and K1 and K2 are the equilibrium constants for the first and second transition. In the case of the three-state unfolding transition with a dimeric intermediate (equation 3), K1 describes the conformational unfolding step while K2 represents the equilibrium dissociation constant (Kd) for dimer dissociation. As in the case of a unimolecular equilibrium described above, we assumed that the free energy of the second-order transition, ΔGi = −RT ln(Kd), varies linearly with denaturant concentration, and the corresponding slope, mi,reflects the effect of the denaturant on dimer dissociation. In the case of a three-state transition with monomeric intermediate (equation 4), K1 corresponds to the Kd for dissociation of the native dimer while K2 describes the unfolding equilibrium of the monomer.
| (3) |
| (4) |
For global fitting and summary plots, the CD data were normalized according to Fu = (Y0 − Yn)/(Yu − Yn), using the fitted intercepts and slopes of the native and unfolded baselines in the raw data to determine Y0 and Yu, respectively. The fluorescence data were scaled relative to the signal of the sample at 0 M GuHCl; the steep slopes of native and unfolded state baselines prevent us from normalizing the fluorescence data.
CD measurements of wt A4, C321S A4, and F283L/C321S A4 were made on an Aviv Model 62DS CD spectrometer (Aviv Associates, Lakewood, NJ). Time dependent measurements were taken at 225 nm, 22° C for 600 sec. GuHCl concentration was varied from 0 to ~7.5 M while the protein concentration relative to total monomer was held constant at 20 μM for wt A4, 5.1, 9.2, 13.7, or 42 μM, for C321S A4, and 5.48, 11.6, 25.4, or 84.8 μM, for F283L/C321S A4, all relative to the monomeric species in 50 mM Na2HPO4 pH 7.5, 100 mM NaCl. Samples were prepared by mixing aliquots of equimolar stock solutions of native and fully unfolded (7.5 M GuHCl) stock solutions. To ensure complete equilibration, samples were incubated overnight at 22 °C prior to measurement. The final GuHCl concentration in each sample was confirmed by refractometry, using a Reichert-Jung Abbe Mark II refractometer (Leica Microsystems Nussloch GmbH, Nussloch, Germany). Data were fitted using Igor Pro version 4.09 (Wavemetrics, Lake Oswego, OR). The protein concentrations were verified with absorbance measurements at 280 nm, using an extinction coefficient of 6210 M− 1 (monomer) cm− 1 along with the NanoOrange® assay (Invitrogen, Carlsbad, CA).
Steady state tyrosine fluorescence measurements were performed at 22° C using a Photon Technologies International (PTI, Lawrenceville, NJ) fluorescence spectrophotometer, using an excitation wavelength of 280 nm (2 nm bandwidth) and an emission bandwidth of 4 nm. The signal at 303 nm was acquired for 600 sec. The protein and GuHCl concentrations were the same as those used for the CD measurements.
Dynamic light scattering
DLS data were recorded on solutions of 75 μM wt A4, 100 μM C321S A4, and 75 μM F283L/C321S A4 in a buffer containing 50 mM Na2HPO4 pH 7.5, 100 mM NaCl, while GuHCl was varied from 0 to ~7.5 M. Samples were prepared by mixing aliquots of equimolar amounts of native and fully unfolded (7.5 M GuHCl) stock solutions, with overnight equilibration prior to measurement. DLS measurements were performed on a Wyatt technologies DynaPro™ Titan molecular sizing system at 23° C operating at a wavelength of 824.7 nm. The data analyzed using DYNAMICS V6, and Igor Pro version 4.09 (Wavemetrics, Lake Oswego, OR) was used to fit autocorrelation curves. Protein concentrations were verified with absorbance measurements at 280 nm, using an extinction coefficient of 6210 cm− 1 M− 1 (monomer) along with the NanoOrange® assay (Invitrogen, Carlsbad, CA).
Analytical ultracentrifugation
AUC experiments were performed using a Beckman Coulter (Fullerton, CA) XL-I AUC unit equipped with Rayleigh interference optics. Sedimentation equilibrium (SE) measurements were performed at 25° C with 201.4 μM C321S A4 in 20 mM HEPES pH 7.4, 100 mM NaCl using absorbance at 280 nm to monitor the protein species during centrifugation at 17,000 or 25,000 rpm, using measurement and fitting methods published previously for studying ribonucleotide reductase.34,35
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed and analyzed according to refs. 19,22. Equation (5) relates the degree of dissociation, α, to the observed anisotropy value, where α = 1 represents the fully dissociated state, and α = 0 represents the fully associated state in a given population of dimeric proteins.19,22 Q is the relative quantum yield of the monomer vs. the dimer, defined as Q = QM/QD. For simplicity, we assume that the quantum yield of the dimeric protein is equal to an equimolar amount of monomer protein, and Q = 1. Anisotropy is abbreviated here as rD for the dimeric value of anisotropy, and rM for the monomeric value.
| (5) |
The α term can be related to the total protein concentration with respect to monomer (c) and the Kd by equation (6). If the right side of equation (5) is substituted for the α term in equation (6), and the positive quadratic solution taken of the expanded combination of terms, we get equation (7), which can be used to fit curves of α versus c, and the Kd determined.
| (6) |
| (7) |
The C321S A4 protein contains four tyrosines and no tryptophans, which did not give sufficient signal in preliminary fluorescence anisotropy measurements. In order to attain sufficient fluorescence, an analog of fluorescein isothiocyanate was used to label the protein, the substrate Alexa Fluor 488 (AF488, Invitrogen, Carlsbad, CA). One 50 μg vial of AF488 was reacted with 400 μM C321S A4 protein for 60 minutes at room temperature in a volume of 0.5 ml. The reaction mixture was then passed through a Superdex™ 75 HiLoad™ 16/60 FPLC column. The protein concentration and labeling ratio were determined according to ref. 18.
Steady-state fluorescence anisotropy measurements were performed with an ISS PC1 spectrofluorimeter (ISS, Champaign, IL). The anisotropy was measured over a range of 7 to 870 nM with a labeling ratio of 0.35 per monomer. Samples were incubated at room temperature for at least 12 h in 20 mM HEPES pH 7.4, 100 mM NaCl prior to measurements.
NMR spectroscopy
NMR spectra were collected in a Bruker DMX-600 spectrometer equipped with a 5-mm x,y,z-shielded pulsed-field gradient triple-resonance probe (Bruker Biospin GmbH, Rheinstetten, Germany), at a total protein concentration of 1.5 mM (monomer basis) at 37° C in 20 mM Na4P2O7 pH 6.0, 100 mM NaCl. Felix (Accelrys, San Diego, CA) and Sparky36 were used for processing and resonance assignments, respectively. Chemical shift values for 1H, 13C and 15N resonances of C321S A4 were assigned on the basis of CBCANH CBCA(CO)NH, 15N-HSQC, 15N-HSQC-TOCSY, and HNHA experiments.37,38,39
The normalized 15NH chemical shift difference (CSD) of double-mutant (F283L/C321S) vs. single-mutant (C321S) A4 was calculated using equation (8),
| (8) |
where the F283L-induced chemical shift difference for 15N and 1H resonances are normalized with respect to the corresponding chemical ranges. Graphical displays of structures were generated using Chimera (www.cgl.ucsf.edu/chimera).
Supplementary Material
Acknowledgments
This study was supported by research grants from the National Institutes of Health, RO1 HL46213 to P.N.W, and R01 GM56250 and CA06927 to H.R., and an appropriation by the Commonwealth of Pennsylvania to the Fox Chase Cancer Center. We are grateful to David M. Jameson (Department of Cell and Molecular Biology, John A. Burns School of Medicine, University of Hawai’i, Honolulu, HI 96822) for advice and assistance with fluorescence anisotropy experiments, to James D. Lear (Department of Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, PA 19104) for advice and assistance with analytical ultracentrifugation experiments, and to Yibai Chen for mass spectrometric analysis. The Biochemistry & Biotechnology, and the Spectroscopy Support Facilities at the Fox Chase Cancer Center provided critical support.
Abbreviations used
- AF488
Alexa Fluor® 488
- AUC
analytical ultracentrifugation
- A4
apple 4 domain of factor XI
- CD
circular dichroism
- DLS
dynamic light scattering
- FXI
factor XI
- FXIa
factor FXIa
- FPLC
fast protein liquid chromatography
- GuHCl
guanidine hydrochloride
- HEPES
(N-(2-hydroxyethl)piperazine-N′-(2-ethanesulfonic acid)
- HSQC
hetronuclear single-quantum correlation spectroscopy
- MALDI
matrix laser desorption ionization
- NMR
nuclear magnetic resonance
- PK
prekallikrein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TOF
time of flight
Footnotes
Supporting information available
Supplementary information on alternative multi-state unfolding models is available free of charge via the internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh PN, Gailani D. Factor XI. In: Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5. Chapter 12. Lippincott Williams & Wilkins; Philadelphia: 2006. [Google Scholar]
- 2.Fujikawa K, Legaz ME, Kato H, Davie EW. The mechanism of activation of bovine factor IX (Christmas factor) by bovine factor XIa (activated plasma thromboplastin antecedent) Biochemistry. 1974;13:4508–16. doi: 10.1021/bi00719a006. [DOI] [PubMed] [Google Scholar]
- 3.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–12. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 4.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–8. [PubMed] [Google Scholar]
- 5.Papagrigoriou E, McEwan PA, Walsh PN, Emsley J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol. 2006;13:557–8. doi: 10.1038/nsmb1095. [DOI] [PubMed] [Google Scholar]
- 6.Meijers JC, Mulvihill ER, Davie EW, Chung DW. Apple four in human blood coagulation factor XI mediates dimer formation. Biochemistry. 1992;31:4680–4. doi: 10.1021/bi00134a021. [DOI] [PubMed] [Google Scholar]
- 7.Wolberg AS, Morris DP, Stafford DW. Factor IX activation by factor XIa proceeds without release of a free intermediate. Biochemistry. 1997;36:4074–9. doi: 10.1021/bi962274y. [DOI] [PubMed] [Google Scholar]
- 8.Gailani D, Ho D, Sun MF, Cheng Q, Walsh PN. Model for a factor IX activation complex on blood platelets: dimeric conformation of factor XIa is essential. Blood. 2001;97:3117–22. doi: 10.1182/blood.v97.10.3117. [DOI] [PubMed] [Google Scholar]
- 9.Kravtsov DV, Monahan PE, Gailani D. A classification system for cross-reactive material-negative factor XI deficiency. Blood. 2005;105:4671–3. doi: 10.1182/blood-2004-05-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meijers JC, Davie EW, Chung DW. Expression of human blood coagulation factor XI: characterization of the defect in factor XI type III deficiency. Blood. 1992;79:1435–40. [PubMed] [Google Scholar]
- 11.Asakai R, Chung DW, Ratnoff OD, Davie EW. Factor XI (plasma thromboplastin antecedent) deficiency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations. Proc Natl Acad Sci U S A. 1989;86:7667–71. doi: 10.1073/pnas.86.20.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujikawa K. Historical perspective of factor XI. Thromb Res. 2005;115:441–50. doi: 10.1016/j.thromres.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325:153–8. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 14.Kravtsov DV, Wu W, Meijers JC, Sun MF, Blinder MA, Dang TP, Wang H, Gailani D. Dominant factor XI deficiency caused by mutations in the factor XI catalytic domain. Blood. 2004;104:128–134. doi: 10.1182/blood-2003-10-3530. [DOI] [PubMed] [Google Scholar]
- 15.Maki K, Cheng H, Dolgikh DA, Shastry MC, Roder H. Early events during folding of wild-type staphylococcal nuclease and a single-tryptophan variant studied by ultrarapid mixing. J Mol Biol. 2004;338:383–400. doi: 10.1016/j.jmb.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 16.Latypov RF, Cheng H, Roder NA, Zhang J, Roder H. Structural characterization of an equilibrium unfolding intermediate in cytochrome C. J Mol Biol. 2006;357:1009–25. doi: 10.1016/j.jmb.2006.01.055. [DOI] [PubMed] [Google Scholar]
- 17.Hobart SA, Meinhold DW, Osuna R, Colon W. From two-state to three-state: the effect of the P61A mutation on the dynamics and stability of the factor for inversion stimulation results in an altered equilibrium denaturation mechanism. Biochemistry. 2002;41:13744–54. doi: 10.1021/bi0265224. [DOI] [PubMed] [Google Scholar]
- 18.Haugland R. The Handbook — A Guide to Fluorescent Probes and Labeling Technologies. 10. Invitrogen, Inc; Carlsbad, CA: 2006. [Google Scholar]
- 19.Jameson DM, Seifried SE. Quantification of protein-protein interactions using fluorescence polarization. Methods. 1999;19:222–33. doi: 10.1006/meth.1999.0853. [DOI] [PubMed] [Google Scholar]
- 20.Weber G. Protein Interactions. Chapman & Hall; New York/London: 1992. [Google Scholar]
- 21.Weber G. Energetics of ligand binding to proteins. Adv Protein Chem. 1975;29:1–83. doi: 10.1016/s0065-3233(08)60410-6. [DOI] [PubMed] [Google Scholar]
- 22.Jameson DM, Sawyer WH. Fluorescence anisotropy applied to biomolecular interactions. Methods Enzymol. 1995;246:283–300. doi: 10.1016/0076-6879(95)46014-4. [DOI] [PubMed] [Google Scholar]
- 23.Privalov PL. Intermediate states in protein folding. J Mol Biol. 1996;258:707–25. doi: 10.1006/jmbi.1996.0280. [DOI] [PubMed] [Google Scholar]
- 24.Sinha D, Marcinkiewicz M, Lear JD, Walsh PN. Factor XIa Dimer in the Activation of Factor IX. Biochemistry. 2005;44:10416–10422. doi: 10.1021/bi050361x. [DOI] [PubMed] [Google Scholar]
- 25.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha D, Marcinkiewicz M, Gailani D, Walsh PN. Molecular cloning and biochemical characterization of rabbit factor XI. Biochem J. 2002;367:49–56. doi: 10.1042/BJ20020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayandharan G, Shaji RV, Nair SC, Chandy M, Srivastava A. Novel missense mutations in two patients with factor XI deficiency (Val271Leu and Tyr351Ser) and one patient with combined factor XI and factor IX deficiency (Phe349Val) J Thromb Haemost. 2005;3:808–11. doi: 10.1111/j.1538-7836.2005.01230.x. [DOI] [PubMed] [Google Scholar]
- 28.Quelin F, Trossaert M, Sigaud M, Mazancourt PD, Fressinaud E. Molecular basis of severe factor XI deficiency in seven families from the west of France. Seven novel mutations, including an ancient Q88X mutation. J Thromb Haemost. 2004;2:71–6. doi: 10.1111/j.1538-7836.2004.00554.x. [DOI] [PubMed] [Google Scholar]
- 29.Saunders RE, O’Connell NM, Lee CA, Perry DJ, Perkins SJ. Factor XI deficiency database: an interactive web database of mutations, phenotypes, and structural analysis tools. Hum Mutat. 2005;26:192–198. doi: 10.1002/humu.20214. [DOI] [PubMed] [Google Scholar]
- 30.Dorfman R, Walsh PN. Noncovalent interactions of the Apple 4 domain that mediate coagulation factor XI homodimerization. J Biol Chem. 2001;276:6429–38. doi: 10.1074/jbc.M010340200. [DOI] [PubMed] [Google Scholar]
- 31.Rudolph R, Bohm G, Lilie H, Janicke R. Protein Function: A Practical Approach. In: Creighton TE, editor. A Practical Approach. 2. Oxford University Press; Oxford, UK: 1997. [Google Scholar]
- 32.Saxena VP, Wetlaufer DB. Formation of three-dimensional structure in proteins. I Rapid nonenzymic reactivation of reduced lysozyme. Biochemistry. 1970;9:5015–23. doi: 10.1021/bi00827a028. [DOI] [PubMed] [Google Scholar]
- 33.Venters RA, Huang CC, Farmer BT, 2nd, Trolard R, Spicer LD, Fierke CA. High-level 2H/13C/15N labeling of proteins for NMR studies. J Biomol NMR. 1995;5:339–44. doi: 10.1007/BF00182275. [DOI] [PubMed] [Google Scholar]
- 34.Scott CP, Kashlan OB, Lear JD, Cooperman BS. A quantitative model for allosteric control of purine reduction by murine ribonucleotide reductase. Biochemistry. 2001;40:1651–61. doi: 10.1021/bi002335z. [DOI] [PubMed] [Google Scholar]
- 35.Kashlan OB, Scott CP, Lear JD, Cooperman BS. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry. 2002;41:462–74. doi: 10.1021/bi011653a. [DOI] [PubMed] [Google Scholar]
- 36.Goddard TD, Kneller DG. Sparky 3. San Francisco, CA.: 2004. [Google Scholar]
- 37.Montelione GT, Wagner G. Conformation-independent sequential NMR connections in isotope-enriched polypeptides by 1H-13C-15N triple-resonance experiments. J Magn Reson. 1990;87:183–188. [Google Scholar]
- 38.Bax A, Clore GM, Driscoll PC, Gronenborn AM, Mitsuhiko I, Kay LE. Practical Aspects of Proto-Carbon-Proton Three-Dimensional Correlation Spectroscopy of 13C-Labeled Proteins. J Magn Reson. 1991;87:620–627. [Google Scholar]
- 39.Kay LE, Ikura M, Bax A. The design and optimization of complex NMR experiments. Application to a triple-resonance pulse scheme correlating Hα, NH, and 15N chemical shift in 15N-13C-labeled proteins. J Magn Reson. 1991;91:84–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.