

Abstract
Prostate cancer (PCa) is the most frequently diagnosed cancer in men worldwide. As PCa is a complex, multigenic disease, it has been challenging to establish its genetic basis, with strong risk factors obscured by the genetically heterogeneous patient populations often available for analysis. Worldwide the PCa phenotype has been further complicated based on the frequent inclusion of cases in which prostate specific antigen (PSA)-tests allow early diagnosis of possibly latent disease. Thus, previous segregation analyses have variously suggested that the familial aggregation of PCa follows autosomal dominance, recessive or X-linked inheritance, but remain inconclusive. The objective of this study was to assess the familial aggregation of PCa in a sample of 1,546 nuclear families ascertained through an affected father and diagnosed during 1988–1993, from the unique, genetically isolated, founder population-based resource of the Finnish Cancer Registry. We performed segregation analysis on two cohorts of 557 early-onset and 989 late-onset families with clinically diagnosed PCa cases, uncomplicated by predictions from PSA screening. We evaluated residual paternal effects, assuming that age at diagnosis followed a logistic distribution after log-transformation. Our results indicate that Mendelian recessive inheritance consistent with the sex-limited X-linked region previously mapped in Finnish families to the HPCX locus best fit the data in each cohort. With a putative high-risk allele frequency qA of 0.09 in the combined analysis, genotype-specific mean ages at diagnosis of 63.6 years for AA and 71.0 years for AB/BB, respectively, were obtained. The significant paternal regressive coefficient was also indicative of a polygenic multifactorial component, suggesting that environmental factors may contribute to the rising incidence of PCa in Finland.
Keywords: hereditary prostate cancer, Mendelian recessive inheritance, segregation analyses, population-based study, Finland
Introduction
Prostate cancer (PCa) is the most frequently diagnosed non-dermatologic cancer among men in the Western World (World Health Organization 2003). In Finland where the incidence of PCa has been rising in the last decade, it is estimated that in the year 2006 there will be 5,485 newly diagnosed PCa cases with the age-adjusted incidence rate of 115.4/100,000 inhabitants (Finnish Cancer Registry, 2006). Familial clustering of PCa was observed as early as the 1950s (Gianferrari et al. 1956), and in about 10% of all cases there is a clear positive family history of the disease. Carter et al (1992) reported that for 40–50% of PCa cases, familial clustering was associated with multiple affected relatives, especially in families of early-onset probands. Hereditary prostate cancer (HPC), which accounts for 5–10% of all PCa, is an etiologically complex disease with several genes implicated in determining risk (Grönberg et al. 1997). In a large Nordic consortium study of twins, Lichtenstein et al (2000) reported an unusually high heritability of 42% for PCa. The clinical phenotype of PCa is complex and heterogeneous, and the arrival of the prostate specific antigen (PSA) era has further complicated the genetic analysis of PCa by allowing the early diagnosis of disease that might remain latent or clinically unimportant. The International Consortium for Prostate Cancer Genetics (ICPCG), which seeks to improve the mapping of PCa genes, has emphasized that one of the major difficulties in studying PCa is genetic heterogeneity, possibly due to multiple, incompletely penetrant PCa-susceptibility genes (Xu et al (2005). Indeed using parametric (dominant and recessive) and nonparametric analyses on 1,233 families, Xu et al (2005) identified five distinct chromosomal regions with “suggestive” linkage (LOD score >1.86) to PCa, namely 5q12, 8p21, 15q11, 17q21, and 22q12. Subsets of the analyzed group of families characterized by large numbers of early-onset (≤65 years) PCa, which are more likely to segregate highly penetrant mutations, provided stronger evidence of linkage in several regions (including the 22q12 locus, with a LOD score of 3.57). Additional PCa susceptibility loci reported to date (Schaid 2004) also include the three cloned genes: HPC1/RNASEL, HPC2/ELAC2 and MSR1 (Rebbeck et al. 2000; Tavtigian et al. 2001; Carpten et al. 2002; Xu et al. 2002).
The Finnish population of five million inhabitants represents a genetically isolated population with a unique gene pool useful for the study of genetic susceptibility to cancer and other complex diseases (de la Chapelle 1993; Peltonen 1997). Reliable population data are obtainable from various linked registries and the population-based Finnish Cancer Registry (FCR) covers virtually all histologically confirmed cancer diagnoses over almost 50 years. In addition, church and parish records enable the identification of familial relationships for individuals over several centuries. In Finland, HPC1/RNASEL, HPC2/ELAC2 and MSR1 loci explain only a small fraction of PCa cases (Xu et al. 1998; Schleutker et al. 2003). Instead, three additional major susceptibility loci have been mapped in Finnish families including the HPCX (Xq27–28), 3p25–26 and 11q14 regions (Xu et al. 1998; Schleutker et al. 2003). Currently, a large proportion of hereditary prostate cancer remains unexplained in Finland. The purpose of this study was to assess the nature of familial aggregation of PCa in a sample of 1,546 Finnish nuclear families using regressive models as employed in complex segregation analysis. Segregation analysis is a statistical method for testing compatibility with Mendelian expectations by estimating the parameters of a given model of inheritance from family data. Previous segregation analyses in diverse populations have variously suggested that familial aggregation of PCa follows autosomal dominance, multifactorial, recessive or X-linked inheritance, but remain inconclusive (Carter et al. 1992; Grönberg et al. 1997; Schaid et al. 1998; Verhage et al. 2001; Conlon et al. 2003; Valeri et al. 2003; Gong et al. 2002; Cui 2001). Families of Icelandic breast cancer probands with PCa-affected men yielded a codominant model (Baffoe-Bonnie et al. 2002). Based on the reported PCa mean age of onset of 61 years in the Finnish Cancer Registry (Mäkinen et al 2002), we collected two cohorts to test the possibility of different modes of inheritance in families of early-onset probands (<61 years) versus late-onset families (≥61 years). We have performed segregation analyses on these two separate cohorts and also analyzed the complete, combined dataset to determine the most parsimonious model for explanation of the familial aggregation of PCa in Finland.
Subjects and Methods
Data sources
The nation-wide population based Finnish Cancer Registry (FCR) was founded in 1952 and reporting of cancer to the FCR was made obligatory in 1961. Currently physicians, hospitals and pathology laboratories send their reports to the registry independently. In addition, the FCR receives information from every death certificate in which cancer is mentioned, registering over 99% of all solid tumors diagnosed in Finland (Teppo et al. 1994). The FCR files can be linked to the registry of deaths and of immigrants issued by the Population Registry Center in Finland. Population registration in Finland has traditions dating back to the 16th century and is considered to be of excellent quality. Since 1964 a centralized, nation-wide, computer-based population registry has been maintained by the National Population Registry Center and is based on unique personal identifiers, which are now used as main keys in every major person registry including the Finnish Cancer Registry.
Probands and relatives
We chose the pre-PSA time period between 1 January 1988 and 31 December 1993 and identified 9,142 men with newly diagnosed PCa nation-wide from the Finnish Cancer Registry. For the current study, probands were selected from three central hospital regions in Mid-Finland (Pirkanmaa) and from East Finland (North Karelia and Kainuu). These regions were selected because the PCa incidence in these regions is close to the average PCa incidence in Finland (Pirkanmaa 58.3/100,000, North-Karelia 48.8/100,000, and Kainuu 49.6/100,000). The first-degree relatives of the 1,546 probands (11,427) were identified from all contributing registries. Of these, 10,650 (93%) were traced successfully, while 777 (7%) were lost to follow-up. The 777 cases lost to follow-up included 603 cases with little or no data from local officials, and 174 whose records were lost during World War II. For the 715 families with persons lost to follow-up, data were missing for a mean of 1.2 persons per family (range one to six persons). Out of the 10,650 relatives with complete follow-up information, 81 per cent (8,628) were alive during the period 1953–1997. This large data set comprised two cohorts independently assembled based on the reported PCa mean age of onset of 61 years in the Finnish Cancer Registry (Mäkinen et al 2002). Of the 989 cohort of older probands (≥61 years), 659 probands came from the Pirkanmaa region together with 320 from the East Finland region. Details of the collection of population-based PCa families and the analyses for other cancers among first-degree relatives have been published elsewhere (Matikainen et al. 2001). Information on the birthplaces of probands was obtained from the Central Population Registry. The local registries (parishes and local authorities) of the communities where probands were born were contacted to obtain the names and birth dates of their parents, siblings, spouses and children. Descriptive statistics of these cohorts and the combined group of 1,546 families are shown in Table I.
Table I.
Descriptive Statistics for Prostate Cancer Cohorts in Finland
| Description | Cohort-1 | Cohort-2 | Combined Cohorts |
|---|---|---|---|
| Probands | 557 | 989 | 1,546 |
| Non-probands | 3,631 | 7,019 | 10,650 |
| Affected non-probands | 51 | 109 | 160 |
| Number of affected | 608 | 1,098 | 1,706 |
| Individuals in cohort | 4,188 | 8,008 | 12,196 |
| Number of Males | 2,418 (57.7%) | 4,664 (58.2%) | 7,082 (58.1%) |
| Number of Females | 1,770 (42,3%) | 3,344 (46.9%) | 5,114 (41.9%) |
| Number of Fathers | 390 | 673 | 1,063 |
| Number of Brothers | 846 | 1,840 | 2,686 |
| Number of Sons | 625 | 1,162 | 1,787 |
| Number of Mothers | 388 | 681 | 1,069 |
| Number of Sisters | 794 | 1,642 | 2,436 |
| Number of Daughters | 588 | 1,021 | 1,609 |
| Mean age of probands (years) | 56.6 ± 3.4 | 74.4 ± 7.4 | 68.2 ±10.7 |
| Range of age at diagnosis of probands (years) | 41.8 – 60.9 | 61 – 96 | 41.8 – 96 |
| Mean age of affected non-probands (years) | 69.6 ± 8.0 | 71.9 ± 7.8 | 71.2 ± 7.9 |
| Range of age at diagnosis of affected non-probands (years) | 48.3 – 85.5 | 48.1 – 88.7 | 48.1 – 88.7 |
| Unaffected men aged ≥ 48 (years) | 909 (18.3%) | 2,141 (23.0%) | 3,050 (21.4%) |
| Mean age of unaffected men (years) | 65.2 ± 9.8 | 67.5 ± 12.3 | 66.6 ± 11.4 |
| Range of ages of unaffected men (years) | 48 – 85.5 | 48 – 104.3 | 48 –104.3 |
| Pedigree sizes (average; range) | 8.9 (3–23) | 9.4 (3–25) | 9.2 (3–25) |
| Percent of pedigrees with ≥ 10 and ≥20 persons | 28% (0.7%) | 37.6% (0.5%) | 34.2% (0.6%) |
Segregation analysis
To test specifically for Mendelian inheritance of PCa in these Finnish pedigrees, maximum likelihood segregation analyses were performed on the age at diagnosis expressed as a censored trait using the REGTL module of the Statistical Analysis for Genetic Epidemiology program (S.A.G.E. 3.1. 1997). Under model 1 of this program, employing class A regressive models (Bonney 1986), the “type” or “ousiotype” (Cannings et al 1978) influences age at diagnosis of PCa through the location and scale parameters of the logistic distribution, but does not influence susceptibility. Specifically, some constant proportion ( ) of the male population is assumed to be at a risk of PCa. The PCa phenotype is defined as a dichotomous variable (Y), where Y = 1 if affected and Y = 0 if unaffected (censored). Parameters estimated in the analysis include: qA, the frequency of the putative high-risk allele ‘A’, βi, baseline parameters, where i represents an individual’s type (AA, AB, BB); αi the age coefficients and γi the susceptibilities (Elston and George 1989). The logistic function describing the probability that an individual is affected by age “a” is given as γi[1/(1+e−Φ], where
| (1) |
The coefficient δF reflects familial influence on risk corresponding to having an affected father. Positive values of δF mean that the individual with an affected father is more likely to have an earlier age at diagnosis, while negative values mean that the individual with an affected father is more likely to have a later age at diagnosis. Nonzero values of δF indicate the effects of polygenic and/or unmeasured shared-familial environmental risk factors on PCa risk. Age at diagnosis for PCa phenotype is assumed to follow a logistic distribution described by two parameters α and β, with the probability distribution function according to Elston and George (1989)
| (2) |
This symmetric distribution is similar to a normal distribution and has a mean –β/α, and variance, π2/3α2, where p has a value of 3.1416. Based on the logistic distribution, the cumulative distribution function (c.d.f.) is given by
| (3) |
The c.d.f. represents the probability that a susceptible person will be affected by a given age. Age-specific penetrances were calculated for each genotype as
| (4) |
If the observed sex-specific ages at diagnosis do not follow a logistic distribution, this model may still be appropriate after transformation. A transformation equation equivalent to: aG1 x ln (age) was considered here, where aG1 is the geometric mean age at diagnosis for PCa, computed from the observed ages at diagnosis among the 160 affected non-probands with PCa, 51 for the early-onset and 109 for the late-onset cohorts (Table 1). Tests for genetic contribution to disease risk were implemented by postulating three types of individuals (AA, AB, BB) with three corresponding transmission parameters (τAA, τAB, τBB) describing the probability that a parent of a given type transmits the disease producing factor ‘A’ to his/her offspring (Elston and Stewart 1971; Elston and Yelverton 1975; Elston 1981). Under the hypothesis of genetic transmission, these τ parameters are constrained to the Mendelian values of τAA = 1.0, τAB = 0.5, τBB = 0.0. Five sub-models of disease transmission were tested against a general model, where the transmission probabilities are estimated but with the restriction of homogeneity of trait distribution across generations to identify the best model for these data (Elston 1981). The “no major gene” model assumes that baseline risk is not influenced by “type” therefore all persons would come from a single distribution of age-specific risk for PCa. Single-locus Mendelian models assume that a major locus with two alleles should act in codominant, dominant or recessive fashion. The dominant and recessive models are special cases of the codominant model, where each genotype has a distinct age at diagnosis distribution. An environmental model with potentially distinct types of individuals was also tested, but here the transmission probability was held constant for all individuals. We present results from the maximum likelihood segregation analyses performed on the log-transformed age at diagnosis of PCa expressed as a censored trait using the REGTL program (S.A.G.E. 3.1. 1997). Log-transformation of ages at diagnosis and ages at examination for all individuals with non-zero ages led to a final model that estimated genotypic baseline parameters (βi) and age coefficient (αi) and lifetime susceptibility for PCa for males along with the frequency (qA) of the high-risk allele A.
Hypothesis Testing
The likelihood ratio test (LRT) was used to test each sub-model against the general model, and was computed as minus twice the natural log likelihood (-2ln (L)) of the general model subtracted from that for a restricted sub-model. This difference is asymptotically distributed as a χ2 distribution with degrees of freedom equal to the difference in the number of independent parameters estimated in the two models.
Another method to compare models uses Akaike’s information criteria (AIC), defined as: AIC = −2ln(L) + 2(number of parameters estimated). The most parsimonious model has the minimum AIC value (Akaike 1974). To correct for ascertainment bias, the likelihood of each pedigree was conditioned on the proband’s affection status, using his age at diagnosis as recorded in the Cancer Registry (Cannings and Thompson 1977; Elston and Sobel 1979).
Results
Descriptive Analyses of Cohort-1
Cohort-1 consisted of 557 nuclear families ascertained through early-onset probands diagnosed before 61 years. Of the 4,188 individuals in the early-onset cohort, 57.7% (2,418) were males. The number of fathers, brothers and sons respectively, were 390, 846 and 625. The corresponding numbers of mothers, sisters and daughters reported were 388, 794 and 588, respectively for a total of 1,770 females. Cohort-1 pedigree sizes ranged between 3 and 23 persons with an average of 8.9. The proportion of pedigrees with 10 or more individuals was 28%. Less than one percent of the pedigrees had more than 20 persons. The range of age at diagnosis of the 557 probands was 41.8 to 60.9 years with a mean of 56.6 (SD ± 3.4) years. The 51 affected non-probands had a mean age of 69.6 (SD ± 8.0) years. The 909 unaffected male relatives aged ≥ 48 years formed 18.3% of the cohort, with a mean age of 65.2 (SD ± 9.8).
Segregation analyses results for Cohort-1 with 557 early-onset PCa families
As shown in Table II, the no major gene model gave a very poor fit to the data in the early-onset PCa cohort and was thus rejected against the general unrestricted model in which all parameters were estimated based on the likelihood ratio test (LRT) (model 1 vs. model 6, χ2 = 18.36, p <0.005 for 6df). The dominant (p < 0.0014), codominant (p < 0.036) and environmental (p < 0.0082) models were all rejected compared to the general model. The final general model reported is almost identical to the recessive Mendelian model (model 4 vs. model 6, χ2 = 1.54, p <0.83 for 4df). The AIC, which takes into account the number of parameters estimated also confirmed that the recessive model was the most parsimonious model. The estimated frequency (± standard error (SE)) for the high-risk allele qA was 0.054 (± 0.01) for the recessive model. Figure 1 presents the predicted cumulative distribution function curves for log-transformed ages at diagnosis for the early-onset families under the recessive model. High-risk homozygous carriers of the putative risk allele AA have predicted age-specific cumulative probabilities greater than the heterozygous AB and BB non-carriers. The predicted mean age at diagnosis (i.e., 50% cumulative risk in Figure 1) for the AA individuals is 60 years and 64.6 years for the non-carriers. The susceptibility parameter was estimated at 1.0 for all male carriers of the risk allele, suggesting that 100% of the male population if they lived to infinity and did not die of competing causes, would express PCa if they were homozygous carriers of the allele A. Under this Mendelian recessive model, the cumulative probability that a male in Finland would be affected by PCa by age 70 was 0.92 for carriers and 0.79 for non-carriers, thus implying that if carriers and non-carriers did not die from competing causes, the estimated risk of being diagnosed with PCa at age 70 years for the homozygote carriers of the deleterious allele (q=0.054), would be 2.7 per 1000 among a hypothetical cohort of 100,000 men.
Table II.
Parameter estimates from segregation analysis of prostate cancer in 557 early-onset Finnish families ascertained through a single prostate cancer proband aged <61 years
| Value of parameter
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypothesis | -2ln L | AIC | df | χ2 | P | qA | τAA | τAB | τBB | βAA | βAB | βBB | α | γ | F(aff) |
| 1. No major gene | 479.02 | 487.02 | 6 | 18.36 | 0.005 | [1.0] | - | - | - | −53.73 (5.31) | =βAA | =βAA | 0.20 (0.014) | 1.0 (0.0) | 3.4 (0.25) |
| 2. Dominant | 473.08 | 485.08 | 4 | 12.42 | 0.014 | 0.9421 (0.597) | [1.0] | [0.5] | [0.0] | −62.27 (5.56) | =βAA | −57.97 (4.32) | 0.24 (0.019) | 1.0 (0.0) | 3.4 (0.24) |
| 3. Codominant | 474.18 | 488.18 | 3 | 13.52 | 0.036 | 0.0000007 (0.0) | [1.0] | [0.5] | [0.0] | −41.41 (3.67) | −63.40 (4.48) | −85.39 (7.01) | 0.25 (0.017) | 1.0 (0.0) | 3.4 (0.33) |
| 4. Recessive | 462.20 | 474.20 | 4 | 1.54 | 0.8266 | 0.0536 (0.01) | [1.0] | [0.5] | [0.0] | −60.11 (4.11) | −64.63 (5.55) | =βAB | 0.25 (0.021) | 1.0 (0.0) | 3.7 (0.41) |
| 5. Environmental | 465.76 | 481.76 | 2 | 5.1 | 0.082 | 0.4183 (0.24) | 1.0 (0.0) | =τAA | =τAA | −77.25 (7.01) | −79.69 (8.34) | −68.55 (6.98) | 0.27 (0.036) | 1.0 (0.0) | 3.3 (0.31) |
| 6. General | 460.66 | 480.66 | - | - | - | 0.7328 (0.45) | 1.0 (0.0) | 0 (0.0) | 0 (0.0) | −65.08 (6.93) | −67.98 (7.01) | −76.79 (7.86) | 0.26 (0.019) | 1.0 (0.0) | 3.0 (0.30) |
χ2 is defined as (-2ln L) of the data under the hypothesis minus (-2ln L) of the data under the general model. Numbers in brackets are fixed at the indicated value; qA : the frequency of the putative high-risk allele; τ: transmission parameter denoting the probability that a parent of a given type transmits the disease-producing factor A to his or her offspring; β: baseline parameter; α: age adjustment parameter; γ: susceptibility parameter describing the cumulative probability of prostate cancer (assuming infinite lifespan).
Figure 1.
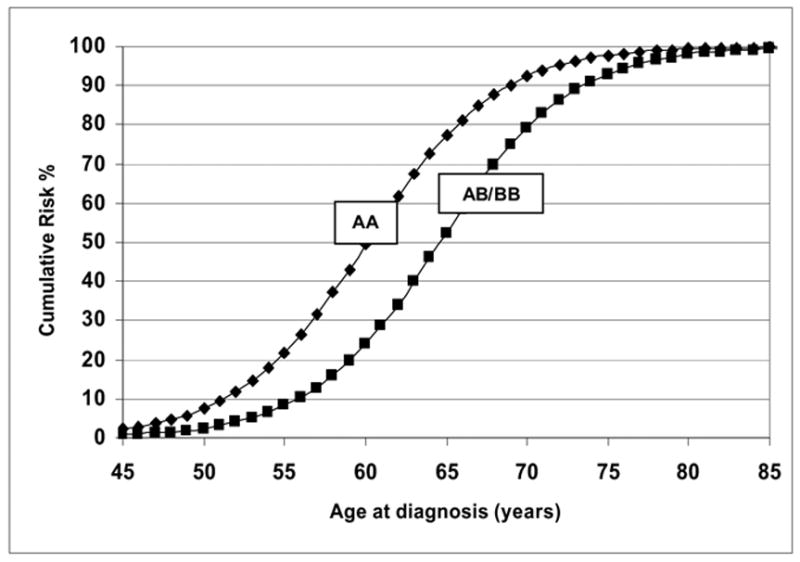
Predicted cumulative risks for recessive AA (carriers) and AB/BB (non-carriers) with an affected father: 557 early-onset Finnish prostate cancer families
Descriptive Analyses of Cohort-2
The 989 families ascertained through older probands diagnosed ≥ 61 years had 8,008 individuals, 4,664 (58.2%) of which were males. The number of fathers, brothers and sons respectively, were 673, 1,840 and 1,162. The corresponding number of mothers, sisters and daughters reported were 681, 1,642 and 1,021, respectively for a total of 3,344 females. Cohort-2 pedigree sizes ranged between 3 and 25 persons with an average of 9.4. The proportion of pedigrees with 10 or more individuals was 37.6%. Less than one percent of the pedigrees had more than 20 persons. The range of age at diagnosis of the 989 probands was 61 to 96 years with a mean of 74.4 (SD ± 7.4) years. The 109 affected non-probands had a mean age of 71.9 (SD ± 7.8) years. The 2,141 unaffected male relatives aged ≥ 48 years formed 23.0% of the cohort, with a mean age of 67.5 (SD ± 12.3).
Segregation analyses results for Cohort-2 with 989 late-onset PCa families
Table III shows the parameter estimates from the segregation analysis for the 989 families ascertained through late-onset probands diagnosed at ≥ 61 years of age. Compared to the unrestricted general model, the no major gene, the Mendelian dominant and environmental models did not fit the data and were rejected at P <0.001. The Mendelian codominant model was also rejected by the LRT with a χ2 of 12.64 and a p value of 0.006. The recessive model was the most parsimonious model according to the LRT (model 4 vs. model 6, χ2 = 6.82, p <0.15 for 4df), and it also had the lowest AIC = 1331.10. Under this recessive model, inheritance of a putative high-risk allele A with an allele frequency (± SE) of 0.086 (± 0.006) had predicted mean ages of onset of 65.6 years for men with the AA genotype and 72.2 years for AB/BB males, respectively. The lifetime risk of being diagnosed with PCa under this model was 5.0 per 1,000 among a hypothetical cohort of 100,000 men.
Table III.
Parameter estimates from segregation analysis of prostate cancer in 989 late-onset Finnish families ascertained through a single prostate cancer proband.
| Value of parameter
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypothesis | -2ln L | AIC | df | χ2 | P | qA | τAA | τAB | τBB | βAA | βAB | βBB | α | γ | F(aff) |
| 1. No major gene | 1371.08 | 1379.08 | 6 | 58.8 | <0.001 | [1] | [1] | [0.5] | [0] | −59.92 (4.16) | =βAA | =βAA | 0.18 (0.013) | 0.26 (0.06) | 2.02 (0.18) |
| 2. Dominant | 1342.72 | 1354.72 | 4 | 30.44 | <0.001 | 0.00148 (0.001) | [1] | [0.5] | [0] | −57.81 (4.53) | =βAA | −62.53 (6.17) | 0.18 (0.012) | 1.0 (0.0) | 3.18 (0.29) |
| 3. Codominant | 1324.92 | 1338.92 | 3 | 12.64 | 0.006 | 0.03137 (0.005) | [1] | [0.5] | [0] | −70.61 (6.88) | −75.26 (7.46) | −79.91 (8.01) | 0.22 (0.016) | 1.0 (0.0) | 4.17 (0.51) |
| 4. Recessive | 1319.10 | 1331.10 | 4 | 6.82 | 0.15 | 0.0855 (0.006) | [1] | [0.5] | [0] | −64.71 (6.12) | −71.30 (6.95) | =βAB | 0.21 (0.015) | 1.0 (0.0) | 3.99 (0.63) |
| 5. Environmental | 1339.66 | 1355.66 | 2 | 27.38 | <0.001 | 0.04486 (0.003) | 0 (0.0) | =βAA | =βAA | −64.20 (5.01) | −62.32 (4.99) | −84.39 (8.15) | 0.19 (0.012) | 1.0) (0.0) | 1.91 (0.09) |
| 6. General | 1312.28 | 1332.28 | - | - | - | 0.9974 (0.014) | 1.0 (0.0) | 1.0 (0.0) | 1.0 (0.0) | −66.93 (5.71) | −70.93 (6.34) | −72.21 (6.94) | 0.21 (0.014) | 0.44 (0.11) | 2.85 (0.45) |
χ2 is defined as (-2ln L) of the data under the hypothesis minus (-2ln L) of the data under the general model. Numbers in brackets are fixed at the indicated value; qA : the frequency of the putative high-risk allele; τ: transmission parameter denoting the probability that a parent of a given type transmits the disease-producing factor A to his or her offspring; β: baseline parameter; α: age adjustment parameter; γ: susceptibility parameter describing the cumulative probability of prostate cancer (assuming infinite lifespan).
Figure 2 shows the predicted cumulative distribution function curves for log-transformed ages at diagnosis for this late-onset cohort in which the AA genotype has a distinctly different mean age at diagnosis of PCa.
Figure 2.
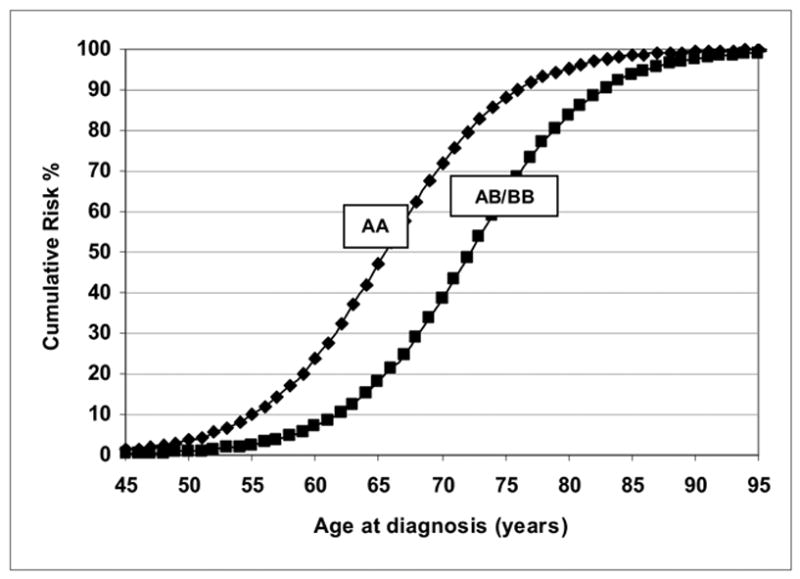
Predicted cumulative risks for recessive AA (carriers) and AB/BB (non-carriers) of high-risk allele A with an affected father for 989 late-onset Finnish prostate cancer families
Descriptive Analyses of Combined Cohort-1 and Cohort-2
The 1,546 families of the combined cohort had 10,650 individuals, 7,082 (58.1%) of which were males. The combined number of fathers, brothers and sons respectively, were 1,063, 2,686 and 1,787. The corresponding numbers of mothers, sisters and daughters reported were 1,069, 2,436 and 1,609, respectively for a total of 5,114 females. The combined cohorts had pedigree sizes ranging between 3 and 25 persons with an average of 9.2. The proportion of pedigrees with 10 or more individuals was 34.2%. Less than one percent of the pedigrees had more than 20 persons. The range of age at diagnosis of the 1,546 probands was 41.8 to 96 years with a mean of 68.2 (SD ± 10.7) years. The 160 affected non-probands had a mean age of 71.2 (SD ± 7.9) years. The 3,050 unaffected male relatives aged ≥ 48 years formed 21.4% of the cohort, with a mean age of 66.6 (SD ± 11.4).
Segregation analyses results for Combined Cohort-1 and Cohort-2
From Table IV, the combined cohorts with 1,546 probands gave parameter estimates very similar to those obtained from Cohort-2 with 989 probands. All other models except the Mendelian recessive model were significantly rejected when compared with the unrestricted general model. The recessive model was the most parsimonious model according to the LRT (model 4 vs. model 6, χ2 = 8.78, p <0.07 for 4df), and it also had the lowest AIC = 1795.12. Under this recessive model, inheritance of a putative high-risk allele A with an allele frequency (± SE) of 0.0903 (± 0.005) had predicted mean ages of onset of 63.6 years for men with the AA genotype and 71.0 years for AB/BB genotype males, respectively. Figure 3 shows that under the recessive model, the predicted cumulative risks for PCa are distinctly different for the AA compared to the AB/BB genotypes. The estimated mean age at diagnosis for the male homozygous carriers of the putative high-risk allele A is 63.6 and it is 71.0 years for AB/BB genotype males. With a cumulative risk of 0.80 for homozygote carriers of the A allele at age 70 years, the estimated risk of being diagnosed with PCa in the absence of competing causes of death was 6.5 per 1,000 among a hypothetical cohort of 100,000 men. All of these models (in each cohort and in the combined cohorts) included a residual paternal regressive coefficient, since inclusion of this coefficient significantly improved the fit of these models. The impact of genotype alone vs. residual effect of having an affected father can be measured by computing the log odds of various combinations.
Table IV.
Parameter estimates from segregation analysis of prostate cancer in combined analysis of two cohorts of 1,546 Finnish families ascertained through a single prostate cancer proband
| Value of parameter
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypothesis | -2ln L | AIC | df | χ2 | P | qA | τAA | τAB | τBB | βAA | βAB | βBB | α | γ | F(aff) |
| 1. No major gene | 1884.38 | 1892.38 | 6 | 110.04 | <0.001 | [1] | [1] | [0.5] | [0] | −54.26 (5.21) | =βAA | =βAA | 0.17 (0.014) | 1.0 (0.0) | 3.47 (0.35) |
| 2. Dominant | 1830.36 | 1842.36 | 4 | 56.02 | <0.001 | 0.0022 (0.001) | [1] | [0.5] | [0] | −61.57 (5.98) | =βAA | −65.77 (5.92) | 0.21 (0.018) | 1.0 (0.0) | 3.50 (0.47) |
| 3. Codominant | 1806.42 | 1820.42 | 3 | 32.08 | <0.001 | 0.1012 (0.007) | [1] | [0.5] | [0] | −70.16 (6.23) | −74.68 (7.26) | −86.40 (8.11) | 0.23 (0.010) | 1.0 (0.0) | 3.20 (0.45) |
| 4. Recessive | 1783.12 | 1795.12 | 4 | 8.78 | 0.07 | 0.0903 (0.005) | [1] | [0.5] | [0] | −63.34 (6.01) | −71.23 (6.75) | =βAB | 0.22 (0.013) | 1.0 (0.0) | 3.90 (0.53) |
| 5. Environmental | 1866.34 | 1882.34 | 2 | 92.0 | <0.001 | 0.4868 (0.061) | 0 (0.0) | =βAA | =βAA | −55.74 (5.64) | −79.41 (8.04) | −56.10 (4.34) | 0.18 (0.011) | 1.0 (0.0) | 3.32 (0.33) |
| 6. General | 1774.34 | 1794.34 | - | - | - | 0.9542 (0.004) | 1.0 (0.0) | 1.0 (0.0) | 1.0 (0.0) | −49.77 (4.7) | −53.49 (5.62) | −57.31 (5.89) | 0.16 (0.014) | 1.0 (0.0) | 3.20 (0.29) |
χ2 is defined as (-2ln L) of the data under the hypothesis minus (-2ln L) of the data under the general model. Numbers in brackets are fixed at the indicated value; qA : the frequency of the putative high-risk allele; τ: transmission parameter denoting the probability that a parent of a given type transmits the disease-producing factor A to his or her offspring; β: baseline parameter; α: age adjustment parameter; γ: susceptibility parameter describing the cumulative probability of prostate cancer (assuming infinite lifespan).
Figure 3.
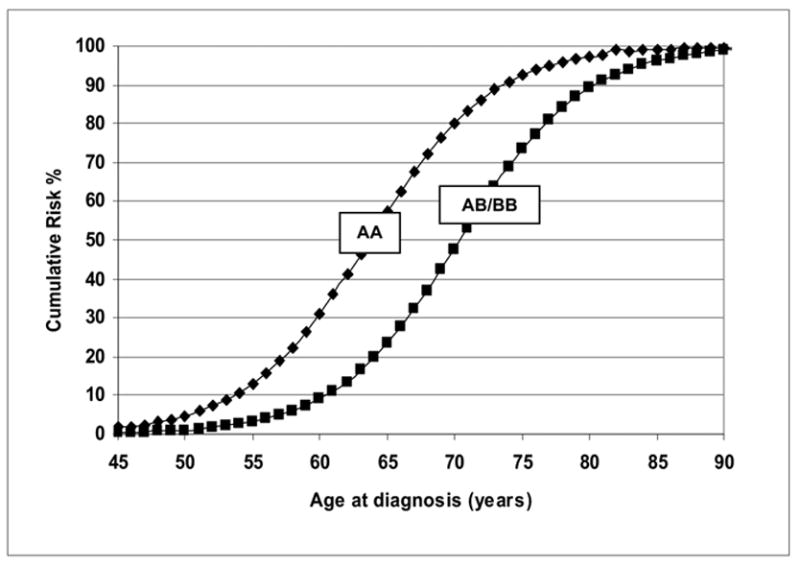
Predicted cumulative risks for recessive AA (carriers) and AB/BB (non-carriers) of the putative high-risk allele A with an affected father for the 1,546 combined Finnish prostate cancer families
In effect, among men of the same age, born in the same cohort, and having the same affected father status, the log odds for being a homozygous carrier of the high risk allele A is computed as the difference between the genotypic baseline coefficients of the homozygous carriers and of the heterozygote and homozygous non-carriers. Using the parameters of the recessive model in the combined cohort as an example:
The odds of PCa in homozygous carriers of the A allele compared to the AB/BB non-carriers is 2670, i.e., the exp (7.89). The log-odds due to an affected father between two individuals with the same genotype is 3.90 and the corresponding odds ratio is 49.40. The increase in log odds for a homozygote for the high-risk allele A with an affected father compared to the heterozygote carrier of the same age born in the same cohort but having an affected father would therefore be (βAA–βAB/BB) + δF(YF)) = 7.89 + 3.90 = 11.79 leading to a high odds ratio.
Discussion
It is evident from the present segregation analysis that after controlling for a residual paternal effect and an age at diagnosis cohort effect, the genetic control of age at diagnosis of PCa in Finland is best explained by Mendelian recessive inheritance. The mathematical beauty of complex segregation analysis is its ability to disentangle genetic from non-genetic contributions to transmission of a phenotype such as PCa from parent to offspring. Recessive ‘A’ allele implies that in the presence of the ‘B’ allele, ‘A’ is not expressed unless in its homozygous form. For an adult onset, sex-limited cancer such as PCa, recessive inheritance with incomplete penetrance and sporadic cases is consistent with X-linked PCa which we previously mapped to the Xq27–28 region using Finnish families characterized by ‘no-male-to-male transmission’ (NMM). A follow-up linkage disequilibrium study utilizing familial/sporadic PCa cases and appropriate healthy controls identified an associated haplotype in the HPCX region (Baffoe-Bonnie et al 2005). The results of this segregation analysis study are therefore consistent with the X-linked PCa transmission described previously (Schleutker et al 2000). Under Hardy-Weinberg equilibrium, the estimated allele frequency of 0.09 for the combined cohort implies that 0.81% of the population in Finland (both genders) would carry this rare putative high-risk allele. However, being a gender limited disease the susceptibility parameter of 1.0 obtained in the analysis suggested that 100% of the homozygous carrier male population at risk would develop PCa if they lived to infinity and did not die of competing causes. In Finland where the incidence of PCa has been rising over a decade, the current study suggests that men live in an environment that makes them susceptible irrespective of genetic susceptibility. Under the most parsimonious recessive hypothesis, the cumulative probability that a man with genotype AA would be affected with PCa by age 70 years was 80% compared to AB/BB genotypes with 50%. This translated into a lifetime risk of 8 per 1,000 among a hypothetical population of 100,000 men with a lower at age diagnosis compared to the heterozygous AB and BB non-carriers of the risk allele making this estimate of the putative PCa risk allele more frequent than reported in some studies. By age 85 years, all males irrespective of genotype have a cumulative risk close to 100 per cent. Compared to non-carriers, homozygous carriers of the risk allele (AA genotype) are diagnosed with PCa at younger mean ages of 60.1, 65.6 and 63.6 years, respectively, among the 557 early-onset and 989 late-onset cohorts or when combined. Since the residual paternal effect was positive, those with affected fathers were at a higher risk for earlier onset PCa with polygenic and/or unmeasured shared-familial environmental risk factors compared to those with unaffected fathers.
The heterogeneity of PCa is supported by the varying results obtained from complex segregation analyses worldwide. The findings of this study are quite different from the previously reported evidence for the segregation of a rare autosomal dominant gene with high penetrance among different populations that included some series of prostatectomy patient families (Carter et al. 1992; Grönberg et al. 1997; Schaid et al. 1998; Verhage et al. 2001; Conlon et al. 2003; Valeri et al. 2003). Grönberg et al. (1997) performed segregation analysis on a population-based sample of 2,857 families selected through an affected father diagnosed with PCa in 1959–1963, and identified from the Swedish nationwide Cancer Registry. The observed clustering of PCa in Sweden was best explained by a dominantly inherited high risk allele with a population frequency (1.67%) and a moderate lifetime penetrance (63%).
Since previous segregation analyses of PCa have rarely reported a recessive mode of inheritance, earlier linkage analyses adopted the autosomal dominant model (Smith et al 1996). We have utilized 52 Finnish families in prior linkage studies using the autosomal dominant model and failed to map susceptibility loci identified in other populations. We note that pedigree analyses of the Finnish families indicated that among half of these families, the disease was observed only in the brothers of probands, with others showing patterns suggestive of maternal inheritance indicative of recessive or sex-limited X-linked patterns of inheritance. Cui et al. (2001) suggested that recessively inherited or X-linked inheritance increased risk at older ages, which was also observed in our linkage analyses of the HPCX locus (Xu et al. 1998; Schleutker et al. 2003). When most of the increase in relative risk in pedigrees was contributed by affected brothers, in the presence of residual brother-brother dependence a recessive or X-linked inheritance of PCa was most likely (Narod et al. 1995; Monroe et al. 1995; Schaid et al. 1998; Valeri et al. 2003). Gong et al. (2002) observed that a multifactorial model with multiple genes, each having low penetrance may be responsible for the familial clustering among 1,719 first degree relatives in American and Canadian families. In a segregation analysis of 389 Icelandic pedigrees that included both breast and PCa, Baffoe-Bonnie et al. (2002), reported that the most parsimonious model was a Mendelian codominant model.
The particular strength of our study is the large population-based data composed of a homogenous Finnish population with registry-based approaches that provide unbiased information of malignancies in families. Prostate cancer was histologically confirmed in all cases in our families, and did not rely on PSA screening. The homogeneity of the Finnish population increases our chances of identifying loci which may be less pronounced in ethnically more diverse populations. Thus, linkage and association analyses of HPC conducted on Finnish families have found loci that are different from those reported in studies from other countries and populations (Schleutker et al. 2003; Seppälä et al. 2003a; Seppälä et al. 2003b).
In conclusion our findings suggest that the inheritance of PCa in the Finnish population is best explained by a Mendelian recessive model with a significant paternal regressive coefficient that is indicative of a polygenic multifactorial component. The rising incidence of PCa in Finland is possibly due to a combination of factors that include socio-cultural and lifestyle changes, environmental factors and the on-going PSA screening (Finnish Cancer Registry, Mäkinen et al, 2003).
Acknowledgments
The authors thank Riitta Vaalavuo for her excellent assistance. Dr. Erica Golemis of the Fox Chase Cancer Center provided beneficial comments on this manuscript. This study was financially supported by the Medical Research Fund of Tampere University Hospital, the Reino Lahtikari Foundation, the Finnish Cancer Organizations, the Sigrid Juselius Foundation and the Academy of Finland (grant # 211123). The program package SAGE supported by the U.S Public Health Service Resource Grant RR03655 from Division of Research Resources, was used for the complex segregation analyses. This work was partially supported by the Intramural Research Program of the National Human Genome Research Institute, the National Institutes of Health (Contract Number N01-HG-55389). A.B.B-B, S.D. and L.O also received support from USPHS grant CA-06927 and an appropriation from the Commonwealth of Pennsylvania.
Footnotes
The original publication is available at http://www.springerlink.com/content/mtk11462490073gj/.
Contributor Information
Sanna Pakkanen, Laboratory of Cancer Genetics, Institute of Medical Technology, University of Tampere and Tampere University Hospital, Tampere, Finland..
Agnes B Baffoe-Bonnie, Division of Population Science, Fox Chase Cancer Center, Philadelphia, PA 19111, USA National Human Genome Research Institute, National Institutes of Health, Baltimore, MD 21131, USA.
Mika P Matikainen, Department of Urology, Tampere University Hospital, University of Tampere, Tampere, Finland.
Pasi A Koivisto, Laboratory of Cancer Genetics, Institute of Medical Technology, University of Tampere and Tampere University Hospital, Tampere, Finland.
Teuvo LJ Tammela, Department of Urology, Tampere University Hospital and Medical School, University of Tampere, Tampere, Finland.
Snehal Deshmukh, Division of Population Science, Fox Chase Cancer Center, Philadelphia, PA 19111, USA.
Liang Ou, Division of Population Science, Fox Chase Cancer Center, Philadelphia, PA 19111, USA.
Joan E Bailey-Wilson, National Human Genome Research Institute, National Institutes of Health, Baltimore, MD 21131, USA.
Johanna Schleutker, Laboratory of Cancer Genetics, Institute of Medical Technology, University of Tampere and Tampere University Hospital, Tampere, Finland.
References
- Akaike H. A new look at the statistical model identification. IEEE Transactions. 1974;19:716–723. [Google Scholar]
- Baffoe-Bonnie AB, Kiemeney LA, Beaty TH, Bailey-Wilson JE, Schnell AH, Sigvaldsson H, Olafsdottir G, Tryggvadottir L, Tulinius H. Segregation analysis of 389 Icelandic pedigrees with breast and prostate cancer. Genet Epidemiol. 2002;23:349–363. doi: 10.1002/gepi.10188. [DOI] [PubMed] [Google Scholar]
- Baffoe-Bonnie AB, Smith JR, Stephan DA, Schleutker J, Carpten JD, Kainu T, Gillanders EM, Matikainen M, Teslovich TM, Tammela T, Sood R, Balshem AM, Scarborough SD, Xu J, Isaacs WB, Trent JM, Kallioniemi OP, Bailey-Wilson E. A major locus for hereditary prostate cancer in Finland: localization by linkage disequilibrium of a haplotype in the HPCX region. Hum Genet. 2005;117(4):307–16. doi: 10.1007/s00439-005-1306-z. [DOI] [PubMed] [Google Scholar]
- Bonney GE. Regressive logistic models for familial disease and other binary traits. Biometrics. 1986;42:611–625. [PubMed] [Google Scholar]
- Cannings C, Thompson E, Skolnick M. Probability functions on complex pedigrees. Adv Appl Probl. 1978;10:26-26–61. [Google Scholar]
- Cannings C, Thompson EA. Ascertainment in the sequential sampling of pedigrees. Clin Genet. 1977;12:208–212. doi: 10.1111/j.1399-0004.1977.tb00928.x. [DOI] [PubMed] [Google Scholar]
- Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992;89:3367–3371. doi: 10.1073/pnas.89.8.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EM, Goode EL, Gibbs M, Stanford JL, Badzioch M, Janer M, Kolb S, Hood L, Ostrander EA, Jarvik GP, Wijsman EM. Oligogenic segregation analysis of hereditary prostate cancer pedigrees: Evidence for multiple loci affecting age at onset. Int J Cancer. 2003;105:630–635. doi: 10.1002/ijc.11128. [DOI] [PubMed] [Google Scholar]
- Cui J, Staples MP, Hopper JL, English DR, McCredie MR, Giles GG. Segregation analyses of 1,476 population-based Australian families affected by prostate cancer. Am J Hum Genet. 2001;68:1207–1218. doi: 10.1086/320114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Chapelle A. Disease gene mapping in isolated human populations: The example of Finland. J Med Genet. 1993;30:857–865. doi: 10.1136/jmg.30.10.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston RC, George VT. Age of onset, age at examination, and other covariates in the analysis of family data. Genet Epidemiol. 1989;6:217–220. doi: 10.1002/gepi.1370060138. [DOI] [PubMed] [Google Scholar]
- Elston RC. Segregation analysis. Adv Hum Genet. 1981;11:63–120. 372–3. doi: 10.1007/978-1-4615-8303-5_2. [DOI] [PubMed] [Google Scholar]
- Elston RC, Sobel E. Sampling considerations in the gathering and analysis of pedigree data. Am J Hum Genet. 1979;31:62–69. [PMC free article] [PubMed] [Google Scholar]
- Elston RC, Yelverton KC. General models for segregation analysis. Am J Hum Genet. 1975;27:31–45. [PMC free article] [PubMed] [Google Scholar]
- Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Hum Hered. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- Finnish Cancer Registry. Cancer incidence in Finland 1995 and 2004. 2006 Cancer statistics at www.cancerregistry.fi last updated on 7 June 2006.
- Gianferrari L, Arrigoni G, Cresseri A, Lovati G, Morganti G. Genetic and clinico-statistical research on neoplasms of the prostate. Acta Gerontol (Milano) 1956;5:224–233. [PubMed] [Google Scholar]
- Gong G, Oakley-Girvan I, Wu AH, Kolonel LN, John EM, West DW, Felberg A, Gallagher RP, Whittemore AS. Segregation analysis of prostate cancer in 1,719 white, African-American and Asian-American families in the United States and Canada. Cancer Causes Control. 2002;13:471–482. doi: 10.1023/a:1015755219674. [DOI] [PubMed] [Google Scholar]
- Grönberg H, Damber L, Damber JE, Iselius L. Segregation analysis of prostate cancer in Sweden: Support for dominant inheritance. Am J Epidemiol. 1997;146:552–557. doi: 10.1093/oxfordjournals.aje.a009313. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- Mäkinen T, Tammela TLJ, Stenman U-H, Määttänen L, Rannikko S, Aro J, Juusela H, Hakama M, Auvinen A. Family History and Prostate Cancer Screening With Prostate-Specific Antigen. Journal of Clinical Oncology. 2002;20(11):2658–2663. doi: 10.1200/JCO.2002.05.006. [DOI] [PubMed] [Google Scholar]
- Mäkinen T, Tammela TLJ, Hakama M, Stenman U-H, Rannikko S, Aro J, Juusela H, Määttänen L, Auvinen A. Tumor Characteristics in a Population-based Prostate Cancer Screening Trial with Prostate-specific. Antigen Clinical Cancer Research. 2003;9:2435–2439. [PubMed] [Google Scholar]
- Matikainen MP, Pukkala E, Schleutker J, Tammela TL, Koivisto P, Sankila R, Kallioniemi OP. Relatives of prostate cancer patients have an increased risk of prostate and stomach cancers: A population-based, cancer registry study in Finland. Cancer Causes Control. 2001;12:223–230. doi: 10.1023/a:1011283123610. [DOI] [PubMed] [Google Scholar]
- Monroe KR, Yu MC, Kolonel LN, Coetzee GA, Wilkens LR, Ross RK, Henderson BE. Evidence of an X-linked or recessive genetic component to prostate cancer risk. Nat Med. 1995;1:827–829. doi: 10.1038/nm0895-827. [DOI] [PubMed] [Google Scholar]
- Narod SA, Dupont A, Cusan L, Diamond P, Gomez JL, Suburu R, Labrie F. The impact of family history on early detection of prostate cancer. Nat Med. 1995;1:99–101. doi: 10.1038/nm0295-99. [DOI] [PubMed] [Google Scholar]
- Peltonen L. Molecular background of the Finnish disease heritage. Ann Med. 1997;29:553–556. doi: 10.3109/07853899709007481. [DOI] [PubMed] [Google Scholar]
- Rebbeck TR, Walker AH, Zeigler-Johnson C, Weisburg S, Martin AM, Nathanson KL, Wein AJ, Malkowicz SB. Association of HPC2/ELAC2 genotypes and prostate cancer. Am J Hum Genet. 2000;67:1014–1019. doi: 10.1086/303096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rökman A, Ikonen T, Seppälä EH, Nupponen N, Autio V, Mononen N, Bailey-Wilson J, Trent J, Carpten J, Matikainen MP, Koivisto PA, Tammela TL, Kallioniemi OP, Schleutker J. Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet. 2002;705:1299–1304. doi: 10.1086/340450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rökman A, Ikonen T, Mononen N, Autio V, Matikainen MP, Koivisto PA, Tammela TL, Kallioniemi OP, Schleutker J. ELAC2/HPC2 involvement in hereditary and sporadic prostate caner. Cancer Res. 2001;61:6038–6041. [PubMed] [Google Scholar]
- S.A.G.E. 3.1. Computer program package available from the Department of Epidemiology and biostatistics, Rammelcamp Center for Education and Research. MetroHealth campus, Case Western Reserve University; Cleveland, Ohio: 1997. Statistical analysis for genetic epidemiology, release 3.1. [Google Scholar]
- Schaid DJ. The complex genetic epidemiology of prostate cancer. Hum Mol Genet. 2004;13(Spec No 1):R103–21. doi: 10.1093/hmg/ddh072. [DOI] [PubMed] [Google Scholar]
- Schaid DJ, McDonnell SK, Blute ML, Thibodeau SN. Evidence for autosomal dominant inheritance of prostate cancer. Am J Hum Genet. 1998;62:1425–1438. doi: 10.1086/301862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleutker J, Baffoe-Bonnie AB, Gillanders E, Kainu T, Jones MP, Freas-Lutz D, Markey C, Gildea D, Riedesel E, Albertus J, Gibbs KD, Jr, Matikainen M, Koivisto PA, Tammela T, Bailey-Wilson JE, Trent JM, Kallioniemi OP. Genome-wide scan for linkage in Finnish hereditary prostate cancer (HPC) families identifies novel susceptibility loci at 11q14 and 3p25–26. Prostate. 2003;57:280–289. doi: 10.1002/pros.10302. [DOI] [PubMed] [Google Scholar]
- Schleutker J, Matikainen M, Smith J, Koivisto P, Baffoe-Bonnie A, Kainu T, Gillanders E, Sankila R, Pukkala E, Carpten J, Stephan D, Tammela T, Brownstein M, Bailey-Wilson J, Trent J, Kallioniemi OP. A genetic epidemiological study of hereditary prostate cancer (HPC) in Finland: frequent HPCX linkage in families with late-onset disease. Clin Cancer Res. 2000 Dec;6(12):4810–5. [PubMed] [Google Scholar]
- Seppälä EH, Ikonen T, Autio V, Rökman A, Mononen N, Matikainen MP, Tammela TL, Schleutker J. Germ-line alterations in MSR1 gene and prostate cancer risk. Clin Cancer Res. 2003a;9:5252–5256. [PubMed] [Google Scholar]
- Seppälä EH, Ikonen T, Mononen N, Autio V, Rökman A, Matikainen MP, Tammela TL, Schleutker J. CHEK2 variants associate with hereditary prostate cancer. Br J Cancer. 2003b;89:1966–1970. doi: 10.1038/sj.bjc.6601425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS, Guo H, Bujnovszky P, Nusskern DR, Damber JE, Bergh A, Emanuelsson M, Kallioniemi OP, Walker-Daniels J, Bailey-Wilson JE, Beaty TH, Meyers DA, Walsh PC, Collins FS, Trent JM, Isaacs WB. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274:1371–1374. doi: 10.1126/science.274.5291.1371. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27:172–180. doi: 10.1038/84808. [DOI] [PubMed] [Google Scholar]
- Teppo L, Pukkala E, Lehtonen M. Data quality and quality control of a population-based cancer registry, experience in Finland. Acta Oncol. 1994;33:365–369. doi: 10.3109/02841869409098430. [DOI] [PubMed] [Google Scholar]
- Valeri A, Briollais L, Azzouzi R, Fournier G, Mangin P, Berthon P, Cussenot O, Demenais F. Segregation analysis of prostate cancer in France: Evidence for autosomal dominant inheritance and residual brother-brother dependence. Ann Hum Genet. 2003;67:125–137. doi: 10.1046/j.1469-1809.2003.00022.x. [DOI] [PubMed] [Google Scholar]
- Verhage BA, Baffoe-Bonnie AB, Baglietto L, Smith DS, Bailey-Wilson JE, Beaty TH, Catalona WJ, Kiemeney LA. Autosomal dominant inheritance of prostate cancer: A confirmatory study. Urology. 2001;57:97–101. doi: 10.1016/s0090-4295(00)00891-8. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Global cancer rates could increase by 50% to 15 million by 2020. 2003 http://www.who.int/mediacentre/news/releases/2003/pr27/en/
- Xu J, Dimitrov L, Chang BL, Adams TS, Turner AR, Meyers DA, Eeles RA, et al. A Combined genome-wide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the International Consortium for Prostate Cancer Genetics. Am J Hum Genet. 2005;77:219–229. doi: 10.1086/432377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zheng SL, Komiya A, Mychaleckyj JC, Isaacs SD, Hu JJ, Sterling D, et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet. 2002;32:321–325. doi: 10.1038/ng994. [DOI] [PubMed] [Google Scholar]
- Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, Wilkens E, Bujnovszky P, Bova GS, Walsh P, Isaacs W, Schleutker J, Matikainen M, Tammela T, Visakorpi T, Kallioniemi OP, Berry R, Schaid D, French A, McDonnell S, Schroeder J, Blute M, Thibodeau S, Trent J. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20:175–179. doi: 10.1038/2477. [DOI] [PubMed] [Google Scholar]