

Abstract
The product of the H19 gene is an untranslated RNA that is expressed exclusively from the maternal chromosome during mammalian development. The H19 gene and its 5′-flanking sequence are required for the genomic imprinting of two paternally expressed genes, Ins-2 (encodes insulin-2) and Igf-2 (encodes insulin-like growth factor-2), that lie 90 and 115 kb 5′ to the H19 gene, respectively. In this report, the role of the H19 gene in its own imprinting is investigated by introducing a Mus spretus H19 gene into heterologous locations in the mouse genome. Multiple copies of the transgene were sufficient for its paternal silencing and DNA methylation. Replacing the H19 structural gene with a luciferase reporter gene resulted in loss of imprinting of the transgene. That is, high expression and low levels of DNA methylation were observed upon both paternal and maternal inheritance. The removal of 701 bp at the 5′ end of the structural gene resulted in a similar loss of paternal-specific DNA methylation, arguing that those sequences are required for both the establishment and maintenance of the sperm-specific gametic mark. The M. spretus H19 transgene could not rescue the loss of Igf-2 imprinting in trans in H19 deletion mice, implying a cis requirement for the H19 gene. In contrast to a previous report in which overexpression of a marked H19 gene was a prenatal lethal, expression of the M. spretus transgene had no deleterious effect, leading to the conclusion that the 20-base insertion in the marked gene created a neomorphic mutation.
Normal mammalian development requires the contribution of haploid genomes from both parents, indicating that the two genomes are not functionally equivalent (1, 2, 3, 4). The nonequivalence is the result of gamete-specific epigenetic modifications of a number of genes that lead to the unequal expression of the two parental alleles during development. To date, 16 such imprinted genes have been identified in the mouse or human (5, 6).
The most likely candidate for the gametic mark or imprint is the methylation of CpG residues in the transcriptional control regions of imprinted genes. Allele-specific DNA methylation has been observed in the vicinity of most imprinted genes. In some instances, the methylation is present on the inactive gene, suggesting a role for DNA methylation in silencing of the gene (7, 8, 9). However, specific methylation of the active alleles of imprinted genes has been described as well (10, 11, 12). Finally, for two imprinted genes, Igf-2r and H19, allele-specific methylation has been shown to originate in the gametes and survive a period of genome-wide demethylation that occurs shortly after fertilization (9, 13). These residual gametic differences remain the best candidates for heritable imprinting signals. The strongest case for a requirement for DNA methylation in maintaining the differential expression of parental alleles of genes comes from the disruption of imprinting in embryos homozygous for the loss of the maintenance methylase, DNA methyltransferase (14).
The H19 gene lies in a cluster of imprinted genes on distal chromosome 7 in the mouse, a region syntenic with chromosome 11p15.5 in humans (15, 16). The genes encoding p57KIP2, a cyclin-dependent kinase inhibitor, and Mash-2, a trophoblast-specific transcription factor, lie at the telomeric end of the cluster and are maternally expressed (17, 18). The two growth factor genes, Ins-2 (encodes insulin-2) and Igf-2 (encodes insulin-like growth factor-2), lie in the middle of the cluster and are both paternally expressed (19, 20), while H19 resides at the centromeric end of the cluster and encodes a maternally expressed RNA (21, 22).
Recent experiments have established a mechanistic link between the imprinting of Ins-2, Igf-2, and H19 that is consistent with a primary role for the H19 gene in the imprinting of both itself as well as the other two genes. First, an internally deleted transgene consisting of 14 kb of DNA surrounding the H19 gene is capable of adopting imprinted expression in heterologous chromosomal locations (7), implying that H19 imprinting is regulated by local signals and does not require either the Igf-2 or Ins-2 gene. Second, a deletion of the H19 5′-flanking sequence and structural gene results in the expression of both Igf-2 and Ins-2 from the maternal as well as the paternal chromosome (23). Thus, the region surrounding the H19 gene that is required for its own imprinting is also required for the imprinting of its neighbors. We have suggested that the mechanistic link between the imprinting of H19, Igf-2, and Ins-2 results from a competition between the genes for the use of shared enhancers (22, 24, 25). On the paternal chromosome, this competition is biased in the direction of Igf-2 and Ins-2 expression by the silencing the H19 gene via DNA methylation (14). In that sense, the imprinting of Igf-2 and Ins-2 can be said to be nonautonomous. On the maternal chromosome, the fully unmethylated H19 gene successfully competes for the enhancers.
This enhancer competition model implies that the regulated event in imprinting at this locus is the establishment and maintenance of the paternal-specific methylation of the H19 gene. In this report, we exploit transgenic mice to begin a genetic analysis of the methylation and imprinting of the H19 gene. Specifically, we have investigated the requirement for the H19 structural gene itself in cis in its own methylation and imprinting.
No role for the H19 RNA in trans has been established to date. The RNA is highly abundant during embryogenesis in mesodermal and endodermal tissues (26, 27). Nevertheless, the only phenotype observed with the deletion of the H19 gene is the loss of imprinting of Igf-2 and Ins-2 (23). Two experimental approaches have attributed biological effects to overexpression of H19 RNA. Hao et al. (28) have shown by transfection that human H19 RNA could suppress the tumorigenicity of rhabdomyosarcoma and Wilms’ tumor cell lines. Additionally ectopic expression of a marked H19 transgene in mice resulted in late embryonic lethality between embryonic day 14 and birth (29). In this report, we also used the transgenic system to clarify the role of ectopic expression in prenatal lethality.
MATERIALS AND METHODS
Mice.
C57BL/6J, DBA/2J, and SJL mice were purchased from The Jackson Laboratory. The strain B6(CAST-H19) has been described previously (13).
Isolation of the Mus spretus H19 Gene.
Genomic DNA prepared from M. spretus liver was digested to completion with EcoRI and size-fractionated by sucrose gradient centrifugation. Fragments of ≈10 kb were isolated and cloned into Lambda Dash II (Stratagene). The library was screened by the method of Benton and Davis (30) using the 3-kb EcoRI–SalI fragment spanning the H19 gene, and positively hybridizing phage were purified. The recombinant phage were subcloned, and the nucleotide sequence of the M. spretus H19 gene was determined by the chain termination method (31) using the Sequenase kit (United States Biochemical).
Transgene Constructions and Microinjection.
The plasmids used for generating transgenic mice include 0.8 kb or 4 kb of DNA 5′ to the structural gene and 8 kb or 11 kb of DNA 3′ to the structural gene cloned into a pBluescript KS vector (Stratagene). In the construct M. spretus H19, the 3-kb EcoRI–SalI fragment spanning the Mus domesticus structural gene is replaced with the 3-kb EcoRI–SalI M. spretus DNA. The construct Δ1H19 has a deletion of a 697-bp DraIII–BsmI fragment from +3 to +701 bp that removes the first half of exon 1. The Luc transgene constructs replace the H19 structural gene from the DraIII site at +3 bp to the unique SalI site 3′ of the gene with the firefly luciferase gene (32).
DNA was microinjected into one of the pronuclei of fertilized one-cell mouse eggs derived from (C57BL/6J × SJL) F1 intercrosses (33). Injected embryos were transferred to the oviducts of pseudopregnant CD1 females. Founder animals were identified by digestion of tail DNA with appropriate enzymes and analysis by Southern blot.
RNA Analysis.
Total RNA was isolated by LiCl-urea extraction (34). An non-allele-specific RNase protection probe for H19 (29) and allele-specific RNase protection probes for H19 and for Igf-2 have been described (24). A 140-bp XbaI DNA fragment spanning the 5′ end of the H19–luciferase fusion gene was subcloned into pBluescript KS (Stratagene), and the resultant plasmid was linearized with KpnI and treated with the Klenow fragment of DNA polymerase I to generate a template for synthesizing a probe specific for H19–luciferase RNA. Radiolabeled probes were incubated with total RNA at 45°C overnight and digested with 40 μg of RNase A per ml and 2 μg of RNase T1 per ml at room temperature for 60 min. The products were separated on 6.0% or 7.5% acrylamide/7 M urea gels and visualized by autoradiography.
RESULTS
Microinjection of the H19 Gene into Mouse Zygotes.
Brunkow and Tilghman (29) had previously attempted to generate stable transgenic lines overexpressing H19 RNA by microinjecting into mouse zygotes H19 transgenes that had been marked with a 20-bp oligonucleotide insertion in the first exon of the gene. This insertion was used to distinguish the transgene from the endogenous gene. The transgene included a 4-kb segment of 5′-flanking DNA that is selectively hypermethylated on the paternal chromosome throughout embryogenesis (13). The 8 or 11 kb of 3′-flanking DNA contained two enhancers, each sufficient for expression of the gene in endodermal cell lines in vitro (35). Surprisingly, no stable lines were obtained that expressed the transgene. Instead, founder transgenic embryos died late in gestation, between embryonic day 14 and birth. The period of embryonic lethality was consistent with the lethal phenotype of mice carrying a maternal disomy of chromosome 7 (36, 37), leading to the hypothesis that extra copies of the H19 gene were lethal in mice.
In that same study, stably expressing transgenic lines were successfully generated using an internally truncated structural gene that carried the same 20-bp insertion in exon 1 (Fig. 1, Δ2H19). However, this transgene also resulted in prenatal lethality, but with incomplete penetrance, based on the fact that the surviving pups represented a minority of the transgenic embryos generated (29).
Figure 1.
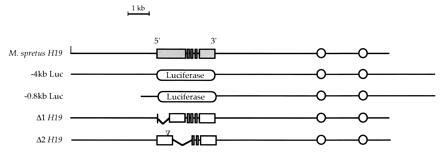
The structures of the transgenes used in this study are depicted. The five exons of the M. spretus structural gene are represented by shaded rectangles and the M. domesticus gene by open rectangles. The firefly luciferase gene is represented as an oval. The two enhancers that lie 5 and 7 kb 3′ of the H19 gene are represented by circles. The position of the 20-bp insertion in Δ2H19 is indicated by the inverted triangle.
To characterize this phenotype further, we wished to determine which elements of the transgene were responsible for the late embryonic lethality. We considered that the phenotype could be due to extra copies of the H19 regulatory elements, such as the promoter, enhancers, or a potential imprinting signal; increased dosage and/or ectopic expression of the H19 gene product itself; or an aberrant gene product created by the insertion of the oligonucleotide at +580 bp. To discriminate among these possibilities, three additional transgenes were generated. In the first two, labeled −4kb Luc and −0.8kb Luc in Fig. 1, the structural H19 gene was replaced with the firefly luciferase gene. These constructs differ only in the amount of 5′-flanking DNA, and they directly test whether the structural gene or its 5′ flank is required for the lethality. To examine the possibility that the oligonucleotide was deleterious, we cloned the M. spretus H19 gene to serve as a wild-type allele that could be distinguished from the endogenous M. domesticus allele in transgenic mice. Sequence comparison of the M. spretus and M. domesticus genes, including part of the promoters and the entire structural gene, revealed >99% sequence identity between alleles (data not shown). However, the small number of base changes was sufficient to distinguish the two at the DNA level using a BamHI polymorphism at +2585 bp in exon 5 and at the RNA level by an allele-specific RNase protection assay (22).
The three transgenes were injected into zygotes, and the presence of transgenic pups was examined at embryonic day 13 and weaning. In contrast to the marked transgene, transgenic pups were identified at the same high frequency at both times (Table 1). Furthermore, these transgenes expressed luciferase and M. spretus H19 RNAs in an appropriate manner in endodermal cells (see below). Together, these experiments demonstrate that neither extra copies of the H19 regulatory domain nor overexpression of wild-type H19 RNA in endoderm interferes with normal development. Rather—and intriguingly—the lethal effect of the original transgene is probably due to the alteration of the gene product brought about by the 20-bp insertion.
Table 1.
Recovery of H19 transgenes
| Transgene | Embryonic day
12–13
|
Postnatal day 21
|
||
|---|---|---|---|---|
| Embryos, no. | No. transgenic (%) | Pups, no. | No. transgenic (%) | |
| M. spretus H19 | 19 | 5 (26) | 31 | 6 (20) |
| −4-kb Luc | 24 | 6 (25) | 61 | 14 (23) |
| −0.8-kb Luc | 31 | 4 (13) | 30 | 6 (20) |
Appropriate Expression and Imprinting of M. spretus H19 Transgenes.
Transgenic lines were established for all constructs by mating founders to DBA/2J mice. As summarized in Table 2, of the seven M. spretus lines analyzed, five expressed the transgene in neonatal liver when inherited from mothers. The two silent transgenes were present at one or two copies in the genome, possibly reflecting a sensitivity of low copy transgenes to position-dependent silencing.
Table 2.
Expression and methylation of H19 and luciferase transgenes
| Transgene | Line no. | Copy no. | Expression† | Methylation pattern‡ |
|---|---|---|---|---|
| M. spretus H19 | S8 | 1 | No | ND |
| S21 | 4 | Mat | Paternal-specific | |
| S25 | 5 | Mat | Paternal-specific | |
| S26 | 2 | No | ND | |
| S28 | 2 | Mat + Pat | Unmethylated | |
| S29 | 2 | Mat + Pat | ND | |
| S30 | 4 | Mat | Paternal-specific | |
| −4-kb Luc | 113 | 21 | Mat + Pat | Unmethylated |
| 138 | 2 | Mat + Pat | Unmethylated | |
| 141 | 2 | Mat + Pat | ND | |
| 142 | 14 | Mat + Pat | Unmethylated | |
| 158 | 1 | No | ND | |
| 167 | 16 | Mat + Pat | Unmethylated | |
| 171 | 17 | Mat + Pat | Unmethylated | |
| −0.8-kb Luc | 82 | 30 | Mat + Pat | ND |
| 94 | 12 | Mat + Pat | ND | |
| 99 | 1 | No | ND | |
| 103 | 8 | Mat + Pat | ND | |
| Δ1H19 | 1–1 | 4 | No | Unmethylated |
| 1–3 | 6 | No | Unmethylated | |
| 1–5 | 3 | No | Unmethylated | |
| 1–18 | 1 | No | Unmethylated |
ND, not determined.
The copy number of each transgene was determined as described.
Mat indicates transgene expression when inherited maternally and Pat indicates expression when inherited paternally. No indicates that the transgene RNA could not be detected.
Two patterns of DNA methlation of the the 5′ flank of the transgene in neonatal liver were noted: paternal-specific when the transgene was methylated only when paternally inherited, or unmethylated when the pattern was unmethylated irrespective of parental origin.
Having established that the transgenes were expressed in five lines, we next examined whether the transgenes could be silenced by passage through a paternal genome. As shown in Fig. 2A for two of three independent M. spretus transgene lines, the M. spretus RNA was readily detected when the transgene was inherited through the maternal germ line but not when inherited through the paternal germ line. Furthermore, the silent transgene in the progeny of a male could be reactivated by passage through the female germ line (Fig. 2B), a hallmark of imprinting. The three lines that displayed imprinted expression of M. spretus H19 carried either four or five copies of the transgene. Two additional lines, both containing only two copies of the transgene each, displayed no parent-of-origin differences in expression (data not shown).
Figure 2.

Maternal-specific expression of the M. spretus H19 transgene. (A) M. spretus H19 transgenic founders of lines 21 and 25 were crossed to DBA/2J mice. Male and female transgenic progeny were backcrossed to DBA/2J and their transgenic (+) and nontransgenic (−) progeny assayed for expression of the transgene. The parental origin of the transgenes are indicated. Five micrograms of total RNA from neonatal livers was assayed by an RNase protection assay (22). Digestion conditions that allow one to distinguish transgene-specific products (arrows) also result in partial digestion of the endogenous RNA. The full-length endogenous H19 RNA is indicated by the closed circle. (B) Male and female siblings of pups inheriting the transgene through the paternal germ line were backcrossed to DBA/2J and RNAs from neonatal livers assayed as described above. Lane and protected fragment designations are as described for A.
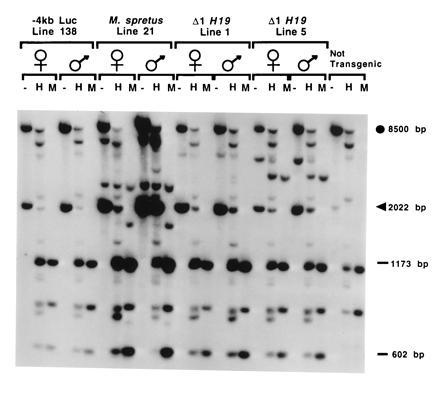
At the endogenous H19 locus, the silent paternal H19 allele is hypermethylated from −5 kb through the structural gene, while the maternal allele is almost completely unmethylated in the same region (7). To determine if the imprinted M. spretus transgene also assumed a differentially methylated state dependent on the parental origin, neonatal liver genomic DNA from animals inheriting the transgene paternally or maternally were digested with HpaII, which is sensitive, and its isoschizomer MspI, which is insensitive, to cytosine methylation. As shown in Fig. 3B, a transgene-specific 2-kb BamHI fragment spanning the 5′-most flanking region of the transgene is almost completely resistant to HpaII digestion at the three clustered sites within it when inherited through a father. In contrast, the same transgene maternally inherited is almost completely digested with HpaII. In Fig. 3C, a 2.5-kb BamHI fragment that is further downstream in the gene and is not transgene-specific displays the same kind of extensive paternal-specific methylation of most copies of the transgene, as indicated by the preponderance of the fully methylated 2.5-kb band in the HpaII lane, whereas the intensity of the band in progeny of females is greatly reduced. The residual signal in that band can be accounted for by the endogenous paternal H19 gene. Consistent with what is observed with the endogenous gene, hypermethylation in the 3′ flank of the gene is weak or undetected (data not shown), indicating that marking of the 5′ flank and gene body by methylation is sequence-specific and does not reflect generalized methylation of exogenous sequences.
Figure 3.

Paternal-specific DNA methylation of the M. spretus transgene. (A) The positions of BamHI (B), HhaI (open circles), and HpaII (open triangles) sites in the endogenous H19 gene (open rectangles) and the relevant transgenes are indicated. The BamHI site at −4 kb is unique to the transgenes; thus, probe I identifies a 2-kb transgene-specific BamHI fragment and a 0.6-kb BamHI–HpaII fragment (asterisks) in all transgenes. Probe II does not distinguish between the endogenous and M. spretus H19 transgene. (B and C) Genomic DNA prepared from neonatal livers from N2 transgenic (+) and nontransgenic (−) pups was digested with BamHI (−) alone or together with HpaII (H) or MspI (M). DNAs were hybridized to probe I (B) or probe II (C). The parental origin of the transgene is indicated at the top of the lanes.
Lack of Imprinting of Luciferase Transgenes.
The luciferase transgenes provided an opportunity to test the function of the 5′ flank and the H19 structural gene itself on the regulated expression and imprinting of H19. Consistent expression of the luciferase transgenes was observed in neonatal endodermal tissues with all lines in which the transgene was present at greater than one copy per genome. Overall, expression correlated well with copy number of the transgene, and the RNA levels were not noticeably reduced by the 3.2-kb truncation of 5′-flanking DNA in −0.8k-b Luc (data not shown). The two nonexpressing lines carrying luciferase genes were both present at single copy in the genome, consistent with the silence of low copy M. spretus H19 transgenes (Table 2).
The expression of maternally and paternally inherited H19–luciferase transgenes in neonatal liver was compared in six lines carrying −4-kb Luc and the three carrying −0.8-kb Luc, as illustrated in Fig. 4. No parental-specific difference in the levels of expression was noted in any instance. Two additional generations of backcrossing were undertaken for three −4-kb Luc lines; however, the expression of the transgene remained independent of its parental origin (data not shown).
Figure 4.
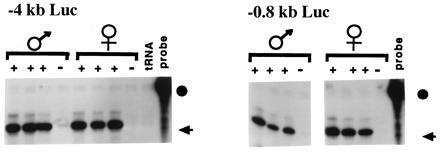
Expression of Luc transgenes. Founders were crossed to DBA/2J and male and female progeny were backcrossed to DBA/2J. The antisense H19-luciferase probe was hybridized to 5 μg of total liver RNA from neonatal transgenic (+) and nontransgenic (−) littermates. Results for lines 142 (−4kb Luc) and 103 (−0.8kb Luc) are presented. The probe (circle) and protected fragment (arrow) are indicated.
Three −4-kb Luc lines were analyzed for evidence of parent-specific DNA methylation in the 5′ flank, using the experimental strategy described above. Representative results are presented in Fig. 5. In contrast to the results obtained for the M. spretus H19 transgene, there were no differences in the pattern of DNA methylation between the maternally and paternally inherited transgenes. Rather, the transgenes were almost completely unmethylated, as is evident from the decrease in intensity of the transgene-specific 2-kb BamHI fragment detected with probe I following HpaII digestion. Thus, by the criteria of both expression and CpG methylation, replacement of the H19 RNA coding sequences results in loss of imprinting of the transgene, with the transgene adopting the “maternal” mode of expression from both chromosomes.
Figure 5.
DNA Methylation of the luciferase and H19 transgenes. Genomic DNA was isolated from N2 neonatal liver of transgenic progeny and was digested with BamHI alone (−), BamHI and HpaII (H), and BamHI and MspI (M). The methylation status of HpaII sites at −3 kb was determined using probe I, as described in the legend to Fig. 3. Probe I identifies a transgene-specific 2-kb BamHI fragment (arrowhead) and an 8-kb BamHI fragment from the endogenous locus (closed circle). The transgenic construct and the parental origins of the transgene are indicated at the top of the lanes.
Defining the Requirements for H19 Imprinting.
The striking difference between the imprinted expression of the M. spretus H19 transgene and the absence of imprinting of the luciferase transgenes could result from the loss of critical DNA sequences, or from interference by foreign firefly sequences. We had previously shown that bases +680 to +1660 are dispensable for imprinting of the internally deleted Δ2H19 transgene (7). To test other sequences within the gene, we created a deletion of the first 700 bp of the structural gene, Δ1H19 (Fig. 1). The deletion encompasses a highly conserved domain within exon 1 that is differentially methylated on the parental chromosomes (7, 21). When transiently transfected into the human hepatoma cell line, Hep3B, Δ1H19 RNA was expressed at ≈5% of the level of M. spretus H19, presumably because the shorter transcript is less stable.
Δ1H19 was microinjected into zygotes and lines were established by crossing to DBA/2J. As expected from the transient transfection results, Δ1H19 RNA was not detected by RNase protection or Northern analysis in the high background of the endogenous H19 RNA expression (data not shown). Therefore, we used the DNA methylation status of the transgenes inherited from both parents as a means to assess the imprinting status of the transgene. Three multicopy lines containing Δ1H19 were examined for methylation of HpaII sites in the 5′-flanking region and at the promoter (Table 2). As shown in Fig. 5 for a representative line, the digestion patterns obtained were indistinguishable for transgenes inherited through the paternal and the maternal germ lines. Furthermore, like −4-kb Luc, they resembled the pattern of the female-inherited M. spretus transgene, which was strongly expressed. Thus, the deletion of 700 bp of the 5′ end of the H19 gene was sufficient to eliminate parental-specific DNA methylation.
Transgene Methylation in Sperm.
The paternally inherited copy of the endogenous H19 gene acquires its methylation during gametogenesis, whereas the female germ line maintains the gene in an unmethylated state (9, 13). That difference is retained during embryogenesis, including a period between fertilization and blastocyst when the majority of the genome is demethylated (38, 39). The failure of the luciferase and Δ1H19 transgenes to maintain paternal methylation of their 5′ flank could reflect a failure to methylate the transgenes during spermatogenesis or a failure to maintain the methylation during embryogenesis. To discriminate between these possibilities, the status of the transgene methylation was examined in testes DNA, which is composed almost entirely of sperm DNA.
As shown for a nontransgenic animal in Fig. 6, the 8-kb BamHI fragment spanning the 5′-distal portion of the flank of the endogenous H19 gene is largely methylated in sperm at all HpaII and HhaI sites contained within it. The two imprinted M. spretus H19 lines are also heavily methylated in sperm, as can be seen by the intensity of the 2-kb transgene-specific band in the HpaII- and HhaI- digested lanes (Fig. 6) and the absence of the fully digested 0.6-kb product.
Figure 6.
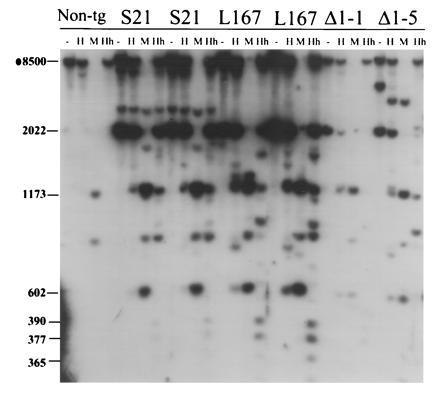
DNA Methylation of the H19 transgenes in testes. Testes DNA was prepared from a nontransgenic male (Non-tg) and males of the transgenic lines indicated. S, M. spretus; L, −4-kb Luc. The DNA was digested with BamHI (−) or BamHI together with HpaII (H), MspI (M), or HhaI (Hh). The DNA was hybridized to radiolabeled probe I. The sizes of the relevant fragments are indicated on the left. The 2- and 0.6-kb fragments are transgene-specific and common to all transgenes.
The luciferase and Δ1H19 lines are inherited from sperm in a slightly less methylated state, as is evident from the appearance of fully digested products in both the HpaII and HhaI digestions using the 5′-specific probe I (Fig. 6). However, in each case, a substantial fraction of the transgenes remain fully methylated within the 5′ region. The methylation patterns of the M. spretus H19, luciferase, and Δ1H19 transgenes also appear essentially identical further downstream, as assessed using probe II (data not shown). Thus the luciferase and Δ1H19 transgenes are inherited in a mostly methylated state but cannot retain their methylation through embryogenesis.
Mutations Disrupting Imprinting Act Only in Cis.
The foregoing experiments establish a requirement for either the DNA sequences within the H19 gene itself, or the RNA product, in transgene imprinting. The experiments were performed in a wild-type H19 genetic background, implying that the endogenous H19 RNA is unable to rescue in trans the loss of imprinting of the mutant transgenes. Previously we reported that the H19 structural gene and its 5′ flank were required for the imprinting of Igf-2 and Ins-2 as well (23). To test whether the loss of imprinting of Igf-2 could be complemented by supplying H19 RNA in trans, we examined the effect of maternally inherited M. spretus H19 transgene expression on the expression of Igf-2 in H19 −/+ heterozygotes. Transgene line S21 was crossed to an H19 deletion homozygote, and then female transgenic progeny were crossed to B6(CAST-H19) males carrying the Igf-2 Mus castaneus allele. An allele-specific Igf-2 RNase protection assay detected both the maternal M. domesticus and the paternal M. castaneus alleles of Igf-2 RNA in neonatal liver in both transgenic and nontransgenic progeny carrying the H19 deletion (Fig. 7A), despite the fact that the wild-type M. spretus H19 RNA was strongly expressed (Fig. 7B). Thus, the transgene does not rescue the loss of Igf-2 imprinting, implying that the chromosomal deletion of H19 is acting in cis to cause misregulation of Igf-2.
Figure 7.

Expression of the M. spretus transgene in H19 deletion maternal heterozygotes. (A) A female heterozygous for a deletion of the H19 gene region (23) and carrying the M. spretus transgene was crossed to a B6(CAST-H19) male. Total liver RNA was prepared from the progeny and analyzed for the expression of Igf-2 RNA using an allele-specific RNase protection assay that distinguishes M. castaneus Igf-2 RNA (+C) from that of M. domesticus (+D). Littermates were genotyped for the presence of the wild-type (+) or mutant (−) endogenous H19 gene (H19 genotype) and the presence (+) or absence (−) of the transgene (Tg). P, probe; M, markers. (B) The same RNAs were analyzed for the expression of the M. spretus H19 gene. The multiple bands in the + Tg lanes are the result of the fact that the probe (P) was derived from the M. castaneus gene.
DISCUSSION
The nonequivalence of the haploid genomes contributed via the egg and the sperm (1, 2, 3, 4) has been attributed to a subset of genes whose expression is restricted to the maternal or to the paternal alleles. Using reciprocal translocations to generate maternal and paternal disomies at a high frequency, a number of chromosomal regions have been identified in the mouse that must contain such imprinted genes (40). The distal portion of mouse chromosome 7 is such a region. Paternal disomic mice for the distal end of chromosome 7 die early in gestation while maternal disomies cause late embryonic lethality (36). The early embryonic lethality observed in Mash-2 null mice could potentially explain the paternal disomy phenotype, as that gene is maternally expressed (41). The gene(s) responsible for the late embryonic lethality of maternal disomies is not yet clear. We had suggested that ectopic expression of H19 might be a phenocopy of the maternal disomy, based on the lethality we observed with expression of a marked H19 transgene. In this study, however, we could provide no evidence for this proposal, as ectopic expression of wild-type H19 is not detrimental to normal development. Rather, the lethality in the earlier study is probably due to a neomorphic mutation generated by the DNA insertion used to mark the transgene.
This study establishes that sequences required for temporally correct expression of the H19 gene in endodermal tissues map between −800 bp and +11 kb relative to the start of transcription of the gene at +1 bp. These transgenic results are consistent with transient transfection studies (35) that had suggested the transcriptional signals in the 5′ flank of the gene were contained within the first 50 bp. The importance of the two 3′ enhancers in directing endoderm-specific expression of both the H19 and Igf-2 genes was recently verified by the targeted deletion of both enhancers in mice (24). In those mice, endoderm expression of H19 and Igf-2 is ablated on the maternal and paternal chromosomes, respectively.
No single-copy M. spretus H19 or luciferase transgene was expressed in vivo, suggesting that the constructs lacked sufficient sequences to insulate the H19 transgene from position effects. On the other hand, transgenes present at three or more copies were uniformly expressed, with an approximate correlation between copy number and expression level. Whether it is the duplication of the enhancers themselves or another element acting as an insulting element that leads to expression of the transgene in two or more copies remains to be determined. In any case, these transgenes allowed us to begin a dissection of the sequences required for imprinting of the H19 gene.
The first expressed imprinted gene identified in mice was not an endogenous gene, but a foreign transgene (42), and transgenic mice have served as a model system for examining the molecular mechanism for genomic imprinting. Essentially, two classes of imprinted transgenes have been investigated. In the first class are transgenes that show imprinting in a position-dependent manner, as manifested by hypermethylation of the maternal allele (43, 44, 45, 46, 47, 48). Imprinting of these transgenes is particularly susceptible to the genetic background of the mouse, a property which has been exploited to map genetic modifiers of transgene imprinting (47, 48). In the second class is the transgene RSVIgmyc, which is a fusion of elements of the Rous sarcoma virus long terminal repeat, the immunoglobulin heavy chain locus, the mouse c-myc gene, and the plasmid vector pBR322 (42). This hybrid fragment is consistently imprinted in a position-independent manner, with expression exclusively from the hypomethylated paternal allele (49, 50). Mutational analysis has begun to identify key sequences required for its imprinting (50).
To date, the only endogenous gene that displays consistent imprinting behavior as a transgene is the H19 gene. Bartolomei et al. (7) had demonstrated in two independent lines that an internally truncated version of the H19 gene, Δ2H19, displayed maternal-specific expression and paternal methylation. Thus, unlike all other transgenes studied to date, the H19 transgene is methylated when inherited from fathers, not mothers, in keeping with the methylation of the endogenous gene itself. This observation has been extended in this study by using the full-length and unmarked M. spretus H19 transgene, which is imprinted in three of five lines that express the gene. The two exceptions were present at just two copies in the genome, suggesting that the transgenes lack the full complement of imprinting signals normally provided at the endogenous locus. In fact, Tremblay et al. (13) have recently identified CpG dinucleotides in the 5′ flank of the H19 gene that lie outside the limits of the M. spretus transgene and display properties that might be expected of a gametic mark. That is, these sites are methylated in sperm but not in eggs and retain their methylation during embryogenesis. Those missing sequences, if important, can be replaced with multiple copies of the sequences between −4 and +11 kb. The fact that the two-copy transgenes are well expressed implies that the imprinting signals can be separated from the transcriptional regulatory elements that are required for high-level expression of the gene.
Even in multiple copies, the imprinting of the M. spretus H19 transgene is incompletely penetrant. In 5% of transgenic pups where the transgene was inherited from the male, the transgene was expressed at levels equal to that seen with maternal inheritance (data not shown). In these pups, methylation of the transgene mimicked that seen with maternally inherited transgenes. These pups represent either a failure to establish the gametic imprint, an early misreading of the imprinting signal, or a failure to maintain it, as the loss of imprinting is complete, rather than intermediate, as would be the case if there was a cell-by-cell error in interpreting the imprinting mark.
A dissection of the imprinting signals on the M. spretus H19 transgene was begun by replacing the structural gene itself with a luciferase reporter gene. By both the criterion of parental-specific expression of the transgene and DNA methylation of its 5′ flank, the fusion gene had lost all imprinting behavior. One can envisage three possible explanations for the loss of imprinting of the luciferase transgenes. The least interesting one is that lack of imprinting did not reflect a requirement for the H19 gene itself, but rather the foreign luciferase DNA interfered with imprinting signals that were present on the transgene. This possibility was ruled out by the lack of methylation imprinting of Δ1H19, where no foreign DNA was introduced. Therefore either the DNA or the RNA it encodes is required either to establish the epigenetic mark in the gametes, and/or to retain that mark in the embryo.
The luciferase transgenes were methylated in sperm, although not to the same degree as the endogenous gene or the imprinted M. spretus H19 gene. The reduction in methylation was most evident in the 5′-most region examined, for both HpaII and HhaI sites. This region contains at least a subset of the sites of exclusive paternal DNA methylation that survive the demethylation that occurs in the embryo (13). It appears that the luciferase transgene cannot maintain the methylation it inherits, as later in development the luciferase 5′ flank had become further undermethylated on the paternally inherited chromosome. Thus the structural H19 gene is required to establish its own transgene imprinting.
Previously Leighton et al. (23) showed that removal of the active maternal H19 gene and its flank is sufficient to completely overcome the silencing of the maternal Igf-2 gene. This result is consistent with the recent demonstration by Penny et al. (51) that removal of the Xist gene, an RNA-coding gene that maps to the X chromosome inactivation center, prevents the inactivation of genes on the X chromosome carrying the deletion. In each case, a genetic conundrum is presented (52). Is it the loss of the DNA sequences or loss of the gene product that results in the failure to silence the neighboring genes? We showed here that expression in trans of M. spretus H19 RNA does not rescue the loss of imprinting of maternal Igf-2, just as the endogenous RNA does not rescue the loss of imprinting of the luciferase transgenes. This suggests that, if there is a role for H19 RNA in either its own silencing or the silencing of Igf-2, the RNA acts in cis. Unlike a protein, a regulatory RNA can act locally—at the site of transcription—so that mutations can have a cis effect, even though the molecular mechanism may involve a gene product.
In this study, we show that a subset of the structural H19 gene itself is required for its imprinting as a transgene. The most straightforward interpretation of these results is that these sequences represent DNA regulatory elements required to mark the locus as paternal and to maintain that mark during embryogenesis. An alternate interpretation, equally consistent with the experimental results, is that an RNA product synthesized from the 5′ third of the H19 gene is required to establish its own methylation imprinting. There are two implications that follow from this interpretation, however. First, the imprinting of Δ2H19 transgene argues strongly that if an RNA product is required, it is not the mature, fully spliced, and folded H19 RNA that accumulates at high levels in many fetal tissues. Rather, the phenotypes of the two H19 deletions can only be reconciled with a role for the RNA itself by proposing that the 5′ end of the RNA acts independently of the rest of the RNA, for example, while the rest of the RNA is still being synthesized. Second, it is paradoxical that the absence of H19 RNA in the Luc and Δ1H19 transgenes is affecting the imprinting of the paternal chromosome, on which the RNA is transiently and weakly expressed only during spermatogenesis (J. Saam and S.M.T., unpublished results). This leaves no apparent role for the RNA on the maternal chromosome, where it is highly expressed. Distinguishing between a role for H19 as a DNA element and H19 as a regulatory RNA will be required to fully understand the regulation of this cluster of imprinted genes on mouse chromosome 7.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (GM51460). K.P. was a fellow of the Damon Runyon–Walter Winchell Cancer Fund, and S.M.T. is an Investigator of the Howard Hughes Medical Institute.
References
- 1.McGrath J, Solter D. J Exp Zool. 1983;228:355–362. doi: 10.1002/jez.1402280218. [DOI] [PubMed] [Google Scholar]
- 2.McGrath J, Solter D. Cell. 1984;37:179–183. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- 3.Surani M A H, Barton S C, Norris M L. Nature (London) 1984;308:548–550. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- 4.Surani M A H, Barton S C, Norris M L. Cell. 1986;45:127–136. doi: 10.1016/0092-8674(86)90544-1. [DOI] [PubMed] [Google Scholar]
- 5.Efstratiadis A. Curr Opin Genet Dev. 1994;4:265–280. doi: 10.1016/s0959-437x(05)80054-1. [DOI] [PubMed] [Google Scholar]
- 6.Barlow D P. Science. 1995;270:1610–1613. doi: 10.1126/science.270.5242.1610. [DOI] [PubMed] [Google Scholar]
- 7.Bartolomei M S, Webber A L, Brunkow M E, Tilghman S M. Genes Dev. 1993;7:1663–1673. doi: 10.1101/gad.7.9.1663. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson-Smith A C, Sasaki H, Cattanach B M, Surani M A. Nature (London) 1993;362:751–755. doi: 10.1038/362751a0. [DOI] [PubMed] [Google Scholar]
- 9.Brandeis M, Kafri T, Ariel M, Chaillet J R, McCarrey J, Razin A, Cedar H. EMBO J. 1993;12:3669–3677. doi: 10.1002/j.1460-2075.1993.tb06041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasaki H, Hamada T, Ueda T, Seki R, Higashinakagawa T, Sakaki Y. Development (Cambridge, UK) 1991;111:573–581. doi: 10.1242/dev.111.2.573. [DOI] [PubMed] [Google Scholar]
- 11.Stoger R, Kubicka P, Liu C-G, Kafri T, Razin A, Cedar H, Barlow D P. Cell. 1993;73:61–71. doi: 10.1016/0092-8674(93)90160-r. [DOI] [PubMed] [Google Scholar]
- 12.Feil R, Walter J, Allen N D, Reik W. Development (Cambridge, UK) 1994;120:2933–2943. doi: 10.1242/dev.120.10.2933. [DOI] [PubMed] [Google Scholar]
- 13.Tremblay K D, Saam J, Ingram R S, Tilghman S M, Bartolomei M S. Nat Genet. 1995;9:407–413. doi: 10.1038/ng0495-407. [DOI] [PubMed] [Google Scholar]
- 14.Li E, Beard C, Jaenisch R. Nature (London) 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 15.Glaser T, Housman D, Lewis W H, Gerhard D, Jones C. Somatic Cell Mol Genet. 1989;15:477–501. doi: 10.1007/BF01534910. [DOI] [PubMed] [Google Scholar]
- 16.Zemel S, Bartolomei M S, Tilghman S M. Nat Genet. 1992;2:61–65. doi: 10.1038/ng0992-61. [DOI] [PubMed] [Google Scholar]
- 17.Guillemot F, Caspary T, Tilghman S M, Copeland N G, Gilbert D J, Jenkins N A, Anderson D J, Joyner A L, Rossant J, Nagy A. Nat Genet. 1995;9:235–241. doi: 10.1038/ng0395-235. [DOI] [PubMed] [Google Scholar]
- 18.Hatada I, Mukai T. Nat Genet. 1995;11:204–206. doi: 10.1038/ng1095-204. [DOI] [PubMed] [Google Scholar]
- 19.DeChiara T M, Robertson E J, Efstratiadis A. Cell. 1991;64:849–859. doi: 10.1016/0092-8674(91)90513-x. [DOI] [PubMed] [Google Scholar]
- 20.Giddings S J, King C D, Harman K W, Flood J F, Carnaghi L R. Nat Genet. 1994;6:310–313. doi: 10.1038/ng0394-310. [DOI] [PubMed] [Google Scholar]
- 21.Brannan C I, Dees E C, Ingram R S, Tilghman S M. Mol Cell Biol. 1990;10:28–36. doi: 10.1128/mcb.10.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartolomei M S, Zemel S, Tilghman S M. Nature (London) 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 23.Leighton P A, Ingram R S, Eggenschwiler J, Efstratiadis A, Tilghman S M. Nature (London) 1995;375:34–39. doi: 10.1038/375034a0. [DOI] [PubMed] [Google Scholar]
- 24.Leighton P A, Saam J R, Ingram R S, Stewart C L, Tilghman S M. Genes Dev. 1995;9:2079–2089. doi: 10.1101/gad.9.17.2079. [DOI] [PubMed] [Google Scholar]
- 25.Bartolomei M S, Tilghman S M. Semin Dev Biol. 1992;3:107–117. [Google Scholar]
- 26.Pachnis V, Brannan C I, Tilghman S M. EMBO J. 1988;7:673–681. doi: 10.1002/j.1460-2075.1988.tb02862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poirier F, Chan C-T J, Timmons P M, Robertson E J, Evans M J, Rigby P W J. Development (Cambridge, UK) 1991;113:1105–1114. doi: 10.1242/dev.113.4.1105. [DOI] [PubMed] [Google Scholar]
- 28.Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Nature (London) 1993;365:764–767. doi: 10.1038/365764a0. [DOI] [PubMed] [Google Scholar]
- 29.Brunkow M E, Tilghman S M. Genes Dev. 1991;5:1092–1101. doi: 10.1101/gad.5.6.1092. [DOI] [PubMed] [Google Scholar]
- 30.Benton W D, Davis R W. Science. 1977;196:180–182. doi: 10.1126/science.322279. [DOI] [PubMed] [Google Scholar]
- 31.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5468. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Wet J R, Wood K V, DeLuca M, Helinski D R, Subramani S. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hogan B, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 34.Auffray C, Rougeon F. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 35.Yoo-Warren H, Pachnis V, Ingram R S, Tilghman S M. Mol Cell Biol. 1988;8:4707–4715. doi: 10.1128/mcb.8.11.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Searle A G, Beechey C V. Genet Res. 1990;56:237–244. doi: 10.1017/s0016672300035333. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson-Smith A C, Cattanach B M, Barton S C, Beechey C V, Surani M A. Nature (London) 1991;351:667–670. doi: 10.1038/351667a0. [DOI] [PubMed] [Google Scholar]
- 38.Monk M, Boubelik M, Lehnert S. Development (Cambridge, UK) 1987;99:371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 39.Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, Cedar H, Razin A. Genes Dev. 1992;6:705–714. doi: 10.1101/gad.6.5.705. [DOI] [PubMed] [Google Scholar]
- 40.Cattanach B M, Jones J. J Inherited Metab Dis. 1994;17:403–420. doi: 10.1007/BF00711356. [DOI] [PubMed] [Google Scholar]
- 41.Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner A L. Nature (London) 1994;371:333–336. doi: 10.1038/371333a0. [DOI] [PubMed] [Google Scholar]
- 42.Swain J L, Stewart T A, Leder P. Cell. 1987;50:719–727. doi: 10.1016/0092-8674(87)90330-8. [DOI] [PubMed] [Google Scholar]
- 43.Sapienza C, Peterson A C, Rossant J, Balling R. Nature (London) 1987;328:251–254. doi: 10.1038/328251a0. [DOI] [PubMed] [Google Scholar]
- 44.Sapienza C, Paquette J, Tran T H, Peterson A. Development (Cambridge, UK) 1989;107:165–168. doi: 10.1242/dev.107.1.165. [DOI] [PubMed] [Google Scholar]
- 45.Reik W, Collick A, Norris M L, Barton S C, Surani M A. Nature (London) 1987;328:248–251. doi: 10.1038/328248a0. [DOI] [PubMed] [Google Scholar]
- 46.Hadchouel M, Farza H, Simon D, Tiollias P, Pourcel C. Nature (London) 1987;329:454–456. doi: 10.1038/329454a0. [DOI] [PubMed] [Google Scholar]
- 47.Allen N D, Norris M L, Surani M A. Cell. 1990;61:853–861. doi: 10.1016/0092-8674(90)90195-k. [DOI] [PubMed] [Google Scholar]
- 48.Engler P, Haasch D, Pinkert C A, Doglio L, Glymour M, Brinster R, Storb U. Cell. 1991;65:939–947. doi: 10.1016/0092-8674(91)90546-b. [DOI] [PubMed] [Google Scholar]
- 49.Chaillet J R, Vogt T F, Beier D R, Leder P. Cell. 1991;66:77–83. doi: 10.1016/0092-8674(91)90140-t. [DOI] [PubMed] [Google Scholar]
- 50.Chaillet J R, Bader D S, Leder P. Genes Dev. 1995;9:1177–1187. doi: 10.1101/gad.9.10.1177. [DOI] [PubMed] [Google Scholar]
- 51.Penny G D, Kay G F, Sheardown S A, Rastan S, Brockdroff N. Nature (London) 1996;379:131–137. doi: 10.1038/379131a0. [DOI] [PubMed] [Google Scholar]
- 52.Pfeifer K, Tilghman S M. Genes Dev. 1994;8:1867–1874. doi: 10.1101/gad.8.16.1867. [DOI] [PubMed] [Google Scholar]