

Abstract
The product of the Schizosaccharomyces pombe rad9+ gene is required for cell cycle arrest at the G2 checkpoints in response to incompletely replicated or damaged DNA. We have identified a human cDNA from an infant brain library that is a structural homolog of S. pombe rad9+, by searching the dBest data base for sequences similar to the fission yeast gene. The human gene encodes a 391-amino acid long, 42,520-Da protein that is ≈25% identical and 52% similar to the yeast protein. The human and yeast gene products demonstrate partial conservation of function, as the human cDNA can rescue to different degrees the sensitivity of S. pombe rad9::ura4+ cells to the DNA synthesis inhibitor hydroxyurea and gamma rays, as well as the associated checkpoint controls. These results suggest an underlying conservation of the molecular mechanisms of S and G2 checkpoint control pathways in most if not all eukaryotes. Fluorescence in situ hybridization using a fragment of the corresponding human genome as a probe, in conjunction with PCR reactions employing DNA from human X rodent somatic cell hybrids, has localized the gene to human chromosome 11q13.1-13.2. This region contains a number of tumor suppressor loci, and based on the biology of checkpoint control genes, HRAD9 should be considered a strong candidate for one of them.
Cell cycle checkpoints are regulatory mechanisms that prevent cell cycle progression in the presence of DNA damage or incompletely replicated DNA (1, 2, 3, 4). In the fission yeast Schizosaccharomyces pombe, loss of any of the rad1+, rad3+, rad9+, rad17+, rad26+, or hus1+ genes abolishes the G2 checkpoint delays which usually follow exposure to DNA damaging or replication blocking agents (3, 4, 5, 6). This suggests that these six genes regulate pathways linking aberrant DNA structures to cell cycle control.
Evidence for the conservation of checkpoint control pathways at the molecular level is suggested by the similarity of the gene product of the human ATM (ataxia telangiectasia mutated) locus and the proteins encoded by the checkpoint genes rad3+ from S. pombe and MEC1 from Saccharomyces cerevisiae (7, 8). The ATM gene product is likely involved in checkpoint control in human cells, as cell lines derived from ataxia telangiectasia (AT) patients exhibit a number of defects indicative of checkpoint deficiency, including sensitivity to ionizing radiation (9), and an increased rate of spontaneous mutation (9). However, the primary defect in AT seems to be at the G1-S rather than at the G2-M checkpoint (10, 11), although both points of cell cycle progression are aberrant in ATM mutated cells (12). We report here the identification of a human homolog of the fission yeast rad9+ checkpoint control gene, and the ability of this gene to partially complement hydroxyurea sensitivity, radiosensitivity, and the checkpoint defects of rad9 null fission yeast.
METHODS
DNA Sequence Analysis.
DNA sequence was determined using Sequenase version 2 (United States Biochemical), according to the instructions of the manufacturer. DNA oligonucleotides with complementary sequences to HRAD9 were synthesized throughout the length of the gene, and were used to prime DNA sequencing reactions. Sequences of the oligonucleotides used were as follows: F1, CTGTTCTGCCCTTCTCT; F2, TGCAGAGTCAGCAAAC; F3, CCAATGACGACATTGA; F4, CTTCCAGCAATACCAG; F5, TGCATTGCAAGTTCGG; F6, AGGCTGAACCAAGAAC; F7, CGTTCCTACCTCTTAT; F8, GACAAGTTTTCCTTGC; F9, TCAAGAGACCAGATGG; T1, GTTTGCTGACTCTGCA; T2, AGAGAAGGGCAGAACAG; T3, CTGGTATTGCTGGAAG; T4, TCAATGTCGTCATTGG; T5, GTTCTTGGTTCAGCCT; T6, CCATCTGGTCTCTTGA; T7, GCAAGGAAAACTTGTC; and T8, ATAAGAGGTAGGAACG. The nucleotide sequence of each strand of the HRAD9 gene was determined at least once. Amino acid sequences were aligned using the Genetics Computer Group (Madison, WI) programs pileup and pretty/consensus (13).
S. pombe Culture, Plasmids, and Manipulations.
S. pombe was cultured by standard techniques (14), unless indicated otherwise. Complete genotypes of strains used are as follows: Sp348, h-S leu1-32 ura4-294 ade6-216; Sp349, h-S rad9::ura4+ leu1-32 ura4-294 ade6-216; Sp296, h-S rad9::ura4+ leu1-32 ura4-D18 ade6-704; Sp18, h-S cdc25-22; Sp286, h-S rad9::ura4+ cdc25-22 leu1-32 ura4-D18. Plasmids used in this study include the expression vector pART1 (15); pHRAD9-1, full-length HRAD9 in pART1; pRAD9-1, S. pombe rad9 in pART1. pHRAD9-1 was made by subcloning the NotI–HindIII fragment containing the full-length HRAD9 gene into the SmaI site of pART1, using standard techniques. S. pombe transformations were performed according to the method of Okazaki et al. (16).
Sensitivity to Hydroxyurea and Induction of Septation.
Cells were cultured to mid-logarithmic phase at 30°C; then hydroxyurea was added to the indicated concentration. Samples were removed at varying times after addition of hydroxyurea, and plated onto PM (pombe minimal) selective media at a density that would eventually yield well-defined colonies. Plates were incubated for 3 to 6 days, until colonies were easily detectable. Relative viability was determined by the ability of drug-treated vs. untreated cells to form colonies.
For determination of septation, which reflects progression past the hydroxyurea-induced checkpoint and entry into mitosis, cells were cultured in the same manner used for testing sensitivity to hydroxyurea, but at the indicated times after drug addition, cells were collected and fixed in 70% ethanol. Subsequently, cells were washed once in 50 mM sodium citrate, then stained with 1 μg of calcofluor (Fluorescent Brightener 28; Sigma) per ml and viewed under a fluorescence microscope.
UV Light and Gamma Ray Irradiation, and Radiation-Induced Cell Cycle Delay.
Gamma irradiation was performed using a Gammacell 220 (Nordion, Ontario, Canada) with a 60Co source, at a dose rate of 1 Gy/sec. UV light treatments were performed at 254 nm, with a dose rate of 2.68 J/m2 per sec. Sensitivity of cells to radiation was assessed by procedures described previously (17). Briefly, cultures were grown to mid-logarithmic phase, then either left unirradiated or exposed to graded doses of gamma rays. Samples were then diluted, plated, and colonies scored as described above for assessing sensitivity to hydroxyurea. For UV light sensitivity, cells were irradiated after plating.
To determine radiation-induced checkpoint control, strains containing cdc25-22 or cdc25-22 rad9::ura4+ were grown as above to mid-logarithmic phase at the permissive temperature of 25°C. Cultures were then shifted to the restrictive temperature of 36°C for 4 hr. Cells were irradiated in the 10 min prior to release from the G2-M block, using a Clinac 2100 C/D with a 6-MV beam, at a dose rate of 0.23 Gy/sec. At time = 0, cells were released to permissive temperature, 25°C. At the indicated times, aliquots were removed and fixed with 70% ethanol. Cells were washed once with 50 mM sodium citrate (pH 7.0) and treated with 250 μg of RNase A per ml in 50 mM sodium citrate (pH 7.0) for 1 hr at 37°C. Cells were stained with 2.5 μg of propidium iodide per ml and examined under a confocal microscope. Binucleate cells were scored as having passed mitosis.
Mapping Studies.
The HRAD9 genomic clone was purified from a human placental DNA library in λ Fix II (Stratagene), by in situ plaque hybridization. Fluorescence in situ hybridization (FISH) was performed using established methods on methotrexate synchronized, phytohemagglutinin stimulated, normal peripheral blood lymphocytes (18). Suppression for 30 min with a mixture of sonicated human DNA (Sigma) and cot1 DNA (GIBCO/BRL) was required to reduce the background. The stained slides were counterstained with 4′,6-diamidino-2-phenylindole and actinomycin D (for a DA-4′,6-diamidino-2-phenylindole banding pattern), mounted in antifade medium and visualized utilizing a Zeiss Axioplan 2 microscope. Approximately 30 metaphase spreads were examined for probe localization. At least one specific probe signal was present in more than 80% of the mitoses examined. Images were captured using a cooled CCD camera (Photometrics PXL1400). Digital alignment of the images from each fluor was done after registration calibration through a triple bandpass filter (fluorescein isothiocyanate/Texas red/4′,6-diamidino-2-phenylindole) to minimize registration error, utilizing commercial software (electronic photography; Biological Detection Systems). Fig. 5A was prepared using photoshop (Adobe Systems, Mountain View, CA) and printed on a Tektronics Phaser 440 dye-sublimation printer. DNA sequencing was performed as described above, using the purified genomic HRAD9 DNA as template, and the leftward oligonucleotide depicted in Fig. 5B. The sequence of the oligonucleotide was: o957, CATGGTGACTGAGATGTG. PCR reactions were performed using DNA obtained from the Coriell Cell Repositories (Mapping panel #2 mini, and the regional mapping panel for chromosome 11) (19, 20). PCRs were carried out using 1 μg of DNA; 100 pmol of primers; 0.2 mM each of dATP, dCTP, dGTP, and dTTP; 2.5 units of Taq polymerase; 1.5 mM MgCl2; and 1× Taq buffer (BRL), in a total volume of 100 μl. PCR was performed for 35 cycles, each consisting of 1 min at 94°C, 30 sec at 57°C, and 2 min at 72°C, with a 5-min preincubation at 95°C and a final extension of 10 min at 72°C. The oligonucleotides used to prime the reaction were: o957, above; o980, CCAAAAATCCTTGATGGTGAAGATGGCGGGCCTCCCTGGAGCATCAAAAT. In Fig. 5D, the subregions of chromosome 11 represented by the various cell lines are as follows: 11936, 11q13.3–11q ter; 11943, 11p13–11q ter; 13400, 11p ter–11q24.2; 10482, 11p ter–11q23.3; 11944, 11p cen–11p ter.
Figure 5.
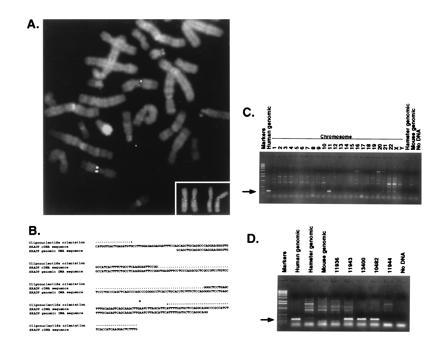
Chromosomal localization of HRAD9 to 11q13.1–11q13.2. (A) Cy3-labeled genomic HRAD9 was hybridized to methotrexate-synchronized normal human lymphocyte metaphase preparations. The white dual chromatid signals are localized to 11q13 (counterstain 4′,6-diamidino-2-phenylindole/actinomycin D). (Inset) Enlarged version of chromosome 11 from two different metaphase spreads. (B) DNA sequence analysis of the HRAD9 genomic clone used in FISH analysis identifies the location of an intron. The location of the primers used for PCR in sections C and D are indicated. (C) PCR analysis using total human, mouse, or hamster genomic DNA, or DNA isolated from somatic cell hybrids containing a single human chromosome only, as indicated. This confirms the chromosomal location indicated by FISH analysis. (D) PCR analysis sublocalizes HRAD9 to 11q13.1–11q13.2, as the genomic DNA is absent in a somatic cell hybrid containing 11q13.3–11q ter (11936), and present in three hybrids bearing the 11q13.1–11q13.2 region (11943, 13400, and 10482). Arrows to the left of C and D indicate position of relevant DNA fragments.
RESULTS
Isolation of a Human RAD9 cDNA.
A search of the dBest data base revealed a sequence expressed in human infant brain (GenBank accession no. R18275R18275) that predicted a 43-aa region with 34% identical and 60% similar amino acid composition when compared with the Schizosaccharomyces octosporus rad9+ gene product. The clone was obtained from the I.M.A.G.E. Consortium, and DNA sequence analysis of the 1.6-kb insert revealed that the protein coding region shared homology with the C-terminal half of the S. pombe and S. octosporus Rad9 proteins, suggesting that the cDNA was not full length. Therefore, the original human cDNA library (21) was screened for a full-length version of the HRAD9 gene, using the truncated clone to generate probes for in situ colony hybridization. Examination of 150,000 colonies yielded 11 clones, of which one contained a 2.1-kb insert; the others contained smaller cDNAs. Sequence analysis indicated that this clone had an insert with a 76-bp 5′ untranslated region, a 1176-bp coding region, and an 850-bp 3′ untranslated region. The open reading frame encodes a protein of 391 amino acids, which shares sequence similarity to the fission yeast Rad9 proteins (Fig. 1). HRAD9 is 25% identical and 52% similar at the amino acid level to S. pombe Rad9, and 27% identical and 54% similar to the corresponding protein from S. octosporus. The predicted Rad9 proteins exhibit sequence similarity over their entire lengths, including the N-terminal end, suggesting that the isolated human cDNA is full length. No motifs indicative of function are present in any of the Rad9 proteins, though a single phosphorylation consensus site for each of casein kinase II and protein kinase C have been conserved among the three proteins.
Figure 1.

Comparison of amino acid sequences predicted for the rad9 gene products from S. pombe and S. octosporus, and the HRAD9 gene product from Homo sapiens. Numbers on the left indicate the next amino acid in the protein sequence. Symbols between amino acids: Line, identical amino acids; Colon, similar amino acids.
Hydroxyurea Resistance and Checkpoint Control.
The HRAD9 cDNA was subcloned into the S. pombe expression vector pART1 (15), and introduced into rad+ and rad9::ura4+ cells to assess the rescue of defects caused by loss of rad9+ function. HRAD9 restores resistance of rad9::ura4+ cells to transient exposure to the DNA synthesis inhibitor hydroxyurea (Fig. 2A). Expression of HRAD9 has no effect on the survival of wild-type cells exposed to hydroxyurea (Fig. 2A). Furthermore, the presence of the control pART1 vector does not influence the hydroxyurea sensitivity of either rad+ or rad9::ura4+ cells (H.B.L. and K.M.H., unpublished observation). The HRAD9 subclone, the S. pombe rad9+ gene, and control pART1 vector were subsequently transformed into rad9::ura4+ mutant cells, and these strains were assayed for changes in mitotic entry in response to hydroxyurea treatment. In cell populations transformed with control pART1 vector, there was an accumulation of septated cells, as the checkpoint deficient strain entered mitosis (Fig. 2B). In contrast, neither the strains carrying HRAD9 nor rad9+ expressing plasmids exhibited this accumulation of septated cells (Fig. 2B). In wild-type cells there is virtually no septation 4 hr after addition of hydroxyurea. The fact that there was ≈20% septation in the rad9::ura4+ strains rescued by either the HRAD9 or rad9+ genes is probably due to the high spontaneous rate of plasmid loss in S. pombe; cells that have lost the rad9+ containing plasmid remain viable for a period of time, but would be functionally checkpoint deficient. In all cases, parallel samples that were not treated with hydroxyurea exhibited 15–20% septation at the final time point. Furthermore, all untreated cultures demonstrated essentially the same cell cycling profile.
Figure 2.
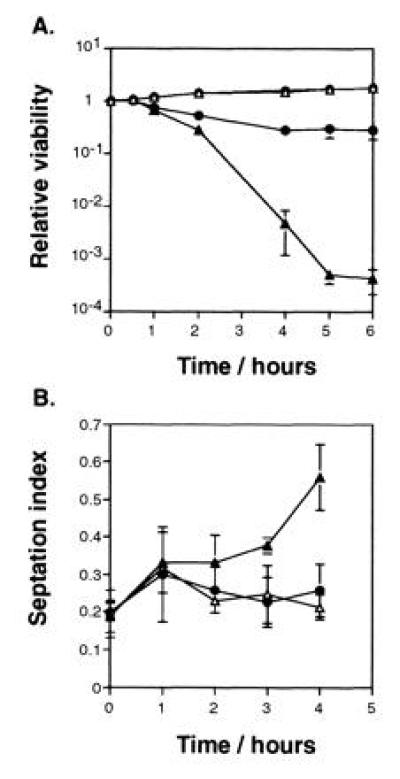
HRAD9 expression increases cell survival and cell cycle delay of rad9 null strains transiently exposed to hydroxyurea. (A) S. pombe cells were cultured to mid-logarithmic phase, and hydroxyurea added to 10 mM. The fraction of the population surviving was determined at the indicated times after hydroxyurea addition, using a colony formation assay, as we have described previously (17). Strains: rad9+ (▵), rad9+ [pHRAD9-1] (○), rad9::ura4+ (▴), rad9::ura4+ [pHRAD9-1] (•). (B) Hydroxyurea-induced cell cycle delay was determined using an asynchronous population of S. pombe exposed to 12 mM hydroxyurea. At the indicated times, aliquots of the culture were taken and fixed with ethanol. Samples were washed once, stained with calcofluor, and examined for septation by fluorescence microscopy. Strains: rad9::ura4+ containing control vector (▴), pHRAD9-1 (•), or pRAD9-1 (▵). Data represent the average of three experiments. Error bars = SD.
Radioresistance and Radiation-Induced Checkpoint Control.
HRAD9 expression also confers a moderate level of resistance to gamma-ray radiation upon rad9::ura4+ mutant cells (Fig. 3A). The same strains described for Fig. 2A were treated with graded doses of ionizing radiation or UV light, and plated to assay cell survival. HRAD9 expression in rad9::ura4+ strains leads to partial restoration of resistance to gamma-ray radiation, while expression of HRAD9 in rad+ cells has no significant effect on survival (Fig. 3A). Expression of HRAD9 in rad+ or rad9::ura4+ mutant cells does not alter sensitivity to UV light (Fig. 3B).
Figure 3.
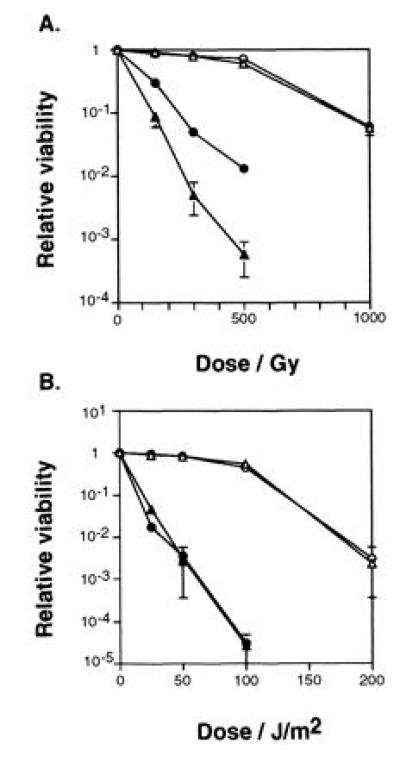
HRAD9 expression increases cell survival of rad9 null strains exposed to ionizing radiation, but not to UV light. (A) Logarithmic phase S. pombe were exposed to the indicated γ radiation doses, or (B) UV light doses, and assayed for viability by the ability to form colonies. Strains: rad9+ (▵), rad9+ [pHRAD9-1] (○), rad9::ura4+ (▴), rad9::ura4+ [pHRAD9-1] (•). Data represent the average of three experiments. Error bars = SD.
HRAD9 expression also induces a detectable mitotic entry delay after exposure to ionizing radiation, which is lost in the rad9::ura4+ mutant (Fig. 4). This is an indication of a weak rescue of the checkpoint deficiency in rad9::ura4+ mutants bearing the HRAD9 plasmid, as reflected by the moderate increase in resistance to ionizing radiation shown by these cells (Fig. 3A). Alternatively, this could be interpreted as representing a reduced rate of mitotic entry by these cells. However, this is unlikely, as rad+ cells containing HRAD9 show radiation resistance and cycling profiles indistinguishable from those observed for cells either carrying the pART1 control vector, or carrying no plasmid. Furthermore, the pART1 control vector does not confer radiation resistance upon rad9::ura4+ cells. Examination of a population of rad9::ura4+ cells bearing HRAD9 and exposed to UV light revealed no significant increase in elongated cells, relative to a similar unirradiated culture. Therefore, based on this result and the lack of increased UV resistance conferred by HRAD9, quantitation of septation index as a measure of checkpoint control post-UV treatment was not pursued.
Figure 4.
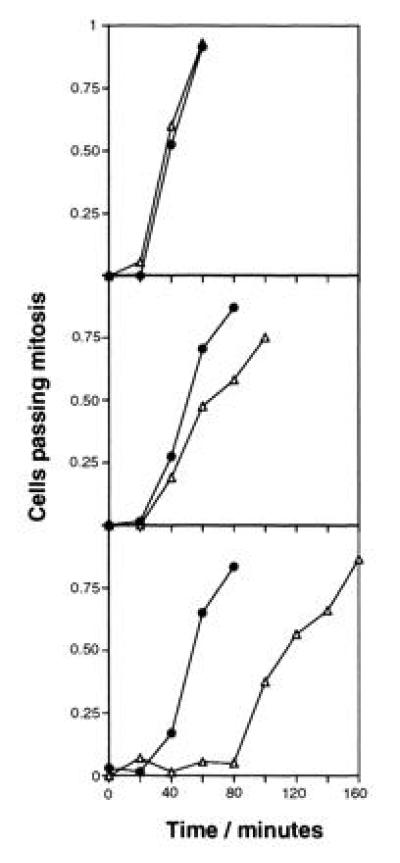
HRAD9 expression partially restores mitotic entry delay in rad9 null strains exposed to ionizing radiation. A cdc25-22 rad9::ura4+ strain was transformed with control vector (Top), or plasmids containing HRAD9 (Middle), or S. pombe rad9+ (Bottom). Strains were synchronized at the G2-M border by a 4 hr incubation at the restrictive temperature for cdc25-22. Synchronized cell populations were mock irradiated (•), or irradiated with 100 Gy ionizing radiation (▵), and released to permissive temperature (time = 0). At the indicated times after release, aliquots were fixed with ethanol. Cells were stained with propidium iodide, and scored under a fluorescence microscope for the fraction of binucleate cells, indicative of passage through mitosis. Data represent the average of three experiments. The profiles presented are reproducible in individual, independent experiments.
HRAD9 Is Located on Chromosome 11q13.1–13.2.
FISH, in combination with the screening of a series of DNAs from a panel of human X rodent somatic cell hybrids, was used to assign HRAD9 to a human chromosomal location. Briefly, a genomic clone of HRAD9 was identified from a human placental DNA library by in situ hybridization to the original HRAD9 cDNA clone. This genomic clone was used as a probe for FISH to metaphase chromosomes, and the chromosomal location was identified as 11q13 (Fig. 5A). Limited DNA sequence analysis of this genomic HRAD9 clone reveals the presence of an intron in the insert, confirming that the bona fide HRAD9 genomic sequence was isolated, and not a processed HRAD9 pseudogene (Fig. 5B). Two oligonucleotides directed against HRAD9 were used to prime PCRs on the genomic clone, total genomic DNA, and on genomic DNA from human X rodent somatic cell hybrid lines. The 275-nucleotide PCR product is only generated in samples containing human chromosome 11 DNA, confirming the chromosomal assignment made by FISH (Fig. 5C). Further PCR analysis sublocalizes HRAD9 to 11q13.1–11q13.2, as the 275-nucleotide PCR product indicative of the genomic DNA is present in three hybrids bearing the 11q13.1–11q13.2 region, but absent from a somatic cell hybrid containing 11q13.3–11q ter (Fig. 5D). Furthermore, the 3′ end of the HRAD9 cDNA contains sequences from the 3′ untranslated region of a human type 1 phosphatase gene, oriented opposite to the direction of translation of HRAD9. This phosphatase has previously been mapped to chromosome 11q13 (22), providing independent confirmation of the assigned map location.
DISCUSSION
A human cDNA encoding a protein that shares significant amino acid sequence homology with the rad9+ checkpoint control gene products from fission yeasts S. pombe (23, 24) and S. octosporus (25) has been isolated. Although a data bank search revealed a limited degree of similarity of Rad9 to several S. cerevisiae protein sequences, none have yet been tested for functional conservation. The similarities of the human and fission yeast proteins at the amino acid level extend through the entire lengths of the molecules, and include a number of highly conserved sequences primarily at the N-terminal regions of the proteins. Interestingly, when compared with the yeast proteins, the protein encoded by HRAD9 is devoid of several amino acids, most notably amino acids 93–114 found in the fission yeast Rad9 proteins. The functional significance of these regions is currently being examined.
Expression of the HRAD9 cDNA in S. pombe rad9::ura4+ cells partially rescues the sensitivity of the mutant to the DNA synthesis inhibitor hydroxyurea, or radiation, as well as the associated checkpoint controls. Complementation of the hydroxyurea sensitivity and cell cycle delay phenotypes was to nearly wild-type levels. There was a moderate rescue of the defects associated with the response of rad9::ura4+ cells to gamma-rays. However, S. pombe rad9::ura4+ cells containing HRAD9 remained sensitive to UV and continued to lack the UV-inducible cell cycle delay at the G2/M border. The different degrees of complementation of S. pombe mutant cells by the human cDNA may reflect differential requirements of rad9+ for achieving increased survival or induction of checkpoint control after exposure to hydroxyurea and each type of radiation. The rad9+ dependent checkpoints in S. pombe respond to signals both during S phase, and in G2 (4, 5). The efficient rescue of one but not both of these defects by HRAD9 expression suggests that these pathways may have diverged in humans, and that HRAD9 mediates the cell cycle delay in response to incomplete DNA replication, but not to DNA damage. Alternatively, if unique protein complexes or DNA sequences serving as specific sites for DNA binding protein interactions mediate DNA damage and incomplete DNA replication signals, HRAD9 may function more efficiently with those involved in the latter signaling mechanism.
Even in the absence of exogenous DNA damage, defects in cell cycle checkpoints lead to genomic instability, as demonstrated for the G2-M checkpoint in S. cerevisiae rad9 mutants (2), and at the G1-S checkpoint in p53 or ATM deficient mammalian cells (9, 26, 27, 28). The resulting widespread genomic abnormalities are typical of cancer cells (29). As noted above, the S. pombe rad3+ gene is homologous to the human ATM gene, and mutations in ATM lead to an increased incidence of cancer (7, 30, 31). AT patients are at an elevated risk for developing numerous types of tumors, and even AT carriers exhibit an elevated risk of developing cancer (30, 31). Both the ATM and p53 genes not only regulate cell cycle checkpoints (26, 27, 32, 33, 34, 35, 36), but also act as tumor suppressors (30, 31, 37, 38, 39). It is likely that other human checkpoint genes will also act as tumor suppressors. The 11q13 region, within which HRAD9 resides, contains at least two tumor suppressor loci. The most prominent is MEN1, which is responsible for type 1 multiple endocrine neoplasia (40). The type 1 phosphatase, which is located near HRAD9, has been excluded as a candidate for the MEN1 gene by fine resolution mapping data (22, 41, 42, 43), suggesting that HRAD9 should also be excluded from consideration as the causative gene for this cancer syndrome. The presence of a second tumor suppressor locus in this region is suggested by studies on cervical cancer derived cell lines (44), and HRAD9 cannot be excluded as a candidate for this gene.
Overall, these results demonstrate that the HRAD9 gene can functionally complement some of the checkpoint defects of S. pombe rad9::ura4+ cells, suggesting that fission yeast rad9-dependent G2 and especially S checkpoint pathways, which promote radiation and hydroxyurea resistance, respectively, are conserved at the molecular level in humans. The extensive genetic information known about G2 checkpoint control mechanisms in yeast will be invaluable toward defining parallel human pathways important for modulating the response of cells to DNA damage or the inhibition of DNA replication.
Acknowledgments
We thank M. B. Soares for providing the cDNA library used to identify the full-length HRAD9 gene, and J. Robins and A. Fung for assistance using the Clinac 2100 C/D. This work was supported by National Institutes of Health Grants CA12536 and GM52493 (H.B.L.), and by grants from the Ontario Cancer Treatment and Research Foundation and the Kingston General Hospital Clare Nelson Bequest (S.D.). H.B.L. was supported in part by a National Institutes of Health Research Career Development Award (CA68446). S.D. is a Career Scientist of the Ontario Cancer Treatment and Research Foundation.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Weinert T A, Hartwell L H. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 2.Weinert T A, Hartwell L H. Mol Cell Biol. 1990;10:6554–6564. doi: 10.1128/mcb.10.12.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley R, Subramani S, Young P G. EMBO J. 1992;11:1335–42. doi: 10.1002/j.1460-2075.1992.tb05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.al-Khodairy F, Carr A M. EMBO J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enoch T, Carr A M, Nurse P. Genes Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- 6.al-Khodairy F, Fotou E, Sheldrick K S, Griffiths D J, Lehmann A R, Carr A M. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 8.Savitsky K, Sfes S, Jagle D A, Zio Y, Sartiel A, Collins F S, Shiloh Y, Rotman G. Hum Mol Genet. 1995;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 9.Taylor A M R, Harnden D G, Arlett C F, Harcourt S A, Lehmann A R, Stevens S, Bridges B A. Nature (London) 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 10.Painter R B, Young B R. Proc Natl Acad Sci USA. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imray F P, Kidson C. Mutat Res. 1983;112:369–382. doi: 10.1016/0167-8817(83)90030-5. [DOI] [PubMed] [Google Scholar]
- 12.Beamish H, Lavin M F. Int J Radiat Biol. 1994;65:175–184. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- 13.Higgins D G, Sharp P M. Comput Appl Biosci. 1989;5:151–153. doi: 10.1093/bioinformatics/5.2.151. [DOI] [PubMed] [Google Scholar]
- 14.Leupold U. Methods Cell Physiol. 1970;4:169–177. [Google Scholar]
- 15.McLeod M, Stein M, Beach D. EMBO J. 1987;6:729–736. doi: 10.1002/j.1460-2075.1987.tb04814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okazaki K, Okazaki N, Kume K, Jinno S, Tanaka K, Okayama H. Nucleic Acids Res. 1990;18:6485–6489. doi: 10.1093/nar/18.22.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lieberman H B. Genetics. 1995;141:107–117. doi: 10.1093/genetics/141.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demetrick D J. In: Fluorescence in Situ Hybridization and Human Cell Cycle Genes. Pagano M, editor. New York: Springer; 1995. pp. 29–45. [Google Scholar]
- 19.Dubois B L, Naylor S L. Genomics. 1993;16:315–319. doi: 10.1006/geno.1993.1191. [DOI] [PubMed] [Google Scholar]
- 20.Drwinga H L, Toji L H, Kim C H, Greene A E, Mulivor R A. Genomics. 1993;16:311–314. doi: 10.1006/geno.1993.1190. [DOI] [PubMed] [Google Scholar]
- 21.Soares M B, Bonaldo M F, Jelene P, Su L, Lawton L, Efstratiadis A. Proc Natl Acad Sci USA. 1994;91:9228–9232. doi: 10.1073/pnas.91.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker H M, Jones T A, da Cruz e Silva E F, Spurr N K, Sheer D, Cohen P T. Genomics. 1990;7:159–166. doi: 10.1016/0888-7543(90)90536-4. [DOI] [PubMed] [Google Scholar]
- 23.Murray J M, Carr A M, Lehmann A R, Watts F Z. Nucleic Acids Res. 1991;19:3525–3531. doi: 10.1093/nar/19.13.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieberman H B, Hopkins K M, Laverty M, Chu H M. Mol Gen Genet. 1992;232:367–376. doi: 10.1007/BF00266239. [DOI] [PubMed] [Google Scholar]
- 25.Lieberman H B, Hopkins K M. Gene. 1994;150:281–286. doi: 10.1016/0378-1119(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 26.Yin Y, Tainsky M A, Bischoff F Z, Strong L C, Wahl G M. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 27.Livingstone L R, White A, Sprouse J, Livanos E, Jacks T, Tlsty T D. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 28.Pandita T K, Pathak S, Geard C R. Cytogenet Cell Genet. 1995;71:86–93. doi: 10.1159/000134069. [DOI] [PubMed] [Google Scholar]
- 29.Nowell P C. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 30.Swift M, Morrell D, Massey R B, Chase C L. N Engl J Med. 1991;325:1831–1836. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 31.Swift M, Reitnauer P J, Morrell D, Chase C L. N Engl J Med. 1987;316:1289–1294. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 32.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 33.Kastan M B, Zhan Q, el-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J., Jr Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 34.Kuerbitz S J, Plunkett B S, Walsh W V, Kastan M B. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connor P M, Jackman J, Jondle D, Bhatia K, Magrath I, Kohn K W. Cancer Res. 1993;53:4776–4780. [PubMed] [Google Scholar]
- 36.Dulic V, Kaufmann W K, Wilson S J, Tlsty T D, Lees E, Harper J W, Elledge S J, Reed S I. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 37.Malkin D, Li F P, Strong L C, Fraumeni J F, Jr, Nelson C E, Kim D H, Kassel J, Gryka M A, Bischoff F Z, Tainsky M A, Friend S H. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava S, Zou Z Q, Pirollo K, Blattner W, Chang E H. Nature (London) 1990;348:747–749. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 39.Donehower L A, Harvey M, Slagle B L, McArthur M J, Montgomery C A, Jr, Butel J S, Bradley A. Nature (London) 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 40.Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Nature (London) 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 41.Larsson C, Weber G, Kvanta E, Lewis K, Janson M, Jones C, Glaser T, Evans G, Nordenskjold M. Hum Genet. 1992;89:187–193. doi: 10.1007/BF00217121. [DOI] [PubMed] [Google Scholar]
- 42.Larsson C, Friedman E. Endocrinol Metab Clin North Am. 1994;23:67–79. [PubMed] [Google Scholar]
- 43.Weber G, Friedman E, Grimmond S, Hayward N K, Phelan C, Skogseid B, Gobl, Zedenius J, Sandelin K, Teh B T, Carson E, White I, Öberg K, Shepherd J, Nordenskjöld M, Larsson C. Hum Mol Genet. 1994;3:1775–1781. doi: 10.1093/hmg/3.10.1775. [DOI] [PubMed] [Google Scholar]
- 44.Jesudasan R A, Rahman R A, Chandrashekharappa S, Evans G A, Srivatsan E S. Am J Hum Genet. 1995;56:705–715. [PMC free article] [PubMed] [Google Scholar]