

Abstract
The Escherichia coli tet-repressor (TetR) operator system was used to develop a variation of the yeast two-hybrid assay in which disruptions of protein–protein interactions can be identified by a positive selection. This assay, designated the “split-hybrid system,” contains a two-component reporter. The first component contains LexA binding sites upstream of the TetR gene and the second contains TetR operator binding sites upstream of HIS3. Interaction of one protein fused to the LexA DNA binding domain with a second protein fused to the VP16 activation domain results in TetR expression. TetR subsequently binds to the tet operators, blocking the expression of HIS3 and preventing yeast growth in media lacking histidine. The utility of the split-hybrid system was analyzed by examining the phosphorylation-dependent interaction of CREB and its coactivator CREB binding protein (CBP). CREB and CBP associate through an interaction that depends upon CREB phosphorylation at Ser-133. Mutation of this phosphorylation site prevents yeast growth in the standard two-hybrid assay but allows growth in the split-hybrid strains. The split-hybrid system was used to identify other CREB mutations that disrupt its association with CBP. These mutations localized around the site of CREB phosphorylation, indicating that only a small portion of the CREB activation domain is required for CBP interaction. The yeast split-hybrid system should be useful in identifying mutations, proteins, peptides, and drugs that disrupt protein–protein interactions.
We have developed a variation of the yeast two-hybrid system (1) that permits identification of factors that abrogate or “split” the association of two interacting proteins. This yeast genetic assay, which we have termed the “split-hybrid system,” can also be used to screen for mutations that block specific associations.
The split-hybrid system converts the disruption of a protein–protein interaction into a positive selection by using the Escherichia coli Tn10-encoded tet-repressor (TetR) operator system (2). We have engineered LexA binding sites upstream of the TetR gene and have placed TetR binding sites (tet operators) upstream from a nutritional reporter gene, HIS3. Interaction of one protein fused to LexA (which contains a DNA binding domain) with a second protein fused to the VP16 activation domain results in the expression of TetR, which binds to the tet operators, blocking expression of HIS3 and preventing yeast growth in media lacking histidine. As with the conventional yeast two-hybrid system, this method allows large numbers of clones to be screened by a selection. In addition, the interaction of TetR with tet operators can be abolished by tetracycline (Tc). The ability to modulate the selection with Tc is useful when the protein fused to LexA has intrinsic activating capability. Furthermore, 3-aminotriazole (3AT), an inhibitor of the histidine pathway, can be used when the interaction of two proteins is relatively weak and unable to produce sufficient TetR to block histidine production fully.
In the present study, we examined two established protein–protein interactions: the serum response factor (SRF) and human T-cell leukemia virus protein Tax and the transcription factor CREB and its binding protein (CBP). Tax activates the c-fos promoter through the serum response element (3). This activation results from a direct interaction between Tax and SRF, as Tax does not bind to the serum response element directly (4, 5). CBP is a coactivator of CREB that mediates the transactivation of cAMP-responsive promoters (6, 7). cAMP activates the cAMP-dependent protein kinase A (PKA), which phosphorylates CREB at Ser-133, located in the kinase-inducible domain (KID). The phosphorylation of Ser-133 allows CREB to associate with CBP (6). In this paper, we demonstrate that these interactions prevent growth in the yeast split-hybrid strains. In contrast, the expression of CBP and a CREB mutant (CREB-M1; Ser-133 → Ala), which is unable to bind CBP, prevents production of TetR and allows the yeast to grow. We used the split-hybrid system to screen for other mutations in CREB that disrupt its association with CBP and determined that most of the mutations were located within a small region immediately adjacent to the PKA phosphorylation site.
MATERIALS AND METHODS
Plasmid Construction.
For pLexA–CBD, a DNA fragment containing the CREB binding domain (CBD) of CBP (amino acids 461–682) was PCR amplified and inserted into the EcoRI and BamHI sites of pBTM116 that carries the TRP1 gene (8). For pLexA–SRF, a DNA fragment containing SRF was digested from pCGN–SRF (gift from M. Gilman, Ariad Pharmaceuticals, Boston) with XhoI and BamHI. The XhoI site was blunt-ended, ligated with BamHI linkers, and inserted into the BamHI site of pBTM116. pLexA–Lamin was a gift from R. Sternglanz (The State University of New York at Stony Brook). For pVP16–CREB, pcDNA3/CREB283, containing the VP16 transactivation domain fused to the rat CREB 341 (amino acids 1–283) (a gift from R. Maurer, Oregon Health Sciences University, Portland), was digested with XbaI, blunt-ended, and ligated with BamHI linkers. The fragment encoding the pVP16–CREB fusion protein was digested with BamHI and HindIII and inserted into pVP16, which carries the LEU2 gene (9). For pVP16–Tax, the DNA sequence encoding Tax was digested from pS6424 (10) with BamHI and inserted into pVP16. For pLeu, pVP16 was digested with HindIII and BamHI to remove the VP16 transactivation domain, blunt-ended, and religated. For pLexA–VP16, the VP16 transactivation domain was PCR amplified, digested with ClaI, blunt-ended, and inserted into the SmaI site of pBTM116. For pRS306/8xLexA op/TetR, the ADH terminator sequence was digested from pBTM116 with SphI and PstI, blunt-ended, and cloned into the blunt-ended NotI site of pRS306 (11) to yield pRS306/Term. PCR was used to link the 5′ promoter sequence of the yeast HIS3 gene (nucleotides −75 to +23) to the translational start of TetR. Sequences encoding the simian virus 40 large T-antigen nuclear localization signal (NLS) were fused to the C terminus of TetR. The PCR product was digested with EcoRI and BamHI and inserted into pRS306/Term. This construct, pRS306/HIS3:TetR/Term, retains the fusion of TetR to the nuclear localization signal, followed by four amino acids generated by the vector backbone (Arg-Ile-His-Asp). The LexA binding site multimer from the plasmid pSH18-34ΔSpe (gift from R. Brent, Harvard Medical School) was PCR amplified (9), digested with EcoRI, and subcloned into pRS306/HIS3:TetR/Term to produce pRS306/8xLexA op/TetR. For pRS303/2xtet op-LYS2, PCR was used to engineer one copy of the tet operator (created by annealing complementary oligonucleotides) into position −53 of pRS303 (11). For the second tet operator site, a MluI restriction site was engineered into position −22 in the HIS3 promoter by PCR. One copy of the tet operator was inserted into the MluI site to yield pRS303/2xtet op. The LYS2 gene was isolated from pLYS2 (9) with EcoRI and BamHI, blunt-ended, ligated with SstI linkers, and ligated into pRS303/2xtet op. For pVP16–CREB(BglII–SacII)–LacZ, β-galactosidase (β-gal) was PCR amplified from pSV-β-gal (Promega) and inserted into the NotI site of pVP16 to produce pVP16–LacZ. A PCR fragment containing CREB (amino acids 1–283) was inserted into pVP16–LacZ at the BamHI site. To generate a cassette vector for subcloning, PCR was used to engineer a BglII site at nucleotides 273–278 and a SacII site at nucleotides 500–505 of CREB.
Yeast Strain Construction.
Yeast were grown in yeast extract/peptone/dextrose or selective minimal medium using standard conditions (12, 13). Yeast strains were derived from AMR69 and AMR70 (9). YI584 was constructed from AMR69 and AMR70 as follows: (i) Ura− derivatives were identified after 5-fluoroorotic acid selection (15); (ii) targeted integration of pRS306/8xLexA op/TetR was carried out by transforming with a plasmid linearized at a NcoI site in URA3 and the resulting strains were examined by Southern blot analysis; (iii) targeted integration of pRS303/2xtet op-LYS2 was carried out by transforming with the plasmid linearized at a HpaI site in LYS2 and the resulting strains were confirmed by Southern blot analysis; (iv) YI584 was constructed by mating the AMR69-derivative MATa strain containing the pRS303/2xtet op-LYS2 with the AMR70-derivative MATα strain containing pRS306/8xLexA op/TetR. The genotype of YI584 is (MATa/MATα, his3Δ200/his3Δ200 trp1-901/trp1-901 leu2-3, 112/leu2-3, 112 ade2/ade2 containing URA::(LexA operator)8-TetR LYS2::(Tet operator)2-HIS3).
Liquid Assay.
YI584 was transformed as described (9) and plated on selective medium plates lacking tryptophan, uracil, leucine, and lysine. After three days growth at 30°C, a pool of colonies was diluted in 5 ml of selective media, vortexed, and sonicated for 10 sec. Cells were counted and seeded at 1000 cells/ml selective media at a final volume of 2 ml. Tc, 3AT, and histidine were supplemented as appropriate. Samples were incubated with shaking for two days at 30°C and were quantitated by measuring the OD600.
β-Gal Liquid Assay.
The β-gal liquid assays were performed as described (14).
PCR Mutagenesis and Creation of Mutant Library.
Mutagenic PCR was a minor modification of that described (15). The reaction mixture contained 20 ng of pVP16–CREB(BglII–SacII)–LacZ, 16 mM (NH4)2SO4, 67 mM Tris·HCl (pH 8.8), 6.1 mM MgCl2, 0.5 mM MnCl2, 6.7 μM EDTA, 10 mM 2-mercaptoethanol, 1 mM primers, 1 mM each dGTP, dTTP, and dCTP, 400 μM dATP, and 2.5 units of Taq DNA polymerase (Promega). After seven cycles of PCR (94°C for 40 sec, 50°C for 40 sec, and 72°C for 40 sec), the PCR product was reamplified with Vent DNA polymerase (New England Biolabs) for 25 cycles. The resultant PCR product was digested with BglII and SacII and inserted into pVP16–CREB(BglII–SacII)–LacZ. Ligations were transformed into DH5α bacterial cells. Transformants were pooled and plasmid DNA was isolated by CsCl gradient centrifugation.
Library Screen.
The yeast strain YI584 expressing pLexA–CBD was transformed with a library of pVP16–CREB(BglII–SacII)–LacZ mutants. 4 ml of cells (OD600 = 0.6) were transformed with 40 μg of library and 4 mg of salmon sperm DNA and plated onto selective medium supplemented with 10 μg/ml of Tc and 1 mM 3AT. β-Gal filter assays were performed as described (16) to identify full-length proteins. DNA from 536 growth(+), 5-bromo-4-chloro-3-indoyl β-d-galactoside(+) colonies was isolated and transformed into E. coli MC1066 cells. The pLexA–CBD plasmid was cured by culturing the MC1066 cells in the absence of leucine (17). The DNA from 193 VP16–CREB(BglII–SacII)–LacZ mutants was isolated and rescreened in YI584 cells, as well as in L40 cells (two-hybrid system; ref. 16). Two-hybrid transformations were plated on selective medium supplemented with 40 mM 3AT. CREB mutants that did not interact with CBD in the rescreening process were sequenced using Sequenase (United States Biochemical, version 2.0).
RESULTS
Characterization of the Split-Hybrid System.
The yeast split-hybrid strain contains two integrated reporter constructs (Fig. 1A). One reporter contains the coding sequence of TetR flanked upstream by the yeast HIS3 minimal promoter and a tandem array of LexA operators. To ensure efficient transport of TetR into the nucleus, the nuclear localization signal of simian virus 40 large T antigen was fused to the C-terminal end of TetR. The other reporter contains tet operator sequences inserted into the promoter region of the HIS3 gene. The expression of TetR in this system depends on the formation of a complex between one protein fused to LexA and another protein fused to the VP16 activation domain. When the fusion proteins interact, the TetR gene is activated. The TetR protein binds to the tet operators and represses promoter activity of HIS3, which causes the yeast to be auxotrophic for histidine. Conversely, when the interaction of two fusion proteins is disrupted (Fig. 1B), no TetR protein is generated, allowing expression of the HIS3 gene and producing yeast prototrophic for histidine.
Figure 1.
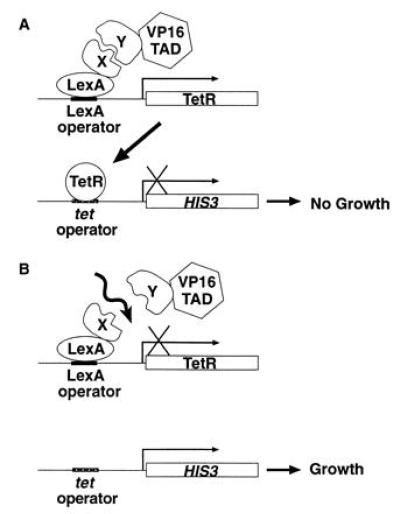
The yeast split-hybrid system. (A) Interaction of protein X fused to LexA with protein Y fused to the VP16 transactivation domain (TAD) promotes the expression of TetR which, in turn, binds to tet operators upstream of the HIS3 gene, preventing the growth of yeast in medium lacking histidine. (B) Disruption (depicted by wavy arrow) of the interaction between proteins X and Y prevents expression of TetR, permitting expression of HIS3 and allowing growth of yeast in medium lacking histidine.
To test the utility of the reporter constructs, we first transformed the yeast split-hybrid strain YI584 with pLexA–VP16. pLexA–VP16 encodes a fusion protein containing the VP16 activation domain and LexA and is a strong transactivator for promoters containing LexA operators. Yeast expressing LexA–VP16 generate TetR protein as detected by gel mobility shift assay (data not shown). These yeast were unable to grow in the absence of histidine, indicating that the overexpressed TetR is capable of binding to tet operators and preventing the expression of HIS3 (Fig. 2 A and B). These strains grew on plates containing histidine, however, indicating that TetR overexpression was not toxic. Yeast expressing pLexA–lamin and pVP16, which do not interact in a yeast two-hybrid system (Fig. 2C) and are thus not expected to lead to the production of TetR, grew in the absence of histidine (Fig. 2 A and B).
Figure 2.
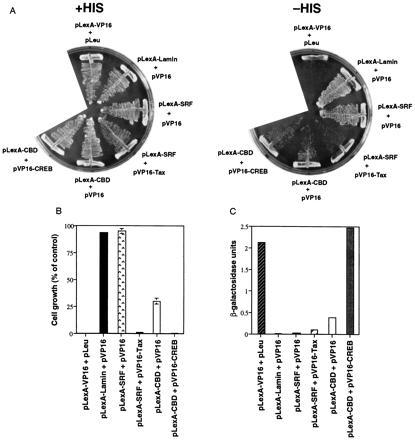
Characterization of the yeast split-hybrid system. The yeast split-hybrid strain YI584 (A and B) are the yeast two-hybrid strain L40 (C) were transformed with the indicated plasmid pairs. Yeast transformants were plated on selective medium plates (lacking tryptophan, uracil, leucine, and lysine) supplemented with histidine and incubated at 30°C for three days. (A) A pool of colonies for each interacting pair of proteins was streaked onto selective medium supplemented with histidine (Left) and onto selective medium lacking histidine (Right). The plates were incubated at 30°C for 3 days. (B) A pool of colonies was seeded in selective media lacking histidine. After incubation of cultures at 30°C for 2 days, growth was measured at OD600. Data represent the mean ± SD of two or three experiments and is presented as a percentage of the OD600 of yeast grown in selective media supplemented with histidine. (C) A pool of colonies was collected and prepared for β-gal liquid assay. Data represent the mean ± SD of two experiments. A β-gal unit = 1000 × OD420/time × protein, where the time is in minutes and the protein is in mg/ml.
To investigate the feasibility of using the split-hybrid system to study protein–protein interactions, a pair of interacting proteins, SRF and Tax, was tested. pLexA–SRF was transformed into strain YI584 along with pVP16–Tax. This transformation failed to yield any colonies in medium lacking histidine (Fig. 2 A and B). In contrast, when LexA–SRF was cotransformed with a vector encoding the VP16 activation domain alone, yeast growth occurred in the absence of histidine. LexA–SRF and VP16–Tax interact relatively weakly in the yeast two-hybrid system (Fig. 2C), yet almost completely abolish growth when expressed in the split-hybrid strain. VP16–Tax is slightly toxic (unpublished observations), which may explain why LexA–CBD and VP16 activate LacZ expression more strongly than LexA–SRF and VP16–Tax in the two-hybrid strain (Fig. 2C) but are unable to fully abolish yeast growth when expressed in the split-hybrid strain (Fig. 2B).
Modulation of the Split-Hybrid System.
A major advantage of using the TetR-operator system is the ability to block TetR binding with Tc. This ability to modulate TetR binding is critical when the LexA fusion protein has intrinsic activating capability, as illustrated in experiments using yeast expressing the CREB-binding domain (CBD) of CBP fused to LexA (LexA–CBD). Yeast expressing LexA–CBD and VP16 had substantially decreased growth in medium lacking histidine (Fig. 2 A and B). Indeed, the levels of growth observed when yeast were cotransformed with pLexA–CBD and pVP16 or pVP16–CREB were almost indistinguishable. To distinguish growth differences between strains expressing VP16 and VP16–CREB, varying concentrations of Tc were added to liquid cultures of yeast grown in the absence of histidine. The repression of yeast growth was relieved by Tc in a dose-dependent fashion (Fig. 3A). These results demonstrate that Tc can overcome basal transactivating capability of the LexA–CBD fusion protein.
Figure 3.
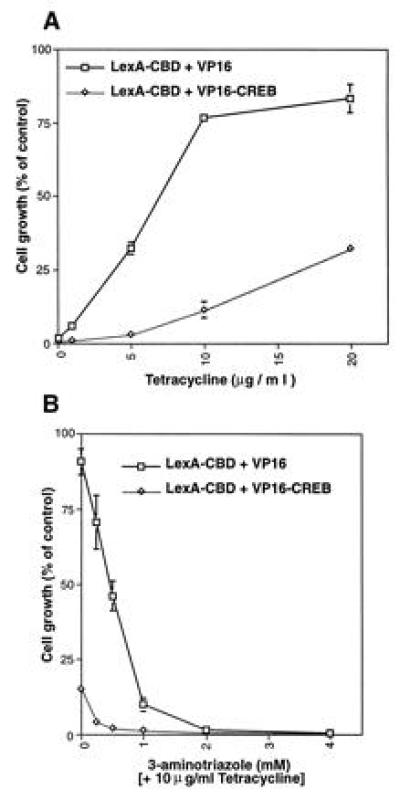
Modulation of the split-hybrid system by Tc and 3AT. Yeast strain YI584 was transformed with pLexA–CBD and either pVP16 (□) or pVP16–CREB (⋄) and plated on selective medium supplemented with histidine. The yeast were grown at 30°C for 3 days and prepared for liquid assay. The cells were seeded in selective media lacking histidine and titrated with Tc (A), or 3AT with 10 μg/ml Tc (B). After incubation of cultures at 30°C for 2 days, growth was measured at OD600. Data represent the mean ± SE of three experiments and is presented as a percentage of the OD600 of yeast grown in selective medium supplemented with histidine.
To use the split-hybrid system most efficiently, it is important to minimize background growth that might be confused with disrupted protein–protein associations. This can be accomplished with 3AT, a competitive inhibitor of the HIS3 gene product. For instance, in the presence of 10 μg/ml Tc, the yeast strain transformed with pLexA–CBD and pVP16–CREB still conferred ≈12% growth in liquid culture (Fig. 3A). To diminish this background, varying concentrations of 3AT were added to the culture media in the presence of 10 μg/ml Tc. At 0.25 mM 3AT, the growth of the yeast strain expressing LexA-CBD and VP16–CREB was <5%, while the growth of the control strain was still maintained at 70% of control levels.
Selection of Mutants Defective in a Protein–Protein Interaction.
One use of the split-hybrid system is to identify point mutants that disrupt protein–protein interactions. The binding of CREB to CBP has been shown to require the phosphorylation of CREB Ser-133, located in the KID (6, 7). Changing Ser-133 to alanine (CREB-M1) abolishes the ability of CBP to activate CREB-mediated transcription. We tested CREB-M1 in the split-hybrid system and verified that this mutation prevents the interaction with CBP (data not shown). Precisely which other residues of the KID of CREB are required for binding to CBP are unknown, however. To define these residues, the KID (amino acids 102–160) of CREB 341 was randomly mutagenized using PCR.
A DNA fragment encoding the β-gal gene was fused to the C-terminal end of VP16–CREB (Fig. 4) to allow identification of clones that contain frame-shift and nonsense mutations. Colonies positive for β-gal are presumed to contain an ORF throughout the mutated region. To facilitate subcloning of mutated sequences, we created a cassette version of the CREB cDNA that contains BglII and SacII sites flanking the 5′ and 3′ ends of the KID, respectively. These modifications altered amino acid 168 from valine to alanine. Primers flanking the KID were used for mutagenic PCR under conditions optimized to achieve 1–3 mutations for the 177-bp region. The PCR products were introduced into pVP16–CREB(BglII–SacII)–LacZ in place of wild-type sequence and a library of mutations was transformed into yeast strain YI584 expressing LexA–CBD. Yeast transformants (n = 27,000) were screened, yielding about 5000 colonies capable of growing on selective media supplemented with 10 μg/ml Tc and 1 mM 3AT. The concentration of 3AT needed to suppress background growth on plates is higher than that needed for liquid culture (Fig. 3B). Control experiments (YI584 transformed with pLexA–CBD and pVP16–CREB(BglII-SacII)–LacZ) indicated that the background colony formation probably represents 5–9% of the transformants plated under these conditions.
Figure 4.

Mutagenesis screening strategy. PCR primers flanking the KID of CREB were used for mutagenesis. KID mutants were ligated into CREB (amino acids 1–283) flanked by the VP16 activation domain and LacZ. The resulting plasmids were transformed into YI584 and plated on selective medium lacking histidine supplemented with 10 μg/ml Tc and 1 mM 3AT. β-Gal assays identified full-length proteins. pVP16–CREB(BglII–SacII)–LacZ mutants were isolated from growth(+) β-gal(+) colonies and rescreened in the split- and two-hybrid systems. Positive mutants were sequenced.
Two additional screening steps were performed to eliminate uninformative mutations and false positives. First, filter β-gal assays were performed on the 5000 growth(+) colonies to eliminate proteins with frame-shift and nonsense mutations. Five hundred thirty-six colonies developed a dark blue color, whereas 412 colonies remained white. The rest of the colonies developed a pale blue color. The white colonies express mutants that contain frame-shift or nonsense mutations. The pale-blue colonies may represent unstable LacZ fusion proteins, and these were not analyzed further. CREB cDNA from 193 dark blue colonies were isolated. These cDNAs were separately retransformed along with LexA–CBD into the split-hybrid strain as well as into the two-hybrid L40 strain (16). These steps eliminated false positives that arise from background colony growth (see above) and confirmed that the mutant CREB protein did not interact with CBP. Among the 193 cDNAs rescreened, 152 did not interact with CBP in the yeast two-hybrid system, 15 interacted weakly, and 26 interacted like wild-type CREB.
Seventy of the 152 CREB mutants were found to contain single amino acid changes, 64 contained two amino acid residue mutations, and 13 contained more than two mutations. Mutants containing more than one amino acid alteration were not analyzed further. The expression levels of mutant proteins with one amino acid change were determined by Western analysis to be identical to that of wild-type VP16–CREB(BglII–SacII)–LacZ. Immunofluorescence assays demonstrated that the CREB mutants were localized in the nucleus (data not shown).
The distribution of CREB mutations identified in the split-hybrid screen centered around the phosphorylation site at Ser-133 (Fig. 5). No disrupting mutations occurred outside of the region between amino acids 130–141. Most of the mutations abrogated the PKA phosphorylation motif, but others were found at Ile-137, Leu-138, and Leu-141. The ability of the split-hybrid system to detect only a limited number of CREB mutants, many of which have been proposed previously to disrupt CREB association with CBP (18), indicates the specificity of the split-hybrid system.
Figure 5.
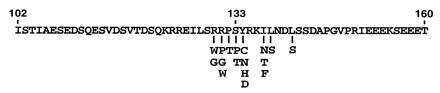
Summary of CREB mutations that disrupt the interaction with CBP. The amino acid residues of the KID are illustrated. Mutants containing single amino acid changes are indicated.
DISCUSSION
In the present study, we describe a genetic system for identifying mutations and/or factors that disrupt the association of two interacting proteins. Because the yeast split-hybrid is a positive selection, it is more efficient than the conventional yeast two-hybrid system for screening a large number of potentially disrupting mutants. The split-hybrid system should be valuable in characterizing interaction domains, as well as in generating dominant negative (19) and compensatory mutants.
The ability to manipulate the system with Tc is particularly useful when the DNA binding component has intrinsic activation properties, as in the case of LexA–CBD. The yeast split-hybrid system can also be modulated with 3AT, an advantage when the interaction between two proteins is relatively weak and incapable of producing sufficient TetR to fully prevent HIS3 gene expression. Furthermore, 3AT decreases background colony formation (Fig. 3B), which is crucial for library screens.
To test the utility of the split-hybrid system, we have identified CREB mutations that disrupt the CREB–CBP interaction. The majority of disrupting mutations occurred in the CREB PKA phosphorylation motif, which is consistent with the observation that nonphosphorylated CREB and CBP do not interact (7). The most common PKA motif is RRX(S/T)X but RX(S/T)X and KRXX(S/T)X are also encountered (20). The arginine residues in this motif are critical for electrostatic interactions with acidic amino acid residues in the catalytic subunit of PKA (21). Thus, as anticipated, several CREB mutations were found at Arg-130 and Arg-131 (Fig. 5). The mutations that occurred at amino acids Pro-132 and Tyr-134 were less expected. It is likely that the mutations at these residues adversely affect the structure of the phosphorylation motif, although these positions are generally thought to be more accommodating. Although they are not part of the “classical” consensus motif, hydrophobic amino acids are commonly found C terminal to PKA sites (22). Further studies are needed to determine whether mutations at Pro-132 and Tyr-134 disrupt phosphorylation of Ser-133 or disrupt the binding of CREB to CBP directly.
Interestingly, substitution of Ser-133 with threonine also prevents the interaction of CREB and CBP. PKA substrates containing a phosphorylatable threonine residue exist in nature (i.e., protein phosphatase inhibitor 1 and myelin basic protein), although they are relatively uncommon (23). Synthetic peptides containing serine to threonine substitutions are poor substrates for PKA phosphorylation (23). Whether the Thr-133 mutation disrupts the CREB–CBP interaction or simply fails to become phosphorylated is unclear. It should also be noted that CREB can be phosphorylated at Ser-133 by a variety of protein kinases besides PKA, including calcium/calmodulin-dependent protein kinase II and IV, protein kinase C, and a nerve growth factor-activated CREB kinase (24, 25, 26, 27). It is not known which, if any, of these particular protein kinases are responsible for the phosphorylation of CREB Ser-133 in yeast. The requirement for the integrity of the entire RRXSX sequence suggests that PKA is a reasonable candidate.
The second category of mutations are adjacent to the PKA phosphorylation motif. Ile-137 and Leu-138 have previously been suggested to be important for hydrophobic interactions with CBP (18). While this is a possibility, it should be noted that most of the mutations at position 137 and 138 converted these hydrophobic residues to polar amino acids that could affect protein folding. The mutation at position 141 also substitutes a polar residue for the hydrophobic leucine. Substitution of Ile-137 with the hydrophobic phenylalanine residue disrupted the interaction between CREB and CBP as well. These results could be due to an effect on folding (because of the considerably larger size of phenylalanine) or could indicate some specificity in the proposed hydrophobic interactions. Structural studies will be required to determine definitively how these mutations affect binding.
Perhaps most surprising was the finding that critical mutations were restricted to such a small portion of the KID. The relatively low affinity of phosphoCREB and CBP, determined to be between 250–400 nM by fluorescence anisotropy measurements (7), is consistent with a fairly limited protein interaction domain, however. The capability of the split-hybrid system to identify a limited number of CREB mutants suggests that the system is highly specific, and thus, should be useful in screening for mutations that disrupt other interacting pairs of proteins.
In addition to mutagenesis screening, a potential use of the split-hybrid system might be in screening for factors that abrogate protein associations, including kinases, phosphatases, proteases, competitive inhibitors, and novel factors that recognize the structural environment provided by particular protein–protein interactions. The split-hybrid system should also permit the screening of drug compounds and derivatives that disrupt clinically important protein–protein interactions.
Acknowledgments
We thank V. K. Tran for assistance with DNA sequencing and W. J. Wolfgang with help with the immunofluorescence assays. This work was supported by National Institutes of Health Grants DK45423, (R.H.G.) and DK09396 (P.S.G).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: TetR, tet-repressor; Tc, tetracycline; 3AT, 3-aminotriazole; SRF, serum response factor; PKA, cAMP-dependent protein kinase A; KID, kinase-inducible domain; CBD, CREB-binding domain; CBP, CREB binding protein; β-gal, β-galactosidase.
References
- 1.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 2.Hillen W, Berens C. Annu Rev Microbiol. 1994;48:345–369. doi: 10.1146/annurev.mi.48.100194.002021. [DOI] [PubMed] [Google Scholar]
- 3.Fujii M, Sassone-Corsi P, Verma I M. Proc Natl Acad Sci USA. 1988;85:8526–8530. doi: 10.1073/pnas.85.22.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujii M, Tsuchiya H, Chujoh T, Akizawa T, Seiki M. Genes Dev. 1992;6:2066–2076. doi: 10.1101/gad.6.11.2066. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki T, Hirai H, Fujisawa J-i, Fujita T, Yoshida M. Oncogene. 1993;8:2391–2397. [PubMed] [Google Scholar]
- 6.Chrivia J C, Kwok R P S, Lamb N, Hagiwara M, Montminy M R, Goodman R H. Nature (London) 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 7.Kwok R P S, Lunblad J R, Chrivia J C, Richards J P, Bächinger H P, Brennan R G, Roberts S G E, Green M R, Goodman R H. Nature (London) 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 8.Bartel P L, Chien C, Sternglanz R, Fields S. In: Cellular Interactions in Development: A Practical Approach. Hartley D A, editor. Oxford: IRL; 1993. pp. 153–179. [Google Scholar]
- 9.Hollenberg S M, Sternglanz R, Cheng P F, Weintraub H. Mol Cell Biol. 1995;15:3813–3822. doi: 10.1128/mcb.15.7.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwok R P S, Laurance M E, Lundblad J R, Goldman P S, Shih H-m, Conner L M, Marriott S J, Goodman R H. Nature (London) 1996;380:642–646. doi: 10.1038/380642a0. [DOI] [PubMed] [Google Scholar]
- 11.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherman F. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 13.Sikorski R S, Boeke J D. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 14.Himmelfarb H J, Pearlberg J, Last D H, Ptashne M. Cell. 1990;63:1299–1309. doi: 10.1016/0092-8674(90)90425-e. [DOI] [PubMed] [Google Scholar]
- 15.Uppaluri R, Towle H C. Mol Cell Biol. 1995;15:1499–1512. doi: 10.1128/mcb.15.3.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vojtek A B, Hollenberg S M, Cooper J A. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 17.Hall M N, Hereford L, Herskowitz I. Cell. 1984;36:1057–1065. doi: 10.1016/0092-8674(84)90055-2. [DOI] [PubMed] [Google Scholar]
- 18.Parker D, Ferreri K, Nakajima T, LaMorte V J, Evans R, Koerber S C, Hoeger C, Montminy M R. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brachmann R K, Videl M, Boeke J D. Proc Natl Acad Sci USA. 1996;93:4091–4095. doi: 10.1073/pnas.93.9.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kemp B E, Pearson R B. Trends Biochem Sci. 1990;15:342–346. doi: 10.1016/0968-0004(90)90073-k. [DOI] [PubMed] [Google Scholar]
- 21.Knighton D R, Zheng J, Ten Eyck L F, Xuong N-h, Taylor S A, Sowadski J M. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 22.Kemp B E, Parker M W, Hu S, Tiganis T, House C. Trends Biochem Sci. 1994;19:440–444. doi: 10.1016/0968-0004(94)90126-0. [DOI] [PubMed] [Google Scholar]
- 23.Zetterqvist O, Ragnarsson V, Engstrom L. In: Peptides and Protein Phosphorylation. Kemp B E, editor. Boca Raton, FL: CRC; 1990. pp. 172–187. [Google Scholar]
- 24.Sheng M, McFadden G, Greenburg M E. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 25.Sheng M, Thompson M A, Greenburg M E. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 26.Xie H, Rothstein T L. J Immunol. 1995;154:1717–1723. [PubMed] [Google Scholar]
- 27.Ginty D D, Bonni A, Greenburg M E. Cell. 1994;77:1–20. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]