

Abstract
Current models of telomere replication predict that due to the properties of the polymerases implicated in semiconservative replication of linear DNA, the two daughter molecules have one end that is blunt and one end with a short 3′ overhang. Telomerase is thought to extend the short 3′ overhang to produce long single-stranded overhangs. Recently, such overhangs, or TG1–3 tails, were shown to occur on both telomeres of replicated linear plasmids in yeast. Moreover, indirect evidence suggested that the TG1–3 tails also occurred in a yeast strain lacking telomerase. We report herein a novel in-gel hybridization technique to probe telomeres for single-stranded DNA. Using this method, it is shown directly that in yeast strains lacking the TLC1 gene encoding the yeast telomerase RNA, TG1–3 single-stranded DNA was generated on chromosomal and plasmid telomeres. The single-stranded DNA only appeared in S phase and was sensitive to digestion with a single-strand-specific exonuclease. These data demonstrate that during replication of telomeres, TG1–3 tails can be generated in a way that is independent of telomerase-mediated strand elongation. In wild-type strains, these TG1–3 tails could subsequently serve as substrates for telomerase and telomere binding proteins on all telomeres.
Keywords: telomere replication, telomerase RNA, yeast
Telomeric DNA of most eukaryotic organisms consists of short tandemly repeated sequences (1, 2). Due to the nature of the repeats, one strand is usually rich in guanines (G-rich strand) and this strand always runs 5′ → 3′ toward the end of the chromosomes (2). The actual amount of double-stranded DNA made up of these repeats varies considerably between different organisms and between different telomeres in the same organism. For instance, in the yeast Saccharomyces cerevisiae the telomeric repeats can be described as C1–3A/TG1–3 sequences and there are 250–350 bp of these repeats on each telomere (3). The physical structure of the very ends of the chromosomes is only known for some protozoans and there consists in a short 10–16 base overhang of the G-rich strand (4, 5, 6). Proteins that bind specifically to this terminal structure have been identified in Oxytricha and related ciliates (7, 8, 9). A similar activity has been detected in extracts of Xenopus eggs (10). Based on these results, it has been proposed that a short overhang of the G-rich strand is a general feature of eukaryotic telomeres (6) and that this structure is bound by terminus- and structure-specific telomere-binding proteins (2, 11).
Telomeres are essential for chromosome integrity in at least two ways: they function as a protection against random fusion events and degradation (12, 13) and they are involved in the completion of the duplication of chromosomal DNA (14). All conventional DNA polymerases require a primer and a template and synthesize DNA in the 5′ → 3′ direction. Given these properties, conventional replication is expected to leave short primer-sized gaps on the 5′ end of the strands that were generated by lagging-strand synthesis (2, 11). Upon successive divisions and in the absence of compensating mechanisms, chromosomal termini are expected to gradually lose terminal sequences until telomere functions are lost, chromosomes become unstable, and the cells die (see, for example, refs. 15, 16, 17). However, an enzymatic activity called telomerase can elongate the short single-stranded 3′ overhangs in a DNA-template-independent manner (ref. 18, and for a review, see ref. 19). The enzyme uses a sequence within its associated RNA as the template for telomeric repeat addition (16, 20). Such long, single-stranded, and G-rich tails (hereafter called TG1–3 tails) are present on yeast telomeres in late S phase, when conventional replication has essentially been completed (21, 22). Fill-in synthesis on TG1–3 tails would complete the replication of telomeres, preventing the loss of sequences and reestablishing short 3′ overhangs.
The telomeres on which the newly synthesized strand is made by leading-strand synthesis are thought to become blunt-ended after conventional replication is complete (2, 11). However, since the terminal structure of the telomeres is expected to be uniform and terminus binding proteins may require a short 3′ overhang, this blunt end would have to be processed into an end with a short 3′ overhang (11). At least in vitro, telomerase requires a single-stranded 3′ end for strand elongation (11, 23). It is not clear whether in vivo, a blunt-ended double-stranded molecule could serve as a substrate for a telomerase holoenzyme and whether this enzyme could generate TG1–3 tails on ends replicated by leading-strand synthesis (24). As alternatives, this conversion could be achieved by telomerase-independent activities such as exonucleases or helicases (24, 25).
Recently, the gene encoding the yeast telomerase RNA TLC1 was cloned (16). Cells lacking this RNA progressively lose telomeric sequences from their telomeres and die after 80–100 generations (16). Introduction of a restriction site by in vitro mutagenesis of the templating region in the TLC1 gene resulted in the incorporation of the restriction site into newly synthesized chromosomal telomeres (16). Moreover, extracts prepared from tlc1::LEU2 yeast strains lack detectable telomerase activity (26). These results demonstrate that the TLC1-encoded RNA is an essential component for yeast telomerase-mediated TG1–3 synthesis in vivo.
The existence of yeast strains lacking telomerase activity provides a unique opportunity to address of the role of telomerase in the formation of telomere replication intermediates in vivo. One of the major predictions of the current models of telomere replication is that the long TG1–3 tails observed at the end of S phase are generated by the yeast telomerase. Using a novel nondenaturing in-gel hybridization technique, we report herein that TG1–3 single-stranded DNA was detected on plasmid and chromosomal telomeres of cells that lacked the TLC1 gene. This single-stranded DNA was sensitive to digestion with a single-strand-specific exonuclease and, therefore, constituted terminal TG1–3 tails. As reported for TLC1+ cells (22), the TG1–3 tails only appeared in S phase and, thus, seem to be a product of a cell cycle-regulated activity. These results demonstrate directly that in yeast, TG1–3 tails can be generated by an activity other than the strand elongating activity of telomerase. We propose that the TG1–3 tails detected on the telomeres at the end of S phase are mostly generated by an exonuclease activity. Such an activity could also explain how a short 3′ overhang is recreated on all yeast telomeres.
MATERIALS AND METHODS
Plasmids and Strains.
The plasmids used in this study were: pAZ1 (27), which is a pRS316 (28) derivative containing genomic copies of the URA3 and TLC1 genes. pBlue61::LEU2 (16) is a plasmid in which most of the genomic sequences of the TLC1 gene were replaced by the LEU2 gene (both plasmids kindly provided by M. Singer and D. Gottschling, University of Chicago). The 7.5-kb yeast linear plasmid YLpFAT10 was derived from YEpFAT10 as described (22) and contains a genomic copy of the TRP1 gene and the 2-μm DNA origin of replication. Single-stranded phagemids pCA75 and pGT75 contain opposite strands of the same 72 bases of yeast telomeric sequences and were produced as described (22). Plasmid pRW75 was obtained by inserting the DNA fragment containing the yeast telomeric repeats from pCA75 into the unique EcoRV site of pRS303 (28) and pVY′K consists of pVZ1 (29) into which a 0.6-kb KpnI fragment from YRp131b (30) was inserted. Yeast transformations, genetic manipulations, and cell propagation were done according to standard techniques (31, 32).
Yeast strains analyzed were RWY12 and RWY14. RWY12 (Mata, ura3-52, lys2-801, ade2-101, his3-Δ200, trp1-Δ1, leu2-Δ1, DIA5-1, tlc1::LEU2) was a haploid derivative of UCC3535 (Mata/Matα, ura3-52/ura3-52, lys2-801/lys2-801, ade2-101/ade2-101, his3-Δ200/his3Δ200, trp1-Δ1/trp1-Δ1, leu2-Δ1/leu2-Δ1, DIA5-1/DIA5-1, TLC1/tlc1::LEU2) (obtained from M. Singer and D. Gottschling, ref. 24). UCC3535 was transformed with the plasmid pAZ1 and sporulated, and Leu+/Ura+ spores were selected. The linear plasmid YLpFAT10 was introduced as described (22). Spores that exhibited a cell death phenotype after 80–100 generations of growth without pAZ1 were selected for study. Strain RWY14 was obtained by deleting genomic TLC1 sequences from strain AR120 (Mata, cdc7, bar1, ura3-52, his6, trp1-289, leu2-3, 112, HMLa, HMRa) (33), using the insert of pBleu61::LEU2 in the one-step gene displacement technique as described (16). Gene disruption was confirmed by Southern blot analysis and the cell death assay after 80–100 generations (data not shown). For long-term viability of the strain, cells were transformed with pAZ1.
Cell Synchronization and DNA Isolation.
RWY14 cells containing pAZ1 were first plated onto YC(FOA) plates (34) to select for cells that had lost the wild-type allele of the TLC1 gene. These cells were then grown in synthetic complete medium and synchronized using two consecutive blocks (α-factor and cdc7), as described (21, 22, 35). Total genomic DNA from these cells was isolated using a modified glass bead procedure (22, 36). Treatment of genomic DNA with Escherichia coli exonuclease I was carried out as described (22) with the inclusion of M13mp18 single-stranded DNA (Pharmacia) as internal control. For the analysis of plasmid telomeres in strain RWY12, cells containing both YLpFAT10 and pAZ1 were plated onto YC(FOA)–Trp plates and then grown in YC–Trp media to midlogarithmic phase (OD660 = 0.6 unit). Low molecular weight DNA was isolated from these cells using a Hirt extraction adapted for yeast cells (37).
Southern Blot Analysis and In-Gel Hybridization.
Agarose gel techniques, Southern blot transfer to nylon membranes, and hybridization conditions were as described (22). For the nondenaturing in-gel hybridizations, a technique described in Counter et al. (38) was modified in the following way: appropriately digested DNA samples were loaded onto 0.75% agarose gels and DNA fragments were separated by electrophoresis for 16 hr at 0.8 V/cm. The DNA was then stained with ethidium bromide for photography. The gels were immersed for 30 min in 2× SSC (0.3 M NaCl/0.03 M sodium citrate, pH 7) at 20°C and then mounted on a Bio-Rad (model 583) gel drier. Drying was carried out for 25–30 min at 20°C. The very thin gels were then placed in sealable plastic bags and hybridized for 16 hr at 37°C to end-labeled oligonucleotides using the hybridization buffer as described (38). After removal of excess hybridization buffer, gels were placed in 0.25× SSC and washed once for 30 min at 20°C, followed by two 1-hr washes at 30°C. Pilot experiments showed that washes in 0.25× SSC at temperatures higher than 32°C dissociate the probe (data not shown). Gels were then exposed to Kodak XAR5 x-ray films for appropriate amounts of time. The oligonucleotide used as probe is a 22-mer of the sequence 5′-CCCACCACACACACCCACACCC-3′ and is referred to as the CA-oligo. Note that if there was single-stranded telomeric DNA of the G-rich strand shorter than the probe CA-oligo (less than 22 nt), the washing conditions used would not allow retention of the probe. For the Y′-specific probe, an in vitro transcription system using T3 RNA polymerase and plasmid pVY′K in the presence of α-32P-labeled CTP was used (39).
RESULTS
An In-Gel Hybridization Technique to Detect Single-Stranded DNA.
Using a nondenaturing Southern blot technique, it was (21, 22) that long 3′ extensions, or TG1–3 tails, appear on yeast telomeres at the end of S phase, when conventional replication is complete. A serious limitation of the previously used technique is the inability to analyze terminal restriction fragments that are larger than 4 kb (40). To resolve this problem, an in-gel hybridization technique using an end-labeled oligonucleotide as probe was adapted from established protocols (38). When phagemid single-stranded DNA derived from pGT75 containing 72 bases of yeast telomeric repeats of the G-rich strand was analyzed with this technique, 0.4 ng of DNA was readily detected (Fig. 1A, lane 2). A linearized plasmid containing the same repeats in double-stranded form did not yield any signal in this gel (Fig. 1A, lanes 3 and 4), even when ≈30 ng was analyzed. However, if the linearized plasmid pMW75 was denatured by heating prior to loading, the DNA was detected (Fig. 1A, lanes 5 and 6). As control, an identical gel as shown in Fig. 1A was hybridized to the probe using the same in-gel hybridization protocol except that the DNA was denatured in the gel prior to hybridization (Fig. 1B). In this gel, the same signals as in the nondenaturing gel were observed for lanes 1, 2, 5, and 6, as expected. In addition and due to the denaturing step, the linearized plasmid pMW75 DNA was now detected (Fig. 1B, lanes 3 and 4). These data show that this in-gel hybridization technique allows highly specific detection of single-stranded DNA with very little background hybridization to a double-stranded DNA of the same sequence. In addition, the technique has no size restrictions for the restriction fragment analyzed. For example, single-stranded DNA extensions on terminal restriction fragments of up to 20 kb and derived from vertebrate telomeres were detectable with high specificity with this technique (R. McElligott and R.J.W., unpublished results).
Figure 1.
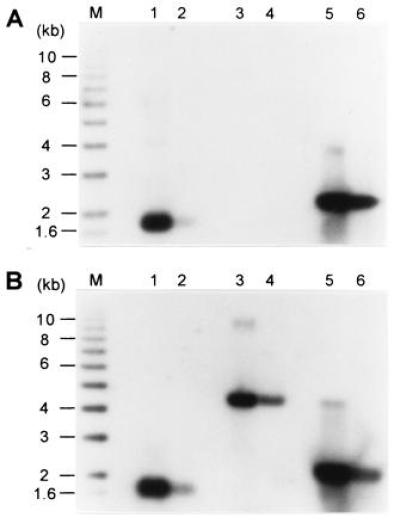
Nondenaturing in-gel hybridization technique to detect single-stranded TG1–3 DNA. In both gels, lanes 1 and 2 contained, respectively, 4 ng and 0.4 ng of single-stranded phagemid DNA derived from pGT75; lanes 3 and 4 contained, respectively, 30 ng and 3 ng of linearized double-stranded pMW75 DNA; and lanes 5 and 6 contained, respectively, 30 ng and 3 ng of linearized pMW75 DNA that was heat-denatured for 10 min at 95°C prior to loading. M denotes end-labeled ladder DNA. The gels were treated for in-gel hybridization. The gel in A was treated as a nondenaturing gel, whereas the gel in B was denatured in the gel prior to hybridization to the end-labeled CA-oligo probe.
Single-Stranded DNA of the G-Rich Strand Occurs on Telomeres in a Yeast Strain That Lacks TLC1.
We next wished to determine if the generation of TG1–3 tails observed on yeast telomeres was dependent on the presence of the yeast telomerase. First and to facilitate the analysis, the telomeres of the high copy linear plasmid YLpFAT10 (22) were examined in strain RWY12, lacking the TLC1 gene (tlc1::LEU2, hereafter called tlc1Δ). Low molecular weight DNA was isolated from these cells using a Hirt procedure and analyzed using the nondenaturing in-gel hybridization technique as described above (Fig. 2). As control, DNA was isolated from the same strain carrying the complementing plasmid pAZ1. A signal for single-stranded TG1–3 DNA was observed for the full-length plasmid and for both terminal restriction fragments irrespective of whether the cells were TLC1+ or tlc1Δ (Fig. 2B). Moreover, the signals obtained for the two strains did not differ significantly. Several lines of evidence argue that telomerase was absent in these cells. (i) At the time of DNA isolation, these cells had been grown for ≥35 generations after plasmid loss. An absence of telomerase should lead to incomplete telomere replication and progressive loss of terminal sequences (16, 41). Indeed, the plasmid telomeres derived from the tlc1Δ strain were considerably shorter than the telomeres in the strain containing a wild-type copy of the TLC1 gene (Fig. 2B). The difference in fragment migration was not due to an artefactual behavior of the DNA since fragments derived from highly repetitive DNA and visible by ethidium bromide staining migrated to the same position, irrespective of whether the DNA was isolated from TLC1+ or tlc1Δ cells (Fig. 2A). (ii) The tlc1Δ cells from which the DNA analyzed in Fig. 2 was isolated died after ≈55 generations of further outgrowth (data not shown). The shortened telomeres and the massive cell death phenotype after further outgrowth confirmed phenotypically that the cells had completely lost telomerase activity. (iii) In cell extracts prepared from analogous tlc1Δ cells, no telomerase activity has been detected in vitro (26). Thus, this evidence strongly suggests that there was no TLC1-supported telomerase activity left in the cells at the time of analysis.
Figure 2.
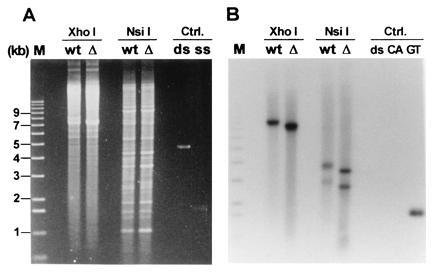
Analysis of the telomeres of the ≈7.5-kb linear plasmid YLpFAT10. Low molecular weight DNA was extracted via a Hirt procedure from TLC1+ (lanes marked wt) and tlc1Δ (lanes marked Δ) cells. The DNA was digested with the indicated enzymes and analyzed on a 0.75% agarose gel. There are no recognition sites for XhoI on YLpFAT10, leaving the plasmid intact in those lanes. There are two recognition sites for NsiI, yielding three fragments for YLpFAT10: the two terminal fragments are ≈2.5 kb and ≈3.5 kb and the internal fragment is ≈1.5 kb (22). (A) The gel stained with ethidium bromide. (B) An autoradiograph of the same gel after hybridization to the end-labeled CA-oligo. Lane M contains end-labeled molecular weight marker DNA. The DNAs loaded under the heading Ctrl (Controls) were as follows: ds, linearized double-stranded pMW75 DNA; CA and ss, single-stranded phagemid DNA derived from pCA75; GT, single-stranded phagemid DNA derived from pGT75.
On this gel, signals for chromosomal telomeres were not observed. This is most likely due to the DNA isolation procedure used since a Hirt extraction preferentially yields low molecular weight DNA and to the fact that the DNA was obtained from nonsynchronized cells (see below for the analysis of chromosomal telomeres).
The results thus indicate that in cells that are deficient for telomerase RNA and that experience telomere attrition, single-stranded TG1–3 DNA was detected on the terminal restriction fragments of a high copy linear plasmid. Signals of about equal intensity were obtained for the TLC1 and the tlc1Δ strains, suggesting that about the same amount of single-stranded DNA was generated in the two strains (Fig. 2B).
In wild-type strains, the single-stranded TG1–3 DNA appears in a cell cycle-regulated manner, namely, at the end of S phase (22). To examine whether the single-stranded DNA observed on plasmid telomeres in tlc1Δ cells also occurred on chromosomal telomeres and with the same temporal regulation as observed for wild-type strains, the TLC1 gene was replaced by the LEU2 gene in the yeast strain AR120 (33), yielding RWY14. These cells were synchronized using two consecutive blocks (α-factor and cdc7) and released into a synchronous S phase by temperature down-shift as described (22, 35). Total DNA derived from cells at different time points was then digested with XhoI and analyzed as described above (Fig. 3A). Due to the presence of conserved middle-repetitive Y′ elements that are found proximal to the telomeric repeats on the majority of yeast chromosomal telomeres, most terminal XhoI restriction fragments are ≈1.3 kb in size (42). Virtually no signal was observed on DNA derived from cells arrested in G1 phase or from cells that were released for 90 min into the synchronous cell cycle (Fig. 3A), reflecting G2/M phase (35). However, single-stranded TG1–3 DNA was detected on telomere-specific restriction fragments derived from cells that were released for 25 min, and the strongest signal was obtained with DNA isolated from cells released for 35 min into S phase (Fig. 3A). This hybridization was not due to variability of DNA loading since the same DNA samples loaded on a parallel gel yielded equivalent signals when examined by conventional Southern blot analysis (Fig. 3B). Note that since these cells have been outgrown in the absence of the TLC1 gene for ≈40 generations, the terminal XhoI restriction fragments derived from Y′ telomeres were again considerably shorter than the expected ≈1.3 kb and migrated to ≈1.1 kb (Fig. 3). The same nondenaturing in gel analysis using an oligonucleotide of the opposite strand as a probe did not yield any signal (data not shown). These data demonstrate that in tlc1Δ cells, single-stranded TG1–3 DNA appeared in a cell cycle-regulated manner on chromosomal telomeres.
Figure 3.
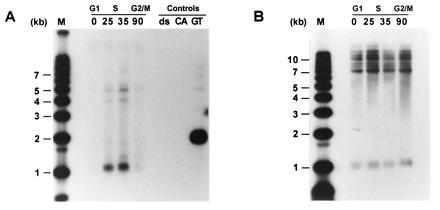
Single-stranded TG1–3 DNA appears in a cell cycle-controlled manner on chromosomal telomeres of tlc1Δ cells. Total DNA was isolated from yeast strain RWY14 (tlc1::LEU2) at the following time points: 0, cells at 37°C were arrested at START due to the temperature-sensitive cdc7 gene; 25, 35, and 90, cells were released into a synchronous S phase for 25, 35, or 90 min, respectively. The control DNA loadings (Controls) were as in Fig. 2B. M, molecular weight standards. Above the actual time points, the corresponding approximate cell cycle phases are indicated. The DNAs were digested with the restriction enzyme XhoI and separated on a 0.7% agarose gel. The gel in A was treated as a nondenaturing gel and hybridized to the end-labeled CA-oligo. The gel in B was identical to the gel in A except that this gel was then treated as a regular Southern blot (i.e., the DNA was denatured in the gel) using a Y′-specific probe.
The Single-Stranded TG1–3 DNA on the Telomeres of tlc1Δ Strains Are Terminal TG1–3 Tails.
It was possible that in the tlc1Δ cells, telomeric DNA metabolism was disturbed in a way that would lead to exposure of internal gaps of the C-rich strand and that the signals observed in the in-gel hybridizations did not detect terminal TG1–3 tails. E. coli exonuclease I is a single-strand-specific DNA exonuclease that degrades DNA from the 3′ end (43). This enzyme was used previously to demonstrate that the signals detecting TG1–3 DNA on telomeres of wild-type strains correspond to TG1–3 tails and not internal gaps of the C-rich strand (22). The same experimental protocol was thus applied to DNA derived from tlc1Δ cells. Using conditions in which a mixed-in single-stranded circular DNA (M13mp18 phage DNA) was not digested by the enzyme, the signal for the TG1–3 DNA disappeared from Y′ and non-Y′ telomeres (Fig. 4A, lane Exo+). Due to some DNA loss in the lower portion of this particular gel during drying, the signal corresponding to Y′ telomeres at ≈1.1 kb is underrepresented (compare the signal at ≈1.1 kb to the signals of the non-Y′ telomeres in Fig. 4B). Therefore, two different exposures of the same two lanes are shown; the longer exposure showing that the signal for the Y′ telomeres completely disappeared after exonuclease treatment (Fig. 4A Right). After the appropriate exposures of the gel were obtained, the DNA in the gel was denatured and the same gel was rehybridized to the CA-oligo. Fragments containing telomeric repeats remained detectable, whether or not the DNA was treated with the exonuclease (Fig. 4B). The commercially available preparations of the exonuclease sometimes contained contaminating single-strand endonuclease activities. However, the preparations we used displayed the expected nuclease specificities with no contaminating activities. Single-stranded circular DNAs (M13mp18 DNA) and a linear double-stranded DNA fragments were insensitive to the exonuclease treatment even after prolonged incubations, while linear single-stranded DNA was rapidly degraded by the enzyme used in our assays (Fig. 4C). These results indicate that the single-stranded DNA detected with the DNA derived from tlc1Δ cells corresponded to TG1–3 single-stranded tails.
Figure 4.
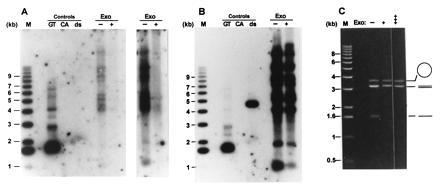
The single-stranded TG1–3 DNAs detected on telomeres of tlc1Δ cells are TG1–3 tails. Total yeast DNA isolated from RWY14 cells was incubated either without (−) or with (+) E. coli exonuclease I for 10 min at 37°C. The DNAs were then precipitated and digested with the restriction endonuclease XhoI. The DNA loadings under Controls are as in Fig. 2. M, molecular weight standards. The gel in A was treated as a nondenaturing gel and probed with the CA-oligo. Two exposures of the lanes containing the genomic DNAs are shown to emphasize the absence of detectable bands in the lane with the exonuclease-treated DNA. After appropriate exposures of the gel in A were obtained, the DNA in the gel was denatured in 0.5 M NaOH and rehybridized to the CA-oligo. (B) An autoradiogram of the rehybridized gel. Note that the double-stranded control DNA now is detected and that the pattern of TG1–3 repeat containing bands is the same in the exonuclease-treated and untreated DNA. (C) The exonuclease used in this experiment displays the expected specificity. Single-stranded circular DNA (M13mp18 phage DNA, open circle), linearized double-stranded pVZ1 DNA (3.2 kb, double lines), and linear single-stranded DNA (heat-denatured pVZ1 DNA, single line) were mixed and incubated in the absence of the exonuclease (−) or in the presence of exonuclease for 10 min (+) or 45 min (+++). The DNAs were then separated on a 0.6% agarose gel and stained with ethidium bromide. Note that the linear single-stranded DNA was completely degraded after 10 min of incubation, while the amount of circular single-stranded M13 DNA remained virtually unchanged even after 45 min of incubation.
DISCUSSION
When considering models for the replication of the eukaryotic telomeric repeat DNA, at least two requirements must be met. (i) The possible gradual losses of telomeric repeats that would occur due to the properties of the conventional replication machinery must be prevented (14). (ii) If terminus-binding proteins require a DNA structure that consists of a short 3′ overhang, such a structure must somehow be recreated on all telomeres (11, 25).
We have recently shown that short linear plasmids can acquire TG1–3 tails on both ends of individual replicated daughter molecules (24). Furthermore, these TG1–3 tails support telomere–telomere interactions via noncanonical base interactions, leading to circular forms of the plasmid (21, 22). Such circular forms were also shown to be present in DNA derived from tlc1Δ cells, providing indirect evidence that even in telomerase-deficient strains, TG1–3 tails are generated at both ends of a linear plasmid (24).
Using a novel nondenaturing in-gel hybridization technique, we show herein directly that in the absence of the TLC1 RNA, single-stranded TG1–3 DNA was detectable on plasmid and chromosomal telomeres (Figs. 2 and 3). The specific disappearance of the signal for single-stranded TG1–3 DNA after treatment of the DNA with exonuclease I of E. coli strongly suggests that the detected TG1–3 DNA formed a single-stranded TG1–3 tail (Fig. 4). Moreover, these TG1–3 tails appeared in a cell cycle-regulated manner: TG1–3 tails were not detected on telomeres when the cells were in G1 or G2/M phases of the cell cycle (Fig. 3A). The probe for our in-gel hybridization experiments was 22 nt long and, to obtain specific signals, the most stringent washing conditions were used. Thus, even though no signal was detected in G1- and G2/M-phase samples, short overhangs of <20 bases may have escaped detection. However, single-stranded DNA was detected 25 min after the release of the cells into S phase and the signal was strongest 35 min after the release (Fig. 3A). When analogous protocols for cell synchronization were used previously, S phase was shown to occur between ≈10 min and ≈40 min after the release (21, 44, 45). Our data thus show that the TG1–3 tails only appeared in S phase on telomeres in tlc1Δ cells. These results are virtually indistinguishable from those obtained when analyzing the cell cycle-regulated appearance of TG1–3 tails in TLC1+ cells: there the tails appear in very late S phase after conventional replication is virtually completed (21, 22).
Given that the tlc1Δ cells have no telomerase-mediated strand elongation activity, the overall lengths of the tails could be expected be shorter in these cells. However, the signals for the TG1–3 tails were very comparable in DNAs isolated from TLC1+ and tlc1Δ cells (Fig. 2). If there is a difference between the two strains, it is apparently not large enough to be detected by the in-gel hybridization technique. Thus, our data suggest that the appearance of TG1–3 tails on yeast telomeres is largely independent from an active elongation activity of the yeast telomerase. Rather, the blunt ends on the leading-strand ends and perhaps also the short overhangs on the lagging-strand ends may be converted into relatively long overhangs by a strand-specific exonuclease. Clearly, telomerase-mediated strand elongation is not essential for passage through the cell cycle (15, 16, 41). Moreover, at least in vitro, the yeast telomerase seems not to be very processive and may add only a few repeats to any given end (26). We thus hypothesize that the bulk of the TG1–3 tails are created by an exonuclease and that the active strand elongation by telomerase only contributes a minor, and in our assay, undetectable addition to those tails. Equally consistent with all the data is the possibility that telomerase-mediated strand elongation may not occur onto the TG1–3 tails generated in S phase (11). In this model, telomerase may or may not be cell cycle-regulated and add only as many telomeric repeats to the telomeres as are necessary to prevent sequence losses. Such very short extension products may well have been undetectable with the experimental approaches used so far and could, therefore, occur at any time in the cell cycle.
The average telomere length in the tlc1Δ cells was ≈150 bp at the time of analysis (Fig. 2). If indeed the length of TG1–3 tails detected in wild-type cells was not considerably different than that detected in the tlc1Δ cells, the length for the TG1–3 tails have an approximate upper limit of ≈150 bases. Furthermore, these same tlc1Δ cells died after ≈90 cell divisions (data not shown). These results demonstrate directly that in the tlc1Δ cells, telomere repeat tracts are shortening due to the absence of telomerase. It would require unreasonably high amounts (>3 × 1010 molecules) of the TLC1 RNA in the initial founder cell for some of these molecules to still be present in the tlc1Δ cell culture after 35 generations. Lastly, in cell-free extracts derived from tlc1Δ cells analogous to the ones analyzed herein, no telomerase activity was found (26) or inconclusive results were obtained (46). Thus, these considerations rule out that some residual TLC1 RNA molecules supporting a telomerase activity were still present in the tlc1Δ cells at the time of analysis.
The exonuclease activity responsible for the creation of the 3′ overhangs could also be involved in recombination and repair (47, 48, 49, 50). Another possibility is that the 5′ → 3′ exonuclease directly associated with the replication machinery accomplishes this function. The Rad27 protein (Rad27p) has been proposed to be the yeast homologue carrying this activity (51) and we are currently investigating whether TG1–3 tails are detected in rad27 cells. Alternatively, a telomere-specific exonuclease may be part of the telomerase holoenzyme complex. Because only the RNA component of the telomerase was missing in our experiments, it is possible that the remaining telomerase complex still possesses an exonuclease activity. Whether or not the reported yeast telomerase-associated nucleolytic activity (26) is the exonuclease predicted from our experiments remains to be clarified. Whatever the origin of the exonuclease, the activity is probably regulated by the Cdc13 protein (Cdc13p) (52). In cdc13 cells at the restrictive temperature, extensive single-stranded DNA containing all of the telomeric repeats and some subtelomeric DNA appears in telomeric areas (52). Moreover, the degraded strands seem to be preferentially the C-rich strands. Thus, by modulating C-strand degradation, this protein may play an important role in telomere maintenance (52).
Creating the proper terminal DNA structure for terminus-binding proteins on all telomeres may turn out to be an essential step in telomere maintenance (see above). The proposed implication of a strand-specific exonuclease to remove some of the telomeric C-strand during replication may serve two purposes: it creates substrates for telomerase and, ultimately, allows the formation of a proper DNA structure on all telomeres. This model accommodates both requirements for telomere repeat maintenance at the ends replicated by leading-strand synthesis and the ends replicated by lagging-strand synthesis. For cells outside of S phase, the model also predicts a uniform DNA structure on all yeast telomeres with a short 3′ overhang. While such an overhang was not detected in our assays, we are currently exploring new strategies to verify this prediction.
Acknowledgments
We thank M. Singer, D. Gottschling, R. Raghuraman, and W. Fangman for generous gifts of plasmids and yeast strains. B. Chabot is thanked for a critical reading of the manuscript. This research was supported by a grant of the Medical Research Council of Canada (Grant MT 12616). R.J.W. is a Chercheur-Boursier Junior II of the Fonds de la Recherche en Santē du Quēbec.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Blackburn E H. Nature (London) 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V A. Science. 1995;270:1601–1607. doi: 10.1126/science.270.5242.1601. [DOI] [PubMed] [Google Scholar]
- 3.Shampay J, Szostak J W, Blackburn E H. Nature (London) 1984;310:154–157. doi: 10.1038/310154a0. [DOI] [PubMed] [Google Scholar]
- 4.Klobutcher L A, Swanton M T, Donini P, Prescott D M. Proc Natl Acad Sci USA. 1981;78:3015–3019. doi: 10.1073/pnas.78.5.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pluta A F, Kaine B P, Spear B B. Nucleic Acids Res. 1982;10:8145–8154. doi: 10.1093/nar/10.24.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson E R, Blackburn E H. Mol Cell Biol. 1989;9:345–348. doi: 10.1128/mcb.9.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gottschling D E, Zakian V A. Cell. 1986;47:195–205. doi: 10.1016/0092-8674(86)90442-3. [DOI] [PubMed] [Google Scholar]
- 8.Price C M. Mol Cell Biol. 1990;10:3421–3431. doi: 10.1128/mcb.10.7.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng H, Hou Z, Schierer T, Dobbs D L, Henderson E. Mol Cell Biol. 1995;15:1144–1153. doi: 10.1128/mcb.15.3.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardenas M E, Bianchi A, de Lange T. Genes Dev. 1993;7:870–882. doi: 10.1101/gad.7.5.883. [DOI] [PubMed] [Google Scholar]
- 11.Lingner J, Cooper J P, Cech T R. Science. 1995;269:1533–1534. doi: 10.1126/science.7545310. [DOI] [PubMed] [Google Scholar]
- 12.McClintock B. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandell L S, Zakian V A. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- 14.Watson J D. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- 15.Lundblad V, Szostak J W. Cell. 1989;57:633–643. doi: 10.1016/0092-8674(89)90132-3. [DOI] [PubMed] [Google Scholar]
- 16.Singer M S, Gottschling D E. Science. 1994;266:404–409. doi: 10.1126/science.7545955. [DOI] [PubMed] [Google Scholar]
- 17.Biessmann H, Mason J M. EMBO J. 1988;7:1081–1086. doi: 10.1002/j.1460-2075.1988.tb02916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greider C W, Blackburn E H. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 19.Greider C W. In: Telomeres. Blackburn E H, Greider C W, editors. M. Plainview, NY: Cold Spring Harbor Lab. Press; 1995. onograph 293568. [Google Scholar]
- 20.Yu G-L, Bradley J D, Attardi L D, Blackburn E H. Nature (London) 1990;344:126–132. doi: 10.1038/344126a0. [DOI] [PubMed] [Google Scholar]
- 21.Wellinger R J, Wolf A J, Zakian V A. Mol Cell Biol. 1993;13:4057–4065. doi: 10.1128/mcb.13.7.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wellinger R J, Wolf A J, Zakian V A. Cell. 1993;72:51–60. doi: 10.1016/0092-8674(93)90049-v. [DOI] [PubMed] [Google Scholar]
- 23.Blackburn E H. Annu Rev Biochem. 1992;61:113–129. doi: 10.1146/annurev.bi.61.070192.000553. [DOI] [PubMed] [Google Scholar]
- 24.Wellinger R J, Ethier K, Labrecque P, Zakian V A. Cell. 1996;85:423–433. doi: 10.1016/s0092-8674(00)81120-4. [DOI] [PubMed] [Google Scholar]
- 25.Zahler A M, Prescott D M. Nucleic Acids Res. 1988;16:6953–6985. doi: 10.1093/nar/16.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohn M, Blackburn E H. Science. 1995;269:396–400. doi: 10.1126/science.7618104. [DOI] [PubMed] [Google Scholar]
- 27.Beeler T, Gable K, Zhao C, Dunn T. J Biol Chem. 1994;269:7279–7284. [PubMed] [Google Scholar]
- 28.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henikoff S, Eghtedarzadeh M K. Genetics. 1987;117:711–725. doi: 10.1093/genetics/117.4.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan C S M, Tye B-K. Cell. 1983;33:563–573. doi: 10.1016/0092-8674(83)90437-3. [DOI] [PubMed] [Google Scholar]
- 31.Schiestl R H, Gietz R D. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 32.Zakian V A, Scott J F. Mol Cell Biol. 1982;2:221–232. doi: 10.1128/mcb.2.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghuraman M K, Brewer B J, Fangman W L. Genes Dev. 1994;8:554–562. doi: 10.1101/gad.8.5.554. [DOI] [PubMed] [Google Scholar]
- 34.Boeke J D, LaCroute F, Fink G R. Mol Gen Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 35.Brewer B J, Fangman W L. Cell. 1987;51:463–71. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- 36.Huberman J A, Spotila L D, Nawotka K A, El-Assouli S M, Davis L R. Cell. 1987;51:473–481. doi: 10.1016/0092-8674(87)90643-x. [DOI] [PubMed] [Google Scholar]
- 37.Livingston D M, Kupfer D M. J Mol Biol. 1977;116:249–260. doi: 10.1016/0022-2836(77)90215-7. [DOI] [PubMed] [Google Scholar]
- 38.Counter C M, Avilion A A, LeFeuvre C E, Stewart N G, Greider C W, Harley C B, Bacchetti S. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wahl G M, Meinkoth J L, Kimmel A L. Methods Enzymol. 1987;152:572–581. doi: 10.1016/0076-6879(87)52064-x. [DOI] [PubMed] [Google Scholar]
- 40.Wellinger R J, Wolf A J, Zakian V A. Chromosoma. 1992;102:S150–S156. doi: 10.1007/BF02451800. [DOI] [PubMed] [Google Scholar]
- 41.McEachern M J, Blackburn E H. Nature (London) 1995;376:403–409. doi: 10.1038/376403a0. [DOI] [PubMed] [Google Scholar]
- 42.Chan C S M, Tye B-K. J Mol Biol. 1983;168:505–523. doi: 10.1016/s0022-2836(83)80299-x. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann I R, Nussbaum A L. J Biol Chem. 1964;239:2628–2634. [PubMed] [Google Scholar]
- 44.McCarroll R M, Fangman W L. Cell. 1988;54:505–513. doi: 10.1016/0092-8674(88)90072-4. [DOI] [PubMed] [Google Scholar]
- 45.Reynolds A E, McCarroll R M, Newlon C S, Fangman W L. Mol Cell Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin J-J, Zakian V A. Cell. 1995;81:1127–1135. doi: 10.1016/s0092-8674(05)80017-0. [DOI] [PubMed] [Google Scholar]
- 47.Cao L, Alani E, Kleckner N. Cell. 1990;61:1089–1101. doi: 10.1016/0092-8674(90)90072-m. [DOI] [PubMed] [Google Scholar]
- 48.White C I, Haber J E. EMBO J. 1990;9:633–670. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H, Treco D, Szostak J W. Cell. 1991;64:1155–1161. doi: 10.1016/0092-8674(91)90270-9. [DOI] [PubMed] [Google Scholar]
- 50.Huang K N, Symington L. Mol Cell Biol. 1993;13:3125–3134. doi: 10.1128/mcb.13.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrington J J, Lieber M R. Genes Dev. 1994;8:1344–1355. doi: 10.1101/gad.8.11.1344. [DOI] [PubMed] [Google Scholar]
- 52.Garvik B, Carson M, Hartwell L. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]