

Abstract
Temperature-sensitive (Ts) mutants of a protein are an extremely powerful tool for studying protein function in vivo and in cell culture. We have devised a method to predict those residues in a protein sequence that, when appropriately mutated, are most likely to give rise to a Ts phenotype. Since substitutions of buried hydrophobic residues often result in significant destabilization of the protein, our method predicts those residues in the sequence that are likely to be buried in the protein structure. We also indicate a set of amino acid substitutions, which should be made to generate a Ts mutant of the protein. This method requires only the protein sequence. No structural information or homologous sequence information is required. This method was applied to a test data set of 30 nonhomologous protein structures from the Protein Data Bank. All of the residues predicted by the method to be ≥95% buried were, in fact, buried in the protein crystal structure. In contrast, only 50% of all hydrophobic residues in this data set were ≥95% buried. This method successfully predicts several known Ts and partially active mutants of T4 lysozyme, λ repressor, gene V protein, and staphylococcal nuclease. This method also correctly predicts residues that form part of the hydrophobic cores of λ repressor, myoglobin, and cytochrome b562.
Temperature-sensitive (Ts) mutants of a gene are ones in which there is a marked drop in the level or activity of the gene product when the gene is expressed above a certain temperature (nonpermissive temperature). Below this temperature, the activity of the mutant is very similar to that of the wild type. Ts mutants provide an extremely powerful tool to study protein function in vivo and in cell culture (1, 2). They provide a reversible mechanism to lower the level of a specific gene product at any stage in the growth of the organism simply by changing the temperature of growth (3). At present, there is no general method to predict which mutations in a protein will give rise to a Ts phenotype. Hence, Ts mutants are generated by random mutagenesis, typically with a chemical mutagen, followed by screening to obtain mutants with a Ts phenotype (4, 5). In the case of prokaryotes and yeast, such procedures work well because simple screens or selections exist and a large number of progeny can be simultaneously screened using simple plate assays. In more complicated organisms, however, such an approach suffers from several drawbacks. Since mutations will be generated throughout the genome, a large number of progeny need to be screened before a Ts mutant is obtained. In the case of the fruitfly, Drosophila melanogaster, this number is typically of the order of several hundred thousand (4). Screening such a large number of progeny can be extremely laborious in situations where a simple screen does not exist. Furthermore, in organisms with long generation times or in cases where it is not possible to obtain large numbers of progeny, random mutagenesis of the entire genome cannot be used to isolate Ts mutants.
In an increasing number of cases, cloned and sequenced versions of a gene are available for manipulation. In both Drosophila (6) and more recently in the medfly Ceratitis capitata (7) procedures now exist for introducing these cloned genes into the germ line of the organism via microinjection. Methods for the production of transgenic organisms also exist for the worm Caenorhabditis elegans (8), as well as for a variety of plant species (9). In all these examples, the exogenous DNA is not incorporated at a specific site in the genome but is either randomly incorporated into the genome (in Drosophila, C. capitata, or plants) or forms a multicopy extrachromosomal array that can be stably transmitted to offspring (in C. elegans). In bacteria, yeast, and the mouse, however, procedures exist for targeted gene replacement (10, 11). Even in an organism as complex as the mouse, it is currently possible to selectively alter just a few base pairs in the entire genome (12). Given the extremely rapid rate of progress in the area of gene targeting and production of transgenic organisms, it should soon be possible to carry out similar sequence replacements in a variety of other organisms in addition to the ones described above. Using PCR-based mutagenesis techniques (13) it is possible to carry out site-directed mutagenesis with a yield of 100%.
We have therefore developed a method to predict those positions in a protein sequence which, if mutated, would be likely to result in a Ts phenotype. Once these positions are identified, a small number of mutants of the gene can be constructed in vitro and reintroduced into the organism. Because only a small number of progeny would need to be examined, these progeny could be extensively characterized. Using this method, it may be possible to examine the effects of Ts mutations even in genes of completely unknown function for which screening procedures do not exist.
A drawback of Ts mutations is that they cannot be used to study gene function in organisms that maintain constant body temperature. However, in many cases, it is also useful to examine partial loss of function mutants in which the mutated gene product displays lower levels of activity than the wild-type gene product (14). We expect that our method will also predict such mutants. The only input required for the method is the protein sequence. No structural information or information from homologous protein sequences is necessary. A recent method for constructing Ts mutants involves making fusions of ubiquitin–TsArg–dihydrofolate reductase to the protein of interest (15). When these constructs were expressed in yeast, the protein of interest exhibited a Ts phenotype. Although this is a powerful and elegant approach, it remains to be seen how well it works in other organisms. In some cases, it is possible that the fusion partners may also affect the normal functioning of the protein at temperatures below the nonpermissive temperature.
It has been observed, from numerous experimental studies on proteins of known structure, that mutations at buried residue positions cause much larger changes in protein stability than mutations at surface positions (16, 17, 18). In the case of T4 lysozyme (19), and more recently in the case of gene V protein (20), substitutions at buried residue positions were shown to result in a Ts phenotype. Decreased protein stability is also correlated with reduction in protein levels in vivo and with generation of a Ts phenotype (21, 22). Our approach has therefore been to predict positions in the protein that are likely to be buried. The two properties that we have chosen to correlate with burial are the average hydrophobicity (23, 24) and the hydrophobic moment (25). We first examined the correlation between these properties and the degree of burial in a data base of 35 nonhomologous proteins of known structure from the Protein Data Bank (26), containing a total of 6143 residues (Table 1, Original data set). These studies were used to derive a set of rules to predict which residues would be buried in a protein of known sequence but unknown structure. These rules were next tested on another set of 30 nonhomologous protein structures from the Protein Data Bank (Table 1, Test1 data set). Finally, these rules were applied to a third set of four proteins for which extensive mutagenesis data exists (Table 1, Test2 data set). Several of the mutants in the last set have been screened for temperature sensitivity, and in many cases the mutant proteins have been purified and the free energy of unfolding has been measured in vitro (18, 20, 27, 28, 29). In all cases our predictions show excellent correlation with experimental results. Neural network procedures (30, 31) have been used to predict exposed and buried residues with overall accuracies of 72–75%. Another recent study (32) has used amino acid substitution patterns and conformational propensities to predict burial in aligned sequence of homologous proteins with an accuracy of about 77%. In these studies, residues with an accessibility (33) of less than either 20% (30, 32) or 15% (31) were considered to be buried. However, the two goals of this study are somewhat different. First, we do not try to identify all buried residues, but rather to identify a subset of these residues that have a high probability of being buried. Specifically we attempt to predict residues with accessibilities less than 15%, with a prediction accuracy of greater than 80%. Second, we identify a set of substitutions at these buried positions that are most likely to result in a Ts phenotype. The precise mechanisms that can give rise to a Ts phenotype are not the focus of this particular study. Neither do we attempt to identify all possible Ts mutants. Instead we identify a small number of sites and indicate a limited set of substitutions that can be feasibly made at each one of these sites. We would judge our method to be successful if at least one of the relatively small number of mutants of a given protein predicted by us to exhibit a Ts phenotype is experimentally shown to do so.
Table 1.
Protein Data Bank data sets used for analysis
| Data set | Protein Data Bank code (chain identifier) |
|---|---|
| Original | 1CCR, 1GD1(O), 1GP1(A), 1HOE, 1MBA, 1MBD, 1OVA(A), 1PAL, 1PPT, 1R69, 1THB(A), 1UTG, 256B(A), 2ALP, 2CSC, 2FBJ(L), 2GBP, 2HMQ(A), 2LHB, 2LTN(A), 2MHR, 2MLT(A), 2PKA(A), 2PRK, 2TEC(E), 2WRP(R), 3BLM, 3FGF, 3GRS, 4CHA(A), 4INS(C), 4TNC, 5RXN, 5TIM(A), 7RSA |
| Test1 | 1ALC, 1ALD, 1BBP(A), 1CSE(E), 1FKF, 1GOX, 1HIP, 1IFB, 1RBP, 1RDG, 1RNH, 1TGN, 1TON, 1UBQ, 1YPI, 2ACT, 2CDV, 2CI2, 2CYP, 2FCR, 2OVO, 2PAB, 2RHE, 2RSP, 2SAR, 2SGA, 2TSC, 351C, 3BCL, 3C2C |
| Test2 | 2LZM, 1LMB, 2SNS, 1YHA |
PARAMETERS AND METHODS
Hydrophobicity Scale.
There are currently more than 35 scales for amino acid residue hydrophobicities (34). For all our calculations we have used the scale of Rose et al. (35), because this most closely correlates with the degree of residue burial. The hydrophobicity values in this scale were chosen to be equal to the average extent of burial of the residue in a data base of 12 protein structures. The average extent of burial Bx is given by
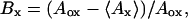 |
1 |
where Aox is the accessible area of residue X in a stochastic standard state, which is analagous to the unfolded state of a protein and <Ax> is the average accessible area of the residue in the data base of 12 proteins. Accessible areas were calculated according to the method of Lee and Richards (33). We have rescaled the hydrophobicity values from this scale to lie between 0 and 100. The numerical values of the rescaled hydrophobicities for the 20 amino acids are as follows: Cys-100, Phe-92, Ile-92, Val-87, Trp-85, Met-85, Leu-85, His-67, Tyr-62, Ala-56, Gly-51, Thr-46, Ser-36, Arg-31, Pro-31, Asn-28, Gln-26, Glu-26, Asp-26, Lys-0. These data show that residues Cys, Phe, Ile, Val, Trp, Met, and Leu are, on an average, buried to a significantly larger extent than the remaining residues. Hence, throughout the rest of this discussion, we designate these residues alone as hydrophobic residues.
Calculations of Average Hydrophobicity and Hydrophobic Moment.
For a given sequence, the average hydrophobicity of a residue (averaged over a seven residue window) is given by:
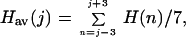 |
2 |
where the H(n)s are the rescaled individual residue hydrophobicities listed above. Plots of the average hydrophobicity along the protein sequence have previously been used to identify the locations of buried and exposed regions (23, 24). Buried segments lie at local maxima in such plots, whereas exposed segments are generally located at local minima. Bi, the percent burial of residue i in a protein of known structure, is given by:
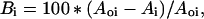 |
3 |
where Ai and Aoi are the accessible surface areas of residue i in the protein and in the extended tripeptide Gly-i-Gly, respectively (33). The hydrophobic moment Hmom is calculated over a nine residue window as follows:
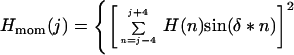 |
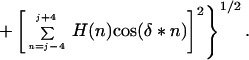 |
4 |
The phase angle δ depends on the periodicity of the secondary structure that the sequence is assumed to adopt. For an α-helix, the phase angle δ = 100°. For a flat β-sheet, it is 180° and for a curved β-sheet it is about 160° (36). Both helices and β strands often have one solvent exposed hydrophilic face and one buried hydrophobic face. Such sequences will be characterized by average Hav and high Hmom values. Buried regions of such sequences therefore cannot be identified using only Hav. Once a sequence with high values of Hmom is identified, it is generally straightforward to determine which residues are buried simply by examining the pattern of hydrophobicity within the sequence. Hmom values have previously been used by Eisenberg (25) to determine the locations of amphiphilic helices within a protein sequence.
Temperature Dependence of ΔG.
The effect of a mutation on the stability of a protein is temperature dependent. The temperature dependence of the change in free energy of protein unfolding (ΔG) is a function of ΔG, as well as of the changes in enthalpy (ΔH) and heat capacity (ΔCp) that occur upon protein unfolding. Although there is extensive information on the effect of a mutation on ΔG, there are few measurements of the effects of a mutation on ΔH and ΔCp at temperatures close to room temperature (37). ΔG for most proteins is in the range of 5–15 kcal/mol at room temperature. For a protein that has not been structurally and thermodynamically characterized, there is thus considerable uncertainity in the value of ΔG at room temperature as well as in the temperature dependence of ΔG. To generate Ts mutants of such uncharacterized proteins, it will therefore be necessary to make several substitutions that affect the protein stability to different extents. It is likely that at least one of these substitutions will affect ΔG to the appropriate extent (see Results and Discussion) to generate a Ts phenotype.
RESULTS AND DISCUSSION
The objective of our studies was to evolve criteria to predict buried residues with greater than 80% accuracy. By accuracy, we mean the ratio of the number of predicted residues that are actually buried in a given protein structure to the total number of predicted buried residues for that protein. It is known (35) that the seven hydrophobic residues listed in the previous section are, on average, substantially more buried than the remaining amino acids. The simplest criteria for picking buried residues would therefore be to select all hydrophobic residues. We examined the correlation between residue burial and whether the residue was hydrophobic for the 35 proteins of known structure listed in Table 1. We used three different definitions of buried residues. Buried residues were ones that had fractional accessibilities less than or equal to (i) 5% (most stringent definition), (ii) 10% (intermediate stringency), and (iii) 20% (least stringent). The results are summarized in Table 2. As expected, the percentage of total hydrophobic residues that are buried is inversely correlated with the degree of stringency. However, even in the least stringent case, less than 75% of the hydrophobic residues are classified as buried. Hence, in addition to the residue hydrophobicity, we need additional criteria to predict buried residues with an an accuracy greater than 80%. In addition, the seven hydrophobic residues comprise approximately one-third of the total number of residues in a protein. It is experimentally unfeasible to make mutations at all hydrophobic residue positions. It is therefore desirable to have additional criteria to restrict the set of predicted buried residues further so that the resulting set is smaller (approximately five predicted buried residues per protein) and the predictions are more accurate. Another advantage of having a small set is that potential Ts mutants can be screened at several temperatures instead of at a single restrictive temperature. The additional criteria were generated by examining the correlation between burial and two additional parameters, Hav (23, 24) and Hmom (25), for reasons discussed in the previous section.
Table 2.
Statistics of buried residues in original data set
| Burial, % | All buried residues, as % of all residues | Buried hydr., as % of all residues | Buried hydr. residues, as % of all hydr. |
|---|---|---|---|
| ≥95 | 28 | 15 | 52 |
| ≥90 | 36 | 18 | 62 |
| ≥80 | 47 | 22 | 74 |
The data set contains a total of 6143 residues of which 1777 residues are hydrophobic. hydr, Hydrophobic residue (Cys, Val, Ile, Leu, Met, Phe, Trp).
Hav(j) is well correlated with burial only in cases where the middle residue, j, is hydrophobic (Table 3). In cases where residue j is not hydrophobic, the correlation of Hav(j) with burial is poor (data not shown). We also examined the likelihood of burial of a residue j as a function of whether residue j − 1 or residue j + 1 or both are hydrophobic. When j − 1, j, and j + 1 are all hydrophobic residues the likelihood of burial increases (Table 3). The data for the cases where only j − 1 and j are hydrophobic and for the cases where only j and j + 1 are hydrophobic are not shown here. In these cases the observed correlation between Hav and burial is slightly less than in Table 3. From this collection of data it is straightforward to derive criteria for prediction of buried residues. As an example, suppose we define buried residues as those that are more than 90% buried and wish to predict such positions with greater than 80% accuracy. From the data in Table 3, we can identify buried residues as those that satisfy either of the following criteria: (i) residue j is hydrophobic and Hav(j) is ≥75 and (ii) residue j, as well as both flanking residues, are hydrophobic and Hav(j) is ≥65. The other criteria, summarized in Table 4, can be similarly derived.
Table 3.
Correlation between Hav (j) and residue burial in original data set
| Hav(j)* | ≥95%
burial
|
≥90% burial
|
≥80% burial
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Bur. | Exp. | Acc. | Bur. | Exp. | Acc. | Bur. | Exp. | Acc. | |
| residue j is hydrophobic | |||||||||
| 0–40 | 35 | 29 | 55 | 42 | 22 | 66 | 50 | 14 | 78 |
| 40–60 | 472 | 532 | 47 | 580 | 424 | 58 | 726 | 278 | 72 |
| 60–65 | 175 | 147 | 54 | 206 | 116 | 64 | 236 | 86 | 78 |
| 65–70 | 115 | 73 | 61 | 136 | 52 | 72 | 149 | 39 | 79 |
| 70–75 | 61 | 23 | 73 | 66 | 18 | 79 | 71 | 13 | 85 |
| 75–80 | 19 | 4 | 83 | 20 | 3 | 87 | 20 | 3 | 87 |
| 80–85 | 6 | 3 | 63 | 7 | 2 | 78 | 8 | 1 | 89 |
| 85–90 | 1 | 0 | 100 | 1 | 0 | 100 | 1 | 0 | 100 |
| residues j − 1, j, j + 1 are hydrophobic | |||||||||
| 0–40 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — |
| 40–60 | 17 | 9 | 65 | 19 | 7 | 73 | 20 | 6 | 77 |
| 60–65 | 19 | 10 | 66 | 19 | 10 | 66 | 22 | 7 | 76 |
| 65–70 | 16 | 6 | 73 | 18 | 4 | 82 | 19 | 3 | 86 |
| 70–75 | 21 | 6 | 78 | 23 | 3 | 85 | 26 | 1 | 96 |
| 75–80 | 11 | 1 | 92 | 12 | 0 | 100 | 12 | 0 | 100 |
| 80–85 | 6 | 2 | 75 | 7 | 1 | 88 | 7 | 1 | 88 |
Bur., number of buried residues with Hav values in the range shown in column 1. Exp., number of exposed residues with Hav values in the range shown in column 1. Acc., percent accuracy = 100*Bur./(Bur. + Exp.).
Hav(j) is calculated from the protein sequence using Eq. 2 with a seven residue window.
Table 4.
Prediction criteria for buried residues
| Burial, % | Predicted fraction,* % | Prediction criteria |
|---|---|---|
| ≥95 | 0.3 | Residue, as well as both flanking residues, are hydrophobic and Hav ≥ 75. |
| ≥90 | 1.3 | Residue is hydrophobic and Hav ≥ 75 or |
| Residue, as well as both flanking residues, are hydrophobic and Hav ≥ 65 | ||
| ≥80 | 6.1 | Residue is hydrophobic and any of the following conditions are met: |
| (i) Hav ≥ 60 and preceding residue is hydrophobic; | ||
| (ii) Hav ≥ 65 and both flanking residues are hydrophobic; | ||
| (iii) Hav ≥ 70; | ||
| (iv) Hmom ≥ 200 and residues at either (−3 and +4) or (−4 and +3) relative to the residue are hydrophobic |
Percent of total residues in a protein that are predicted to be buried. These numbers are based on the total number of predicted buried residues found for a given burial cutoff in the original data set of 6143 residues.
Hmom is not as well correlated with residue burial as Hav. For the two most stringent burial criteria we were not able to find any values of Hmom that were accurate predictors of the degree of burial. For the least stringent burial criterion (≥80% buried), we were only able to obtain a sufficiently high degree of accuracy if we imposed the following additional constraints (Table 4): (i) δ = 100° (assume that the sequence is α helical), (ii) the central residue j of the window is hydrophobic, and (iii) either residues j − 3 and j + 4 or residues j − 4 and j + 3 are hydrophobic. It thus appears that Hmom is only useful for predicting burial for sequences that adopt an amphiphilic α helical structure. Because an α-helix has 3.6 residues per turn, residues j − 3, j, and j + 4, as well as residues j − 4, j, and j + 3 will all lie on the same face of the helix. Hence, if either of the two sets of residues is hydrophobic, it is likely that this face is the buried face of the helix and consequently that residue j is a buried residue.
Once a site is identified as buried, the next step is to specify the nature of the substitution to be made at that position to generate a Ts phenotype. A Ts phenotype will arise if the amount of the active gene product in vivo is significantly decreased at the nonpermissive temperature relative to the amount present at permissive temperatures of growth. In addition to the free energy of folding of the protein, the amount of protein present in vivo may depend on a variety of complex factors, such as the rate of protein synthesis, susceptibility to proteolysis, and whether chaperones are involved in degradation or folding of the protein. The relative importance of these factors will in general be unknown and case specific. Our approach is therefore to suggest five different substitutions at each predicted buried site that differ in the stereochemistry and polarity of the substituted residue. These substitutions will span a wide range of free energy and, we assume that at least one of these substitutions will destabilize the protein to an extent appropriate to generate a Ts phenotype. The free energies of unfolding of typical globular proteins are in the range of 5–15 kcal/mol at room temperature. A Ts mutation should destabilize the protein by an amount that is an appreciable fraction of the free energy of folding at the nonpermissive temperature.
Previous studies have shown that addition or deletion of a single methylene group at a buried site destabilizes a protein by about 1 kcal/mol (17, 27, 37, 38, 39). If the wild-type protein is only marginally stable (with a free energy of unfolding of less than 5 kcal/mol), then addition or deletion of up to two methylene groups (a conservative substitution) or addition of a single β-branched methyl group should be sufficient to result in a Ts phenotype. Replacement of buried hydrophobic residues with charged or polar residues or with glycine (nonconservative substitutions) will destabilize the protein to a significantly larger extent, of the order of 5–10 kcal/mol (18, 40, 41, 42). The exact amount of destabilization produced by a mutation will depend on the effect of the mutation on ΔG, ΔH, and ΔCp. In general, these will not be known for the protein of interest. It is therefore desirable to make both conservative and nonconservative substitutions at predicted buried sites, so that at least one of these will result in a Ts phenotype. Based on the above discussion, a suggested set of substitutions for each of the seven hydrophobic residues is listed in Table 5. To minimize the total number of substitutions at each position the charged and polar amino acids listed in Table 5 involve introduction of a single negative charge and a hydroxyl group, respectively, and are chosen to be as similar in size to the wild-type residue as possible.
Table 5.
Amino acid substitution table for buried hydrophobic residues
| Buried residue | Conservative substitutions | Nonconservative substitutions |
|---|---|---|
| Val | Ala, Phe | Gly, Asp, Thr |
| Ile | Phe, Cys | Gly, Glu, Thr |
| Leu | Phe, Val | Gly, Glu, Thr |
| Met | Phe, Val | Gly, Glu, Thr |
| Phe | Leu, Met | Gly, Glu, Thr |
| Cys | Val, Met | Gly, Asp, Ser |
| Trp | Phe, Leu | Gly, Glu, Thr |
We next used the criteria listed in Table 4 to perform predictions of buried residues for the three data sets of nonhomologous proteins listed in Table 1. The results of these predictions are summarized in Table 6. As expected, the predictions on the original data set have an accuracy of greater than 80% in all cases. It is also encouraging to note that for the second and third data sets the accuracy of prediction, in most cases, is close to or greater than 80%. For comparison, we have also indicated accuracies of prediction using the simplest burial criterion, namely assuming all hydrophobic residues to be buried. These accuracies are consistently lower than the accuracies obtained by using the additional criteria listed in Table 4. The increase in accuracy from the additional criteria is greater than 50% for the highly buried residues. In addition, the number of predicted residues is greatly reduced by using the additional criteria making it experimentally feasible to make site directed mutants at each predicted buried residue site.
Table 6.
Prediction accuracies for the three data sets shown in Table 1
| Data set | Prediction accuracy,*
%†
|
Prediction accuracy,* % assuming all
hydrophobic residues are buried
|
|||||
|---|---|---|---|---|---|---|---|
| ≥95% burial | ≥90% burial | ≥80%‡ burial | ≥80%§ burial | ≥95% burial | ≥90% burial | ≥80% burial | |
| Original | 80 | 80 | 84 | 78 | 52 | 62 | 75 |
| Test1 | 100 | 60 | 75 | 89 | 46 | 54 | 69 |
| Test2 | None found | 100 | 83 | 100 | 44 | 52 | 65 |
Buried residues in each data set are predicted using the criteria in Table 4 and the accuracies are calculated from the percentage of predicted residues that are actually buried in the protein crystal structure.
Percent accuracy = 100% (number of predicted buried residues actually buried)/(total number of predicted buried residues).
Using criteria from Table 4.
Based solely on Hav criteria.
Based solely on Hmom criteria.
We next examined the agreement between our predictions (based on the criteria for ≥80% burial in Table 4 and the substitutions of Table 5) and the known Ts mutants in the Test2 set of proteins. The complete set of predicted buried residues for T4 lysozyme, λ repressor, and gene V protein is summarized in Table 7. Fourteen of the 16 predicted sites are greater than 80% buried and for each of the three proteins at least 50% of the predicted sites are ≥99% buried. The average extent of burial is 89%. Ts mutants have been experimentally found at 6 of the 16 predicted sites. In five of these six cases, the substitution giving rise to a Ts phenotype was among the ones listed in Table 5. In the one remaining case the predicted substitution differed from the actual one by a single methylene group. It is thus highly probable that the predicted substitution would also exhibit a Ts phenotype. In T4 lysozyme, two of the mutants predicted by us were isolated experimentally by random mutagenesis (19). In λ repressor, no published information was available regarding the Ts behavior of the predicted mutants listed in Table 7. However, several of the mutants were shown experimentally to exhibit a partial loss of activity, relative to the wild type under similar conditions. The prediction results for gene V protein were particularly encouraging. Gene V protein has been the object of a careful and detailed study of the relationship between in vivo activity and in vitro stability measurements (20). A total of 68 single and double mutants at buried sites were studied. Three of the four sites identified by us correspond to known Ts mutants of the protein. In each case the exact substitution found experimentally was in the predicted list. Phages with these mutant proteins formed plaques similar in size to the wild type at 34°C but did not form any plaques at 40.5°C, and thus correspond to tight Ts mutants.
Table 7.
Predicted set of Ts mutants for T4 lysozyme, λ repressor, and gene V protein
| Residue/substitution predicted | Burial in wild type, % | Experimental result |
|---|---|---|
| Phe-4-Val* | 54 | NS |
| Met-6-Val* | 100 | Met-6-Ile is Ts, Met-6-Val NS |
| Leu-7-Val* | 100 | NS |
| Met-102-Thr* | 99 | Met-102-Thr is Ts |
| Val-103-Ala* | 94 | Val-103-Ala is Ts |
| Phe-104-Leu* | 82 | NS |
| Val-87-Ala* | 98 | NS |
| Phe-51-Leu† | 99 | Phe-51-Val, Ile partially active, Phe-51-Gly, Asp inactive |
| Leu-65-Thr† | 100 | Leu-65-Thr partially active, Leu-65-Gly, Glu inactive |
| Ile-68-Phe† | 82 | NS |
| Phe-76-Thr† | 100 | Phe-76-Ser partially active, Phe-76-Thr NS |
| Ile-84-Thr† | 100 | Ile-84-Ser partially active, Ile-84-Thr NS |
| Val-35-Phe‡ | 100 | Val-35-Phe is Ts |
| Val-44-Thr‡ | 41 | NS |
| Val-45-Thr‡ | 100 | Val-45-Thr is Ts |
| Ile-78-Cys‡ | 82 | Ile-78-Cys is Ts |
It should be appreciated that our method identifies only a small fraction (0.3–6.1%) of the total residues in the protein as sites for potential Ts mutants. It is therefore experimentally feasible to make a limited set of mutations at each of these sites and devise detailed screening procedures at several temperatures to look for possible Ts mutants. Residue substitutions at predicted sites are made solely on the basis of stereochemical criteria. T4 lysozyme and gene V protein are 164 and 87 amino acids long, respectively. There are thus 3116 and 1653 theoretically possible single-site mutants for each of these proteins. It is therefore encouraging that several of the small set of 16 mutants predicted by our method were actually isolated through mutagenesis and shown to have a Ts phenotype.
The predictions of our method for staphylococcus nuclease are listed in Table 8. Five of the six predicted residues are buried. The average extent of burial of these five residues is 97%. In the case of staphylococcus nuclease, data on temperature sensitivity were not available. However, five of the six predicted mutant sequences corresponded to known mutants with a decrease in free energy of unfolding of over 3 kcal/mol. This is a significant fraction of the free energy of the unfolding of wild-type staphylococcus nuclease (5.5 kcal/mol) and we expect that a large fraction of these mutants would display a Ts phenotype.
Table 8.
Predicted buried residues for staphylococcus nuclease
| Residue/substitution predicted | Burial observed in wild-type protein, % | ΔΔG, kcal/mol |
|---|---|---|
| Ile-15-Gly | 50 | −3.3 |
| Leu-36-Gly | 99 | −5.3 |
| Leu-37-Gly | 97 | −3.8 |
| Leu-38-Gly | 98 | −0.6 |
| Val-39-Gly | 100 | −4.7 |
| Leu-108-Gly | 90 | −7.2 |
The experimental value for the change in free energy of unfolding (ΔΔG) when the wild-type residue is substituted by Gly is taken from ref. 18.
A recently developed method identifies hydrophobic cores in proteins of known structure by an automated procedure (43). Core residues are highly buried, interacting sets of residues, and comprise approximately 10% of the total number of residues. Three protein structures (1MBD, 256B, and 1LMB) were common to both that study and this one. We compared the residues identified in these three proteins by our sequence-based prediction criteria, with the core residues identified using the structure-based automated procedure. Of 21 residues identified by our method, 10 were also part of the hydrophobic core. Our criteria were developed solely to predict residue burial. No additional information about the hydrophobic core or about Ts phenotypes was used to generate the criteria. However, approximately one-half of the residues predicted by us, in addition to being buried, also form part of the hydrophobic core. This may explain why a significant fraction of the small set of predicted residues are also experimentally found to be sites for Ts mutations.
We have thus developed a procedure for the accurate prediction of a fraction of the total buried residues within a globular protein of known sequence but unknown structure. We have also indicated a set of substitutions that are likely to confer a Ts phenotype when made at such positions. We have been able to correctly predict the locations, as well as the amino acid replacements, present in several known Ts mutants. Further experimental work is required to assess the generality of the method. Our criteria were derived from a data base of globular proteins and, therefore, will only be applicable to globular proteins. Given the protein sequence, the prediction of buried residue locations is straightforward. Substitutions at buried locations will certainly destabilize the protein. Whether substitutions at such buried positions will consistently result in a Ts phenotype in vivo remains to be seen. Too small a destabilization will not have any effect and too large a destabilization will result in loss of function of the protein at all temperatures at which the organism is viable. The effect of a mutation on in vivo levels of the protein will depend on a number of factors. These may include the exact location of the residue in the three-dimensional structure of the protein, the stability of the wild-type protein, and the effects of the mutation on the ΔG, ΔH, and ΔCp of unfolding, as well as on protein dynamics. These effects are poorly understood even in proteins of known structure (37). Despite these caveats, in the cases we have analyzed, the predictions of our method show good agreement both with experimental data on Ts phenotypes observed in vivo and with in vitro measurements of protein stability. A Fortran program, implementing the method that predicts buried residues given the amino acid sequence, is available from the authors on request.
Acknowledgments
We wish to thank K. Krishnan, L. Iyer, and S. Varadarajan for useful discussions. We are grateful to the Supercomputer Education and Research Center, the Interactive Graphics facility, and the Distributed Information Centre, Indian Institute of Science for use of their computational and computer graphics facilities. This work was supported by grants from Council for Scientific and Industrial Research (37(0813)/93/EMRII) and Department of Biotechnology (BT/R&D/15/09/93).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: Ts, temperature sensitive.
References
- 1.Horowitz H N. Adv Genet. 1950;3:33–71. doi: 10.1016/s0065-2660(08)60082-6. [DOI] [PubMed] [Google Scholar]
- 2.Fried M. Proc Natl Acad Sci USA. 1965;53:486–491. doi: 10.1073/pnas.53.3.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couso J P, Bate M, Martinez-Arias A. Science. 1993;259:484–489. doi: 10.1126/science.8424170. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki D T, Grigliatti T, Williamson R. Proc Natl Acad Sci USA. 1971;68:890–893. doi: 10.1073/pnas.68.5.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson H C M, Sauer R T. Cell. 1985;42:549–558. doi: 10.1016/0092-8674(85)90112-6. [DOI] [PubMed] [Google Scholar]
- 6.Spradling A C, Rubin G M. Cell. 1983;34:47–57. doi: 10.1016/0092-8674(83)90135-6. [DOI] [PubMed] [Google Scholar]
- 7.Loukeris T G, Livadaras I, Arcà B, Zabalou S, Savakis C. Science. 1995;270:2002–2005. doi: 10.1126/science.270.5244.2002. [DOI] [PubMed] [Google Scholar]
- 8.Hodgkin J, Plasterk R H A, Waterston R H. Science. 1995;270:410–414. doi: 10.1126/science.270.5235.410. [DOI] [PubMed] [Google Scholar]
- 9.Schell J, Van Montagu M. Bio/Technology. 1983;1:175–180. [Google Scholar]
- 10.Nash H. Annu Rev Genet. 1981;15:143–167. doi: 10.1146/annurev.ge.15.120181.001043. [DOI] [PubMed] [Google Scholar]
- 11.Scherer S, Davies R W. Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez-Solis R, Zheng H, Whiting J, Krumlauf R, Bradley A. Cell. 1993;73:279–294. doi: 10.1016/0092-8674(93)90229-j. [DOI] [PubMed] [Google Scholar]
- 13.Sarkar G, Sommer S S. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 14.Simon M A, Bowtell D D L, Dodson G S, Laverty T S, Rubin G M. Cell. 1991;67:701–716. doi: 10.1016/0092-8674(91)90065-7. [DOI] [PubMed] [Google Scholar]
- 15.Jürgen Dohmen R, Wu P, Varshavsky A. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- 16.Pakula A A, Young V B, Sauer R T. Proc Natl Acad Sci USA. 1986;83:8829–8833. doi: 10.1073/pnas.83.23.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kellis J T, Nyberg K, Fersht A R. Biochemistry. 1989;28:4914–4922. doi: 10.1021/bi00437a058. [DOI] [PubMed] [Google Scholar]
- 18.Shortle D, Stites W A, Meeker A K. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 19.Alber T, Dao-pin S, Nye J A, Muchmore D C, Matthews B W. Biochemistry. 1987;26:3754–3758. doi: 10.1021/bi00387a002. [DOI] [PubMed] [Google Scholar]
- 20.Sandberg W S, Schlunk P, Zabin H B, Terwilliger T C. Biochemistry. 1995;34:11970–11978. doi: 10.1021/bi00037a039. [DOI] [PubMed] [Google Scholar]
- 21.Parsell D A, Sauer R T. J Biol Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 22.Pakula A A, Sauer R T. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 23.Rose G D, Roy S. Proc Natl Acad Sci USA. 1980;77:4643–4647. doi: 10.1073/pnas.77.8.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopp T P, Woods K R. Proc Natl Acad Sci USA. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg D. Annu Rev Biochem. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- 26.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 27.Matthews B W. Adv Protein Chem. 1995;46:249–277. doi: 10.1016/s0065-3233(08)60337-x. [DOI] [PubMed] [Google Scholar]
- 28.Hecht M H, Nelson H C M, Sauer R T. Proc Natl Acad Sci USA. 1983;80:2676–2680. doi: 10.1073/pnas.80.9.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim W A, Sauer R T. Nature (London) 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 30.Holbrook S R, Muskal S M, Kim S-H. Protein Eng. 1990;3:659–665. doi: 10.1093/protein/3.8.659. [DOI] [PubMed] [Google Scholar]
- 31.Rost B, Sanders C. Proteins. 1995;23:295–300. doi: 10.1002/prot.340230304. [DOI] [PubMed] [Google Scholar]
- 32.Wako H, Blundell T M. J Mol Biol. 1994;238:682–692. doi: 10.1006/jmbi.1994.1329. [DOI] [PubMed] [Google Scholar]
- 33.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 34.Cornette J L, Cease K B, Margalit H, Spouge J L, Berzofsky J A, DeLisi C. J Mol Biol. 1987;195:659–685. doi: 10.1016/0022-2836(87)90189-6. [DOI] [PubMed] [Google Scholar]
- 35.Rose G D, Geselowitz A R, Lesser G L, Lee R H, Zehfus M H. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 36.Eisenberg D, Wesson M, Wilcox W. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman G D, editor. New York: Plenum; 1985. pp. 635–647. [Google Scholar]
- 37.Varadarajan R, Connelly P R, Sturtevant J M, Richards F M. Biochemistry. 1992;31:12315–12327. doi: 10.1021/bi00164a005. [DOI] [PubMed] [Google Scholar]
- 38.Lim W A, Farruggio D C, Sauer R T. Biochemistry. 1992;31:4324–4333. doi: 10.1021/bi00132a025. [DOI] [PubMed] [Google Scholar]
- 39.Sandberg W S, Terwilliger T C. Science. 1989;245:54–57. doi: 10.1126/science.2787053. [DOI] [PubMed] [Google Scholar]
- 40.Varadarajan R, Lambright D G, Boxer S G. Biochemistry. 1989;28:3771–3781. doi: 10.1021/bi00435a022. [DOI] [PubMed] [Google Scholar]
- 41.Dao-pin S, Anderson D E, Baase W A, Dahlquist F W, Matthews B W. Biochemistry. 1991;30:11521–11529. doi: 10.1021/bi00113a006. [DOI] [PubMed] [Google Scholar]
- 42.Stites W E, Gittis A G, Lattman E E, Shortle D. J Mol Biol. 1991;221:7–14. doi: 10.1016/0022-2836(91)80195-z. [DOI] [PubMed] [Google Scholar]
- 43.Swindells M B. Protein Sci. 1995;4:93–102. doi: 10.1002/pro.5560040112. [DOI] [PMC free article] [PubMed] [Google Scholar]