

Abstract
Cytosines in single-stranded DNA deaminate to uracils at 140 times the rate for cytosines in double-stranded DNA. If resulting uracils are not replaced with cytosine, C to T mutations occur. These facts suggest that cellular processes such as transcription that create single-stranded DNA should promote C to T mutations. We tested this hypothesis with the Escherichia coli tac promoter and found that induction of transcription causes ≈4-fold increase in the frequency of C to U or 5-methylcytosine to T deaminations in the nontranscribed strand. Excess mutations caused by C to U deaminations were reduced, but not eliminated, by uracil-DNA glycosylase. Similarly, mutations caused by 5-methylcytosine to T deaminations were only partially reduced by the very short-patch repair process in E. coli. These effects are unlikely to be caused by differential repair of the two strands, and our results suggest that all actively transcribed genes in E. coli should acquire more C to T mutations in the nontranscribed strand.
Keywords: cytosine deamination, strand bias, uracil-DNA glycosylase, cytosine methylation
Cytosine is the most unstable of the four bases in nucleic acids and deaminates at a rate of 3 to 7 × 10−13 sec−1 in double-stranded DNA (1, 2). At this rate, ≈40 to 100 deamination events should occur in a human genome per day. This “spontaneous” hydrolytic deamination of cytosine creates uracil, a base not commonly found in DNA. If unrepaired, uracil can pair with an adenine during replication, causing a C to T mutation. For this reason, cells contain uracil-DNA glycosylase (UDG), an enzyme that removes the uracil and initiates its replacement with cytosine. Despite this, C:G to T:A transitions dominate spectra of spontaneous base substitutions in Escherichia coli and in mammalian cells (3, 4, 5, 6). In primates, C:G to T:A transitions are thought to account for 42% of all base substitutions within the β-globin cluster (7, 8). These observations show that C:G to T:A mutations play a major role in evolution and suggest that deamination of cytosine may be the underlying cause of the mutations.
Methylation at position 5 of cytosine is known to increase the rate of cytosine deamination. Rate of deamination of 5-methylcytosine (5meC) in DNA is 2 to 4 times higher than that for cytosine (2, 9, 10). Additionally, deamination of 5meC creates T:G mismatches that are more difficult to repair than U:G mismatches. Probably because of these reasons, sites of cytosine methylation are hot spots for C to T mutations (3, 11, 12, 13).
The deamination rate of cytosine also increases when the two strands of DNA are separated or when there is disruption of Watson–Crick base pairing of cytosine with guanine. The rate is 140 times higher when the target cytosine is present in single-stranded DNA than in double-stranded DNA (1), and the rate is at least 8- or 26-fold higher when cytosine is in a C:C mismatch or in a C:T mismatch than when it is in a C:G pair (14). Hydrolytic deamination of cytosine is thought to be initiated by the addition of a water molecule across the 5, 6 double bond followed by electronic rearrangements that create an iminium group that is susceptible to hydrolysis (15, 16). In double-stranded cannonically paired DNA, access to position 6 of cytosine is restricted by the deoxyribose sugar, and this restricts the ability of water to attack C6 of cytosine to initiate deamination.
If separation of DNA strands increases the risk of hydrolytic deamination, cellular processes such as transcription, replication, conjugation, and recombination have the potential of promoting C to T mutations. For example, in a simple model for transcription elongation, the nontranscribed strand should be transiently in single-strand form when the transcription bubble passes through. Such potential deamination risk for cytosines in the nontranscribed strand during transcription has been noted before (17, 18, 19, 20), but has not been investigated in depth. A possible reason for this inattention is the existence of strand bias in nucleotide excision repair. In E. coli (21) and in mammalian cells (22), transcription-blocking lesions are repaired preferentially when present in the transcribed strand. Transcription-coupled nucleotide excision repair has helped explain the observed bias in mutations caused by mutagens such as UV light in favor of the nontranscribed strand (23, 24, 25), and has raised the possibility that all observations of strand bias in mutations may be explained by strand bias in DNA repair. In fact, Skandalis et al. (19) have argued that there is a strand bias in 5meC to T mutations in the human hprt gene and have suggested that this is the result of strand bias in a base excision repair process that repairs T:G mismatches.
We have examined the question of strand bias in the deamination of cytosine in an E. coli genetic system in which transcription was regulated. Further, in this system the same cytosine could alternately be placed in the nontranscribed or in the transcribed strand, and its susceptibility to deamination could be studied. Our results show that transcription of the gene promotes deamination of this cytosine only when it is present in the nontranscribed strand.
MATERIALS AND METHODS
Bacterial Strains and Plasmids.
E. coli strain GM30 (thr-1 hisG4 leuB6 rpsL ara-14 supE44 lacY1 tonA31 tsx-78 galK2 galE2 xyl-5 thi-1 mtl-1) and GM31 (GM30 dcm-6) were obtained from M. G. Marinus (University of Massachussetts School of Medicine). GM30ung (GM30 ung-1 tyrA::Tn10) and GM31ung (GM31 ung-1 tyrA::Tn10) were gifts from M. Lieb (University of Southern California School of Medicine).
A cassette containing the mutant allele (kanS-D94) of the kanamycin-resistance gene was excised from the construct pKanS-D94 (26) with HindIII and cloned into pKK223-3 (Pharmacia) to generate plasmids pTKanS11 and pTKanS12 with direct and reverse orientation of the kan gene with respect to the tac promoter, respectively. To isolate lacIQ gene, plasmid pUCIQ2 (gift of P. Cunningham, Wayne State University) was amplified by PCR using primers LacIQ-F61 (5′-GCGAATGCGCATATGAGTGAGCTAACTCACATT-3′) and LacIQ-R21 (5′-CGTCCGGACATATGACAGCTATGACCATGATTAC-3′). Sites for restriction enzymes BsmI and BspEI sites are underlined in the sequences. The 1.2-kb PCR product was isolated from low melting point agarose gel, digested with BsmI and BspEI, and subcloned into the pTKanS11 and pTKanS12 plasmids to create pIQS11 and pIQS12, respectively. pDCM33 (dcm+ vsr+) and pDCM72 (dcm+) have been described previously (26, 27).
RNA Isolation and Dot Blot Analysis.
For RNA isolation, 5-ml cultures of GM30ung− cells carrying either pIQS11 or pIQS12 plasmid were grown to OD550 of ≈0.3 in Luria–Bertani medium (LB) containing carbenicillin (50 μg/ml). One-hundred microliters of each culture was added to 10 ml of prewarmed LB medium containing carbenicillin (50 μg/ml) with isopropyl β-d-thiogalactoside (IPTG) (1 mM) or without inducer. Cells were grown to OD550 of ≈0.3 and harvested, and total cellular RNA isolation was essentially performed as described (28). Total RNA samples were dissolved in 200 μl of 2× SSC and quantified by gel electrophoresis and optical density measurements. Normalized quantities of total RNA, based on RNA concentration in samples, were used in dot blot hybridization of RNA.
Dot blot analysis of total RNA was performed by using Bio-Dot Microfiltration Apparatus (Bio-Rad) and GeneScreen Plus membrane (DuPont/NEN) according to the manufacturer’s instructions. Probes dcm94U (5′-GACTGGCTGCTACUAGGCGAAGTGCC-3′), complementary to the product of transcription of the coding strand of kanS-D94 allele, and dcm94B (5′-GGCACTTCGCCTGGTAGCAGCCAGTC-3′), complementary to kanS-D94 mRNA (resulting from noncoding strand transcription), were used for hybridization. These oligomers hybridize with the kanS-D94 DNA at codon 94 and hence detect RNA transcripts from this segment of the gene. The oligomers were end-labeled with 32P and purified on Sephadex G-50 (Pharmacia) spin-columns before hybridization. Dot blot hybridization was carried out with seven successive 2-fold dilutions of the RNAs. Prehybridization, hybridization, and washing conditions were performed according to the protocols supplied by the manufacturer (DuPont/NEN). The filter with RNAs was first hybridized to probe dcm94B and a PhosphorImager scanner was used to detect and quantitate the intensity of bound radioactivity. The filter was then stripped of the bound probe and rehybridized to probe dcm94U. The amount of bound radioactivity was again quantitated with the PhosphorImager. The phosphorescence intensity of different RNA spots from PhosphorImager were plotted against the total concentration of RNA applied in each spot, and the intensity of the spots was calculated by regression analysis of the linear portion of each plot.
Assay for C to T Mutations.
To determine reversion frequencies, an appropriate E. coli strain was electroporated with pIQS11 or pIQS12. Following transformation and plating, three independent colonies were picked from each plate and grown in 5 ml LB containing appropriate antibiotics at 37°C in a shaker at 300 rpm until the OD550 reached 0.3. Fifty microliters of each cell culture was transferred to 5-ml prewarmed LB containing appropriate antibiotics with (1 mM) or without IPTG. Cells were again grown until OD550 reached 0.3 (≈3.5 h). Addition of IPTG to the growth medium caused only slight slowing of growth (15 to 30 additional min). Cells were centrifuged at 2600 g for 10 min, and the cell pellet was resuspended in 1 ml LB broth. Dilutions of these cells were spread on LB plates to determine the number of viable cells, and the remaining culture was spread on kanamycin plates to determine the number of revertants. The revertant frequency was the ratio of the number of KanR colonies to the total number of viable cells.
RESULTS
The Genetic System.
To study the effects of transcription on mutagenesis, a mutant allele (kanS-D94) of the kanamycin-resistance gene was cloned under the control of the tac promoter and kanamycin-resistant (KanR) revertants were scored in different genetic backgrounds and under different conditions of cell growth. The mutant allele codes for a protein with Leu-94 to Pro-94 change and the wild-type protein is restored by a C to T change in the middle of the codon (ref. 26 and Fig. 1). This cytosine is the target for methylation by E. coli cytosine methyltransferase, Dcm, and hence it is possible to study C to U to T or 5meC to T conversions at the same position. Codon 94 in kanS-D94 is also part of a BstNI recognition site (CCAGG), and the occurrence of mutations in this codon can be conveniently monitored by studying the loss of this BstNI site. We have previously used this kan allele (26, 29, 30) and a related allele containing a HpaII site (30, 31) in studies in which several dozen KanR revertants were characterized by restriction mapping or DNA sequencing (26, 30). Results of these studies have shown that KanR revertants found in this reversion system arise as a result of C to T mutations at second position in codon 94 and not due to “second-site” mutations.
Figure 1.
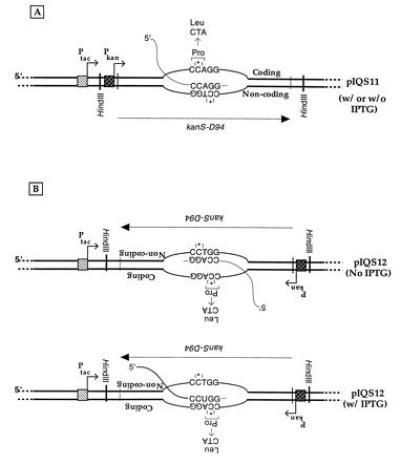
Predicted transcription patterns of pIQS11 and pIQS12. The expected dominant transcription patterns of pIQS11 (A) and pIQS12 (B) in the presence or absence of IPTG are shown. The figure is not drawn to scale, and exaggerates the size of the transcription bubble. Positions and directions of the tac and the kan promoters are shown. The strands of DNA are distinguished as being coding and noncoding, and the 5′ to 3′ directionality of the kan gene is indicated by an arrow below the DNA. The HindIII sites used to clone the kanS-D94 allele in pKK223-3 are marked. The sequence of the Dcm site at codon 94 of the gene is shown and the mutation that would restore KanR phenotype is indicated above it. The asterisk above the second cytosine within the Dcm site indicates of methylation of this base when Dcm is present in cells.
Two plasmids containing kanS-D94 were constructed, pIQS11 and pIQS12, that differ from each other only in the orientation of the kan gene with respect to the tac promoter (Fig. 1). Both plasmids contain the lacIQ gene, and hence the tac promoter is strongly repressed in the absence of IPTG in the growth medium. For pIQS11, transcription from either the tac or the kan promoter is expected to result in the copying of the “noncoding” strand (Fig. 1A). In contrast, for pIQS12 the choice of the template strand for transcription is expected to depend on the state of induction of the tac promoter. In the absence of IPTG, a majority of the transcription should initiate from the kan promoter, resulting in the copying of the noncoding strand. However, when IPTG is added to the growth medium, transcription from the kan promoter should be inhibited and copying of the “coding” strand by the RNA polymerase should occur (Fig. 1B).
The levels of transcripts from each of the two strands in cells containing these plasmids were consistent with the scheme presented in Fig. 1. In cells containing pIQS11, addition of IPTG to the growth medium increased transcription of the noncoding strand by a factor of ≈9 (Fig. 2), while decreasing transcription of the coding strand by a factor of ≈2. Presumably there is a low level of adventitious transcription of the coding strand and induction of transcription of the noncoding strand from the tac promoter suppresses it. IPTG had the opposite effect on transcription in cells containing pIQS12. Induction of the tac promoter decreased the transcription of the noncoding strand by a factor of ≈4, while increasing transcription from the coding strand by a factor of ≈26 (Fig. 2). The smaller magnitude of increase of the tac promoter-specific RNA from pIQS11 (9-fold) compared with that from pIQS12 (26-fold), probably reflects different background levels of transcription in the two constructs. Specifically, in pIQS11 presence of the constitutive kan promoter in the same orientation as tac creates a higher basal level transcription from the noncoding strand. It was necessary to include the kan promoter in these plasmids to assure that when the expected C to T mutation occurred, kan+ revertants would grow on plates with kanamycin.
Figure 2.
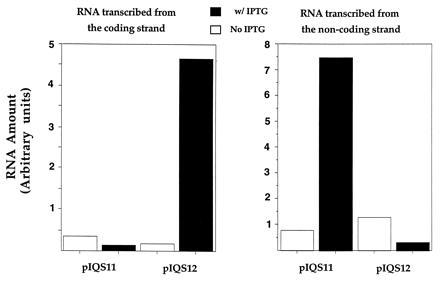
Amounts of transcripts produced from the two strands of pIQS11 and pIQS12. Amounts of RNAs calculated from analysis of PhosphorImager intensities of RNAs hybridized with two different probes are shown. The intensities are proportional to amounts of RNAs from the two strands. Because no effort was made to calculate the absolute RNA amounts from these numbers, the units for the x axis are marked as “arbitrary units.” Amounts of RNAs from the two strands are represented in separate histograms.
Relationship between Transcription and KanR Revertant Frequency.
The changes in KanR revertant frequency correlate well with the level of transcription of the noncoding strand. These experiments were done by dividing a culture started from a bacterial colony of an ung− strain containing the appropriate plasmids into two cultures and adding IPTG to one of the two cultures. Hence, results obtained for members of a pair of cultures could be compared with each other. Induction of the tac promoter in pIQS11 caused ≈4-fold increase in the revertant frequency (Table 1). In similar experiments, induction of pIQS12 caused a 2- to 4-fold decrease in the revertant frequency, consistent with the observed decrease in transcription of the noncoding strand (Fig. 2). Similar changes in revertant frequency were found in a number of different genetic backgrounds in response to induction of the tac promoter and the magnitude of the change was consistent from experiment to experiment (Table 1 and see below).
Table 1.
Effect of transcription on revertant frequency
| Plasmid | Colony | Without IPTG | With IPTG | Ratio* |
|---|---|---|---|---|
| pIQS11 | 1 | 6.3 × 10−7 | 1.1 × 10−6 | 1.8 |
| 2 | 2.2 × 10−7 | 1.2 × 10−6 | 5.2 | |
| 3 | 2.2 × 10−7 | 1.0 × 10−6 | 4.5 | |
| Mean† | (3.6 ± 2.4) × 10−7 | (1.1 ± 0.1)×10−6 | 3.8 ± 1.8 | |
| pIQS12 | 1 | 3.0 × 10−7 | 7.1 × 10−8 | 0.24 |
| 2 | 1.4 × 10−7 | 5.8 × 10−8 | 0.41 | |
| 3 | 1.3 × 10−7 | 7.9 × 10−8 | 0.61 | |
| Mean† | (1.9 ± 1.0) × 10−7 | (6.9 ± 1.0) × 10−8 | 0.42 ± 0.19 |
Ratio of revertant frequency in the presence of IPTG to revertant frequency in the absence of IPTG for the same colony.
Mean (± SD) of revertant frequencies or ratios for the three colonies.
These results are qualitatively similar to the increases and decreases in the level of transcription from the noncoding strand of the plasmids (Fig. 2), establishing a direct correlation between transcription of this strand and mutation frequency. However, it should be noted that the magnitude of the increase in revertant frequency with pIQS11 was always lower than the magnitude of the level of increase in transcription. The likely reason for this discrepancy is discussed below.
Eight independent KanR colonies were selected and plasmid DNAs were isolated from the cells. When the BstNI digestion patterns of these DNAs were compared with the pattern for a plasmid with the kanS-D94 allele, the BstNI site at codon 94 was found to be lost in the revertants (data not shown). This is the expected result if the predicted mutation occurs in codon 94 of the kan gene.
Repair of Transcription-Induced Uracils by UDG.
To show that the observed reversions occurred by a C to U to T pathway, we compared the KanR revertant frequencies in ung+ and ung− backgrounds. Because UDG coded by ung+ removes uracil from single-stranded or double-stranded DNA (32), it is expected to reduce the KanR revertant frequency. Regardless of whether or not the tac promoter was induced, UDG-mediated repair process substantially reduced the revertant frequency (Fig. 3). It reduced the revertant frequency by a factor of 10 to 15, consistent with previous studies of this DNA repair process (33).
Figure 3.
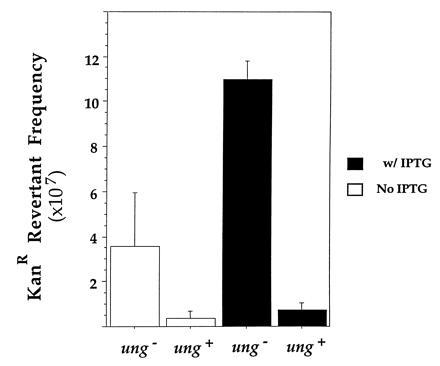
Effect of UDG-mediated repair on revertant frequency. Mean reversion frequencies in cultures from three independent colonies of GM31 or GM31ung containing pIQS11 are shown. The procedure used for this experiment was similar to that used for generating data presented in Table 1 and Fig. 3. Thus, cultures grown with and without IPTG originated from same bacterial colonies. The data have been regrouped and presented in a fashion that allows comparison of cultures with the same state of the tac promoter induction, but which were in different genetic backgrounds.
It should be noted that although UDG-mediated repair reduced the transcription-induced increase in frequency of C to T mutations, it did not eliminate the increase. Following induction of the tac promoter, the frequency of KanR revertants still increased by a factor of ≈2 (Fig. 3). To confirm this, nine independent cultures of ung+ cells were tested for transcription-induced increase in mutations using the procedure described above. In these cultures, there was a net increase in KanR revertant frequency as a result of induction of transcription [(3.6 ± 2.3) × 10−8; P < 0.01]. These results confirm that the observed mutations are caused by the deamination of target cytosine to uracil and suggest that induction of the tac promoter creates more uracils than UDG is able to repair.
Methylation of Target Cytosine Increases KanR Revertant Frequency.
Because rate of deamination of 5meC is 2- to 4-fold higher than that of C (2, 9, 10), if deamination of cytosine to uracil in single-strand DNA were the cause of transcription-induced increase in revertant frequency, then methylation of this cytosine should increase the revertant frequency. To test this, pIQS11 was introduced in a dcm− ung− strain. A second plasmid that codes for Dcm (pDCM72) or the vector pACYC184 was also introduced in these cells. Cells were grown with or without IPTG, and KanR reversion frequencies in the different strains were compared.
Regardless of the state of induction of the tac promoter, methylation caused a 2- to 3-fold increase in the KanR revertant frequency (Fig. 4). Although the effect was small, it was reproducible. In four separate experiments, cytosine methylation increased the revertant frequency by a factor between 1.7 and 3.0 (data not shown). This result further supports the assumption that the KanR revertants arise due to deamination of target C (or 5meC).
Figure 4.
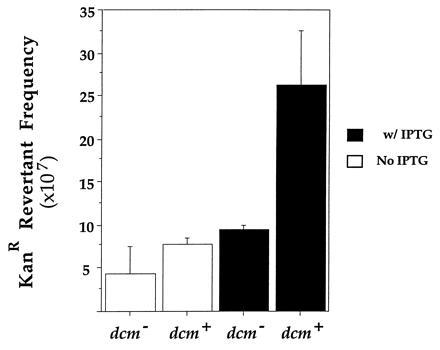
Effect of cytosine methylation on revertant frequency. Mean reversion frequencies in cultures from three independent colonies of GM31ung containing pIQS11 and pACYC184 or pDCM72 are shown. Experiments were carried out and the data processed in a manner similar to that described in legend to Fig. 3.
In E. coli, a specialized mismatch correction process [called very short patch (VSP) repair] corrects T:G mismatches that arise due to deamination of 5meC within Dcm sites to C:G (34). Repair is initiated as a result of endonucleolytic cleavage of the phosphodiester linkage immediately 5′ of the T by the product of the gene vsr (27, 35). DNA polymerase I is thought to replace the mismatched thymine with cytosine (36), and DNA ligase is expected to seal the nick to complete the reaction. At a lower efficiency, VSP repair also corrects U:G mismatches to C:G (37).
To demonstrate that the pathway by which transcription induces 5meC to T mutations involves T:G (or U:G) intermediates, KanR reversion frequencies were determined in vsr+ and vsr− backgrounds and compared. In both backgrounds, cells contained Dcm and hence the target cytosine was methylated. We found that regardless of the state of induction of the tac promoter, VSP repair reduced the revertant frequency by a factor of 2 to 3 (Fig. 5). Once again, although the effect was small, it was reproducible (data not shown). These results confirm the prediction that when the target cytosine is methylated, a majority, and probably all, KanR revertants arise due to deamination of 5meC to T, creating T:G mismatches.
Figure 5.
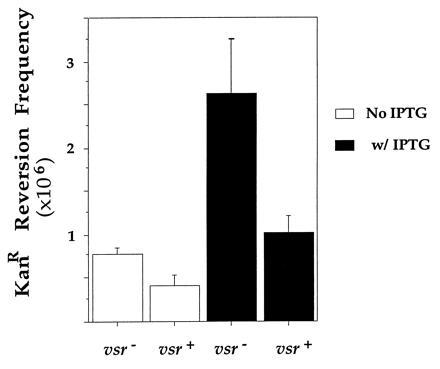
Effect of VSP repair on revertant frequency. Mean reversion frequencies in cultures from three independent colonies of GM31ung containing pIQS11 and pDCM72 or pDCM33 are shown. Experiments were carried out and the data processed in a manner similar to that described in legend to Fig. 3.
DISCUSSION
We have shown here that the frequency of C or 5meC to T mutations increases when the target base is in the nontranscribed strand with respect to an induced tac promoter. There are two reasons for attributing this increase to deaminations of these bases. First, methylation of target cytosine increases the revertant frequency by a factor of 2 to 3- a number consistent with the higher rate of 5meC deamination compared with that of C. Second, DNA mismatch correction systems that are known to repair products of deamination of C and 5meC reduce the mutation frequency without affecting the strand bias.
The simplest way to explain the increase in deamination of C and 5meC is to suggest that when a transcription bubble passes over the target base, the nontranscribed strand is present in single-stranded form, whereas the transcribed strand is present as a RNA/DNA hybrid or is protected by the RNA polymerase (RNAP). Thus, the target cytosine, when present in the nontranscribed strand, is exposed to the hazard of hydrolytic deamination.
Because transcription-coupled repair (21) creates a strand bias in mutations similar to that reported here, it may be argued that a transcription-coupled mismatch correction process creates the observed mutational bias. We consider this to be unlikely for several reasons. Preferential repair of the transcribed strand has only been demonstrated for nucleotide excision repair (38) and base mismatches are poor substrates for this kind of repair (39, 40). Also, because U:G or T:G mismatches do not block transcription, it is difficult to imagine how the mismatch correction process could acquire a strand-bias. Further, we observe a net increase in the revertant frequency as a result of induction of transcription and no DNA repair process should increase the occurrence of mutations. Finally, we have seen the same strand bias even when the host was deficient in repair processes that correct U:G and T:G mismatches. Therefore, if the observed bias in mutations is the result of a bias in DNA repair, it must be due to a repair process that is yet to be described.
We have found that while induction of transcription increases deamination of C and 5meC in the nontranscribed strand, the extent of increase is consistently less than the increase in the level of mRNA. This is probably because the rate of initiation of the tac promoter does not keep up with the rate of elongation of the transcript. As a result, the target cytosine spends a substantially higher fraction of the time in double-strand state than in single-strand state. The rate of deamination of the target cytosine (k) during transcription can be described by the following equation:
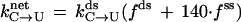 |
1 |
where kds is the rate in double-strand DNA, and where fds and fss are the fractions of time during each cycle of transcription that the target cytosine spends in double-strand and single-strand state, respectively. Under physiological conditions, the rate of deamination of C in single-strand DNA is 140 times the rate in single-strand DNA (14). For the lacUV5 promoter, the half-life of the initiation complex is ≈1 min (41) and the tac promoter is ≈5 times stronger than lacUV5 (42). Hence it is reasonable to assume that the time between each passage of RNAP over target C to be ≈12 sec in the induced state and ≈108 sec (12 × 9) in the uninduced state. If the transcription bubble is assumed to contain a 12 nucleotide single-strand region (43), at a rate of 50 nucleotides per sec, RNAP would clear this bubble in 0.24 sec. In other words, the target C will be in single-strand form for 0.24 sec during each passage of RNAP. Using these numbers in Eq. 1, the knet (induced) is 3.8× kds and knet (uninduced) is 1.3× kds. Thus, the observed ratio for the frequencies of C to T mutations in the induced versus uninduced state of the tac promoter (3.8; Table 1) is comparable to the predicted ratio (2.9).
The foregoing analysis suggests that strand bias in C to T mutations during transcription could be used to study the process of transcript elongation. If the same gene is transcribed from different promoters, the observed mutational strand bias should directly depend upon the strength of the promoter and should inversely depend on the rate of elongation. For example, the rate of elongation by the T7 RNAP is eight times the rate by the E. coli RNAP (44). Therefore, if promoter strengths were similar, genes transcribed by T7 RNAP would be less likely to show strand-biased mutations than E. coli RNAP transcribed genes. Other factors may also influence the strand-biased mutations. For example, the length of DNA in single-strand state is likely to be different for different RNAPs. Different polymerases may create different sizes of transcription bubbles and expose varying stretches of nontranscribed DNA strand. It may be possible to use the strand-biased C to T mutations to study these characteristics of RNAPs or their mutants.
Our observation that the magnitude of the increase in C to T mutations in the nontranscribed strand is significantly less than the magnitude of increase in transcription suggests a reason why this effect may have been largely overlooked before. Most conclusions regarding spontaneous mutations have been based on analyses of mutation spectra of genes. Because such analyses are complicated by base distribution in the target gene and by sequence preferences in mutagenesis as well as DNA repair, small differences in frequency of mutations between the two strands may not be easily apparent. The approach used in our study does not have such limitations and hence is more sensitive.
There is considerable interest in the idea that genes that are highly transcribed may accumulate more spontaneous mutations (17, 18, 45). The results presented here support this idea and predict that during evolution genes should acquire more C to T mutations in the nontranscribed strand. Recently, this was shown to be correct for some enterobacterial genes (20). When the sequences of several genes were compared among numerous E. coli and Salmonella enterica strains, more C to T than G to A mutations were found in the coding strand. Although the authors of that study attributed the differences to strand-bias in DNA repair (20), we propose that they arise from excess C to U conversions in the coding strand. Our results also predict that in different tissues, different genes should mutate at different rates and that the genes whose products are in greatest demand in the cell are most susceptible to spontaneous mutations. Thus, strand bias in cytosine deamination could play a major role in the acquisition of deleterious mutations and in genome evolution.
Acknowledgments
This work was initiated in collaboration with Dr. M. Lieb. We thank Dr. M. Lieb, J. Roberts (Cornell University, Ithaca, NY) and S. Jinks-Robertson (Emory University, Atlanta) for useful discussions. We acknowledge the help of P. Modrich (Duke University, Durham, NC) and R. Ebright (Rutgers University, Piscataway, NJ) in the preparation of the manuscript. This work was supported by National Institutes of Health Grants HG00004, GM45860, and GM53273.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: 5meC, 5-methylcytosine; LB, Luria–Bertani; UDG, uracil-DNA glycosylase; RNAP, RNA polymerase; VSP, very short patch.
References
- 1.Frederico L A, Kunkel T A, Shaw B R. Biochemistry. 1990;29:2532–2537. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- 2.Shen J-C, Rideout W M, III, Jones P A. Nucleic Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coulondre C, Miller J H, Farabaugh P J, Gilbert W. Nature (London) 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 4.de Jong P J, Grosvosky A J, Glickman B W. Proc Natl Acad Sci USA. 1988;85:3499–3503. doi: 10.1073/pnas.85.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halliday J A, Glickman B W. Mutat Res. 1991;250:55–71. doi: 10.1016/0027-5107(91)90162-h. [DOI] [PubMed] [Google Scholar]
- 6.Douglas G R, Gingerich J D, Gossen J A, Bartlett S A. Mutagenesis. 1994;9:451–458. doi: 10.1093/mutage/9.5.451. [DOI] [PubMed] [Google Scholar]
- 7.Gojobori T, Li W-H, Graur D. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- 8.Li W-H, Wu C-I, Luo C-C. J Mol Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 9.Lindahl T, Nyberg B. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 10.Ehrlich M, Norris K F, Wang R Y-H, Kuo K C, Gehrke C W. Biosci Rep. 1986;6:387–393. doi: 10.1007/BF01116426. [DOI] [PubMed] [Google Scholar]
- 11.Lieb M. Genetics. 1991;128:23–27. doi: 10.1093/genetics/128.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper D N, Youssoufian H. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 13.Greenblatt M S, Bennett W P, Hollstein M, Harris C C. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 14.Frederico L A, Kunkel T A, Shaw B A. Biochemistry. 1993;32:6523–6530. doi: 10.1021/bi00077a005. [DOI] [PubMed] [Google Scholar]
- 15.Hayatsu H. Prog Nucleic Acid Res Mol Biol. 1976;16:75–124. doi: 10.1016/s0079-6603(08)60756-4. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro R. In: Chromosome Damage and Repair. Seeberg E, Kleppe K, editors. New York: Plenum; 1981. pp. 3–18. [Google Scholar]
- 17.Fix D F, Glickman B W. Mol Gen Genet. 1987;209:78–82. doi: 10.1007/BF00329839. [DOI] [PubMed] [Google Scholar]
- 18.Davis B D. Proc Natl Acad Sci USA. 1989;86:5005–5009. doi: 10.1073/pnas.86.13.5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skandalis A, Ford B N, Glickman B W. Mutat Res. 1994;314:21–26. doi: 10.1016/0921-8777(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 20.Francino M P, Chao L, Riley M A, Ochman H. Science. 1996;272:107–109. doi: 10.1126/science.272.5258.107. [DOI] [PubMed] [Google Scholar]
- 21.Mellon I, Hanawalt P C. Nature (London) 1989;342:95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 22.Mellon I, Spivak G, Hanawalt P C. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 23.Vrieling H, Van Rooijen M L, Groen N A, Zdzienicka M Z, Simons J W, Lohman P H, van Zeeland A A. Mol Cell Biol. 1989;9:1277–1283. doi: 10.1128/mcb.9.3.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koehler D R, Awadallah S S, Glickman B W. J Biol Chem. 1991;266:11766–11773. [PubMed] [Google Scholar]
- 25.Sockett H S, Romac S, Hutchison F. Mol Gen Genet. 1991;230:295–301. doi: 10.1007/BF00290680. [DOI] [PubMed] [Google Scholar]
- 26.Wyszynski M, Gabbara S, Bhagwat A S. Proc Natl Acad Sci USA. 1994;91:1574–1578. doi: 10.1073/pnas.91.4.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohail A, Lieb M, Dar M, Bhagwat A S. J Bacteriol. 1990;172:4214–4221. doi: 10.1128/jb.172.8.4214-4221.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers W C. Anal Biochem. 1970;33:459–463. doi: 10.1016/0003-2697(70)90316-7. [DOI] [PubMed] [Google Scholar]
- 29.Yebra M J, Bhagwat A S. Biochemistry. 1995;34:14752–14757. doi: 10.1021/bi00045a016. [DOI] [PubMed] [Google Scholar]
- 30.Bandaru B, Gopal J, Bhagwat A S. J Biol Chem. 1996;271:7851–7859. doi: 10.1074/jbc.271.13.7851. [DOI] [PubMed] [Google Scholar]
- 31.Bandaru B, Wyszynski M, Bhagwat A S. J Bacteriol. 1995;177:2950–2952. doi: 10.1128/jb.177.10.2950-2952.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindahl T. Proc Natl Acad Sci USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan B K, Weiss B. J Bacteriol. 1982;151:750–755. doi: 10.1128/jb.151.2.750-755.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieb M, Bhagwat A S. Mol Microbiol. 1996;20:467–473. doi: 10.1046/j.1365-2958.1996.5291066.x. [DOI] [PubMed] [Google Scholar]
- 35.Hennecke F, Kolmar H, Bründl K, Fritz H-J. Nature (London) 1991;353:776–778. doi: 10.1038/353776a0. [DOI] [PubMed] [Google Scholar]
- 36.Dzidic S, Radman M. Mol Gen Genet. 1989;217:254–256. doi: 10.1007/BF02464889. [DOI] [PubMed] [Google Scholar]
- 37.Gabbara S, Wyszynski M, Bhagwat A S. Mol Gen Genet. 1994;243:244–248. doi: 10.1007/BF00280322. [DOI] [PubMed] [Google Scholar]
- 38.Scicchitano D A, Hanawalt P C. Proc Natl Acad Sci USA. 1989;86:3050–3054. doi: 10.1073/pnas.86.9.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas D C, Kunkel T A, Casna N J, Ford J P, Sancar A. J Biol Chem. 1986;261:14496–14505. [PubMed] [Google Scholar]
- 40.Huang J C, Hsu D S, Kazantsev A, Sancar A. Proc Natl Acad Sci USA. 1994;91:12213–12217. doi: 10.1073/pnas.91.25.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stefano J E, Gralla J. Biochemistry. 1979;18:1063–1067. doi: 10.1021/bi00573a020. [DOI] [PubMed] [Google Scholar]
- 42.Amann E, Brosius J, Ptashne M. Gene. 1983;25:167–178. doi: 10.1016/0378-1119(83)90222-6. [DOI] [PubMed] [Google Scholar]
- 43.Kainz M, Roberts J. Science. 1992;255:838–841. doi: 10.1126/science.1536008. [DOI] [PubMed] [Google Scholar]
- 44.Iost I, Guillerez J, Dreyfus M. J Bacteriol. 1992;174:619–622. doi: 10.1128/jb.174.2.619-622.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta A, Jinks-Robertson S. Science. 1995;268:1616–1619. doi: 10.1126/science.7777859. [DOI] [PubMed] [Google Scholar]