

Abstract
The locus control region of the β-globin gene is composed of four erythroid-specific hypersensitive sites. Hypersensitive site 2 has been shown to be a powerful enhancer and contains a tandem repeat sequence for the transcription factors AP1 and NFE2 (activating protein 1 and nuclear factor erythroid 2, respectively). The human NRF2 (NFE2 related factor 2) has been isolated by bacterial expression screening using this core sequence as a probe. p45-NFE2, NRF1, and NRF2 belong to the CNC (“cap ‘n’ collar”) subfamily of the basic region–leucine zipper transcription factors, which exhibits strong homology at specific regions such as the “CNC” and the DNA binding and leucine zipper domains. Although the erythroid-specific p45-NFE2 has been implicated in globin gene regulation, p45-NFE2 null mice succumb to bleedings due to lack of platelets and those that survive exhibit only a mild anemia. To determine the function of NRF2, which we found to be widely expressed in vivo, we have characterized the genomic structure of the mouse NRF2 gene, disrupted the Nrf2 gene by homologous recombination in mouse embryonic stem cells (ES cells), and generated NRF2−/− mice. Homozygous mutant mice developed normally, were not anemic, reached adulthood, and reproduced. Our studies indicate that NRF2 is dispensable for mouse development.
Keywords: locus control region, hypersensitive site, gene knockout, CNC-bZip protein
A wealth of knowledge has been accumulated by the study of the human β-globin gene expression generated since the characterization of the locus control region (LCR) (1, 2, 3), which consists of four erythroid specific DNase I hypersensitive sites (HS1–HS4) and is responsible for the erythroid and developmental stage specific expression of the globin genes (4, 5, 6, 7, 8, 9). Work centered on hypersensitive site 2 (HS2) carried out through in vitro and in vivo footprinting experiments (10, 11) identified a tandem AP1-NFE2 (activating protein 1 and nuclear factor erythroid 2, respectively) repeat sequence (GCTGAGTCATGATGAGTCA). Experiments using transient transfections and transgenic mice (10, 12, 13, 14) suggested that this dimeric motif was essential for strong enhancer activity and that the proteins that bind to this sequence play a critical role in the expression of β-globin and β-globin-like genes in humans.
Work undertaken by several groups to clone the transcription factors that bind to this NFE2 site so far identified three genes that are members of the novel CNC (“cap ‘n’ collar”) subfamily of basic region–leucine zipper (bZIP) transcription factors. CNC (15) is a homeotic gene involved in the development of the head and neck structure in Drosophila. NFE2 is a heterodimer consisting of the hematopoietic-specific p45-NFE2 subunit (16) and a ubiquitously expressed p18-MAF subunit (17). Two p45-NFE2-related genes NRF1 (or LCR-F1) and NRF2 (18, 19, 20) have also been cloned in our laboratory and others. These two genes are not restricted to the erythroid cells; they are widely expressed in different tissues, though with different levels of expression. p45-NFE2, NRF1, and NRF2 constitute a new subfamily transcription factors, for they share homology at several protein domains including the CNC and bZIP. Most notably their DNA binding domains are virtually identical in amino acid sequence with only conservative amino acid substitutions. Although these three genes are mapped to three different chromosomes (21), each is situated within loci that contain a member of gene families such as integrin, collagen, and Hox genes. NRF1 and NRF2 have been shown to be capable of transactivating reporter genes in cell culture (10, 20, 22), but their function in vivo are still unknown.
Targeted disruption of p45-NFE2 (23, 24) results in blockage of maturation of megakaryocytes, but only mild impairment of erythropoieis. Mice lacking p18-MAF show no detectable phenotype and are indistinguishable from their normal littermates (25). Although NRF1 and NRF2 are not up-regulated in the p45-NFE2 null mice (24), they may complement the lack of p45-NFE2. To investigate the function of NRF2 in vivo, we used homologous recombination in embryonic stem cells (26) to generate NRF2 mutant mice.
MATERIALS AND METHODS
cDNA Library Screening.
The human NRF2 cDNA was used to screen a mouse lung cDNA library in λZAPII (Stratagene). Several clones were purified, excised with helper phage, and recircularized to generate subclones in Bluescript SK− phagemid vectors (Stratagene). To determine the sequence of the 5′ end of cDNA, rapid amplification of cDNA 5′ ends (RACE) was carried out according to the manufacturer’s instructions (GIBCO/BRL). poly(A) RNA was isolated from mouse kidney using the FastTrack 2.0 kit (Invitrogen) and used as a template for the synthesis of the first strand cDNA.
Genomic Clones Isolation.
Several phage clones were isolated from a 129SVJ library in λFIX II (Stratagene) through several rounds of screening, first with a mouse mNRF2 cDNA probe and then with oligonucleotides. The whole inserts from the phage clones were subcloned into pBluescript KSII at the NotI site to facilitate their mapping.
Sequence Analysis.
Sequence analysis was performed using the chain termination methods with cycle sequencing kits (Promega and Amersham) and the sequence analysis package from Genetics Computer Group (Madison, WI).
In Situ Hybridization.
Tissues from 15.5-day-old embryos of normal or knockout mice were harvested and fixed immediately in PBS containing 4% paraformaldehyde. After fixation for 24 hr at 4°C, the specimens were washed in PBS, dehydrated through a series of ethanol washes, cleared in xylene, and embedded in paraffin. Sections were cut at 7 microns and mounted on Superfrost/plus (Fisher Scientific) treated slides. Riboprobes were prepared from an EcoRV–HindIII fragment of the cDNA that is in the region deleted by the targeting vector (see below). This fragment is subcloned in pBluescript KSII. [35S]UTP-labeled riboprobes were synthesized with T3 or T7 RNA polymerase from linearized plasmid DNA. Hybridization and washes were carried out according to established protocols (27). The slides were then dipped in NTB2 emulsion (Kodak), and the emulsion was exposed for 4–7 days at 4°C. After development, the slides were stained with eosin and hematoxylin for histology and then mounted with Permount. Microphotography was performed using a Zeiss Axiophot microscope.
Targeting Vector Construction.
The targeting vector was based on the pPNT vector (28), which contains both the positive selection neomycin resistance and the negative selection thymidine kinase markers under the control of the phosphoglycerate kinase promoter and polyadenylylation signal. The 5′ homology region consists of a 5.5-kb NheI–EcoRV fragment that encompasses the 3′ end of intron 1 to part of exon 4. This 5′ fragment was first fused in-frame to a LacZ reporter cassette containing the last intron and polyadenylylation signal of protamine 1 (29) and is subcloned into the NotI–XhoI sites of pPNT. The 3′ homology region consists of a 6.2-kb XbaI–NheI fragment downstream of the NRF2 gene. The construct is linearized with NotI before electroporation.
ES Cell Line and Electroporation.
The JM1 ES cells derived from 129SVJ mice were cultured on embryonic fibroblast feeder layers in DMEM supplemented with 15% FBS/0.1 mM of 2-mercaptoethanol/2 mM glutamine/0.1 mM nonessential amino acid/purified leukemia inhibiting factor (GIBCO/BRL) at 103 units/ml. Cells (n = 107) were electroporated with 20 μg of linearized targeting construct DNA using an electroporator (Bio-Rad) in 1 ml of complete ES cell culture medium at 240 V and 25 mF. Transfected cells were first cultured in nonselective medium overnight, and then subjected to selection with G418 at 200 μg/ml (GIBCO) and ganciclovir at 2 μM (Syntex, Palo Alto, CA). Single colonies were picked after 10–12 days, and cells were expanded in 24-well dishes. Aliquots of each clone were frozen for stock and the rest were expanded for DNA analysis. The ES cells with homologous recombination were injected into blastocysts, and chimeric animals were then bred to produce F1 mice. Southern blots were used to identify germ-line transmission, and the heterozygous animals were interbred to produce homozygous knockout animals.
Northern and Southern Blot Analyses.
Northern and Southern blots analyses were performed according to standard procedures (30). Total RNA was extracted from intestine, kidney, stomach, and testis using RNAzol B (Biotex Laboratories, Edmonton, Canada), and poly(A) RNA was subsequently isolated. Multiple tissue RNA blots and mouse embryo blots were purchased from CLONTECH.
Hematologic Analysis.
Blood was collected from adult mice by retroorbital puncture. The hematocrit, hemoglobin concentration, mean corpuscular volume, mean corpuscular hemoglobin, reticulocyte count were done (CVD, Sacramento, CA).
Reverse Transcription–PCR Analysis.
Total RNA was used as template for the first strand cDNA synthesis using Superscript II reverse transcriptase (GIBCO/BRL) and random hexamers. Primer pairs spanning at least one intron were used for PCR analysis to distinguish amplification products of cDNA from those of genomic DNA.
RESULTS
mNRF2 cDNA.
Sequence analysis of 13 clones isolated from a mouse lung cDNA library revealed that they were all cDNA fragments derived from the same messenger RNA. Two of the overlapping clones covered 2005 bases, excluding the poly(A) tail. RACE using gene specific primers on exon 2 extended the final clone to 2380 bases. After we obtained the mouse sequence, the mNRF2 sequence was reported by Chui et al. (31). Our sequence data is similar to that published one with several differences. (i) The translation start site in our sequence is extended by 16 amino acids at the amino end. We believe that these 16 amino acids are translated as they are present in a human cDNA sequence of Nrf2 (GenBank accession no. T39964T39964) (Fig. 1A). The difference between our translation sequence and that of Chui et al. (31) could be due to a G nucleotide missing in their sequence. This G nucleotide is present in both our cDNA and genomic sequences. (ii) The 5′ untranslated region of our cDNA, determined from the analysis of six RACE clones, is shorter than that of Chui et al. (31), whose sequence extends for approximately 105 nucleotides further. This difference is unexplained. This extension is present in our genomic sequence with a few mismatches (Fig. 1B).
Figure 1.

Sequence of NRF2. (A) Comparison of the putative translation start site of NRF2. The translation of the murine cDNA at the exon 1 and exon 2 junction is compared with the translation of the human NRF2 sequence T39964T39964 found in the EST data base. The extra amino acids are italicized, the methionine described in previous publications is in boldface type, and the G nucleotide underlined is missing in entry U20532 (GenBank) (31). (B) Alignment of the 5′ ends of the cDNAs (Nrf2cDNA and U20532) and genomic (Nrf2GEN) sequences of the first exon of mNrf2. The numbers above the cDNA represent the number of RACE clones that start at that base. ∗, Putative translation start sites derived from our sequence; •, the published translation start site (31). The sequence of intron 1 is between parentheses with gt representing the splice donor junction; sequence differences are underlined; the missing nucleotide G in U20532 is in boldface italicized type in our cDNA and genomic sequences.
Genomic Structure of mNRF2.
The transcriptional unit of mNRF2 was mapped within 30 kb segment of genomic DNA. The NRF2 gene consists of five exons interrupted by four introns, the first of which spans ≈25 kb. The sequences at all the exon–intron boundaries were determined and are shown in Table 1. The DNA sequence was generated by a combination of subcloning and primer walking. The promoter of NRF2 is very GC rich and contains neither a TATA box nor a CCAAT box. Several SP1, EKLF, and AP-2 binding sites are found within the first 500 bp upstream of exon 1. Similar to the p45-NFE2 locus (32), a gene coding for an heterogenous nuclear ribonucleoprotein (hnRNP) is found 10 kb downstream of NRF2 with its transcriptional direction opposite that of NRF2. This is further evidence suggesting that the CNC-bZIP family of gene evolved from one ancestral gene.
Table 1.
Sequences at the exon–intron boundaries of murine Nrf2
| Exon no. | Exon size | Sequences at exon–intron junctions (splice acceptor//splice donor) |
|---|---|---|
| 1 | 184 | Promoter //TCCCAGCAGgtgctgccc |
| 2 | 267 | taaacatagGACATGGAT//TACTCCCAGgtacactcg |
| 3 | 90 | ttcctccagGTTGCCCAC//GACCATGAGgtataaaaa |
| 4 | 168 | gcatttcagTCGCTTGCC//GAATTACAGgtaagagag |
| 5 | 1671 | ctttcctagTGTCTTAAT//ACTTAAACTACTTTGG.. |
Sequences of exons are in uppercase, introns are in lowercase, invariant dinucleotide splicing junction are in boldface, and genomic sequence downstream of polyadenylation site are italicized.
mNRF2 Expression in Tissues.
Northern blot analysis showed that like the human NRF2, mNRF2 is also expressed in a wide range of tissues at different levels in different tissues (Fig. 2 Left). Northern blot analysis of mouse fetal RNA shows significant amounts of NRF2 mRNA from developmental stage of day 7 to 17. (Fig. 2 Right). In addition, reverse transcription–PCR analysis of ES cells RNA revealed expression of NRF2, indicating that NRF2 is expressed from cells at the blastocyst stage (data not shown). In situ hybridization (Fig. 3) using 35S-labeled riboprobe on section of 15.5-day-old embryos shows that although NRF2 is expressed at low levels in many tissues, it is expressed at high levels in several organs such as the liver, lungs, and kidneys. The highest level of expression is found in the cells lining the digestive tract, extending from the esophagus to the small and large intestines. In the central nervous system, NRF2 is expressed in the lateral ventricle and its medial wall and the choroid plexus in the fourth ventricle, which is the site of the future pituitary. NRF2 expression can also be found in the olfactory epithelium in the nasal cavity, the thyroid and submandibular glands, and the brown fat layer in the back. On higher magnification, NRF2 expression is most pronounced in the luminal cells of the stomach and intestine, the lining of the bronchi and alveoli and the renal tubules (Fig. 4).
Figure 2.
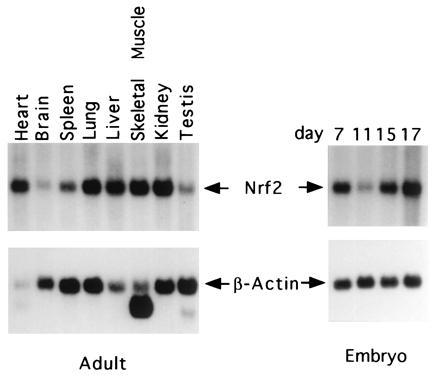
Expression of the mNRF2. Adult mouse multiple tissue RNA blots (Left) and mouse embryos RNA blots (Right) hybridized with mNRF2 and β-actin.
Figure 3.
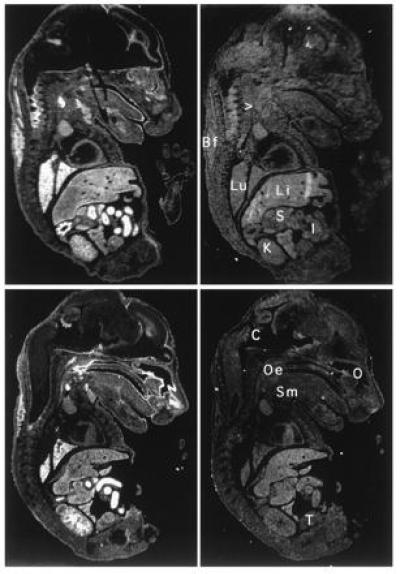
Localization of NRF2 mRNA in the mouse embryo. In situ hybridization was performed on neighboring sections using antisense (Left) and sense (Right) probe. Two saggital sections are shown under darkfield illumination. Li, liver; Lu, lung; K, kidney; I, intestines; S, stomach; Oe, esophagus entrance (nasopharynx); C, choroid plexus; Sm, submandibular gland; O, Olfactory epithelium; T, testis; Bf, brown fat; > points to the thyroid.
Figure 4.
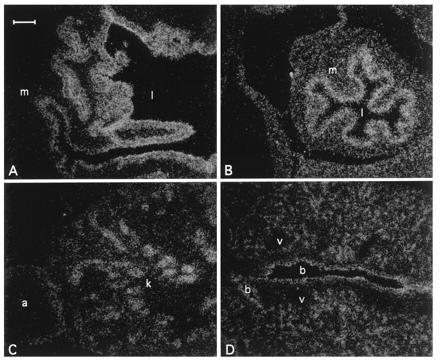
Localization of Nrf2 mRNA in the day 16 embryo developing stomach (A), intestine (B), kidney (C), and lung (D) under darkfield illumination. m, Muscle; l, lumen; k, kidney; a, adrenal gland; b, bronchus; v, blood vessel. (Bar = 100 μm.)
Production of NRF2−/− Mice.
A replacement type targeting vector was constructed as shown (Fig. 5A). The 5′ and 3′ homologous genomic sequences were 5.5 kb and 6.2 kb long, respectively. In this targeting vector, a 4.2-kb segment of DNA containing part of exon 4 and all of exon 5 of NRF2 is replaced by a 5.5-kb fragment of DNA coding for the bacterial gene LacZ followed by the positive selection neomycin resistance cassette. This replacement vector was engineered to delete the carboxyl 457 amino acid of the NRF2 molecule and to splice in the bacterial reporter gene LacZ. This design would effectively nullify the NRF2 gene function, since the CNC bZIP regions are deleted, and the construct provides a means of monitoring the NRF2 promoter activity. DNA from ES cell clones was analyzed by Southern blotting to screen for homologous recombinants (Fig. 5 B and C). Of the 60 clones analyzed, 10 NRF2+/− heterozygous ES clones were identified, thus a targeting efficiency of 1 in 6 was achieved in JM1 ES cells.
Figure 5.
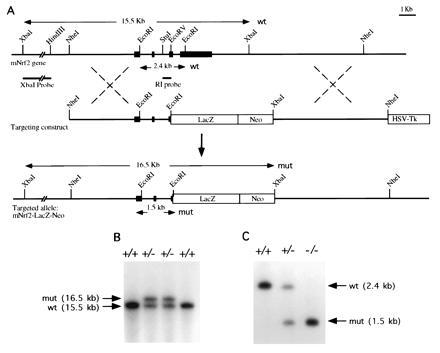
(A) Genomic sequence and targeting constructs with the resultant NRF2 knockout gene. (B) Southern blot analysis of ES cells. An external probe XbaI–HindIII fragment gave a 15.5-kb band for the wild-type and 16.5-kb band for the homologous recombinant. (C) Southern blot analysis of F2 offspring. A StuI–EcoRV probe gave a 2.4-kb band for the wild type and a 1.5-kb band for the recombinant.
Chimeric mice were generated, and germ-line transmission was detected. Heterozygous F1 mice appeared normal and were interbred to generate homozygous NRF2−/− mice. Of 63 offspring, 17 (27%) were homozygous for the disrupted allele and 17 (27%) were wild type, indicating a normal Mendelian inheritance pattern.
Analysis of NRF2−/− Mice.
The homozygous mice were normal in appearance and showed no discernible difference in their development or in their behavior compared with their wild-type littermates. At 6 weeks of age, wild-type, NRF2+/−, and NRF2−/− were killed for gross and histological examination. No obvious anatomical differences in the major organs were seen. Histological examination of the stomach, intestine, and kidney did not reveal any abnormality in the heterozygous and homozygous animals (data not shown). Hematological indices of NRF2−/− mice were all within the range of heterozygous and wild-type littermates (data not shown). Attempts at staining adult tissues with 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) failed to detect any β-galactosidase activity. Homozygous and heterozygous embryos isolated at 13.5 and 15.5 days of gestation appeared to have developed normally and synchronously with wild-type littermates up to 9 months of age.
RNA Analysis of NRF2−/− Mice.
In situ hybridizations were carried out on paraffin section of adult stomach and intestine from wild-type and NRF2−/− mice using a riboprobe synthesized from a fragment of cDNA that was deleted in the targeted allele. No hybridizations were detected on sections of stomach and intestine from NRF2−/− mice (data not shown). poly(A) RNA from intestine, kidney, stomach, and testis were isolated from wild-type, NRF2+/−, and NRF2−/− mice and analyzed by Northern blotting. A 500-bp cDNA probe (BsrDI–EcoRV) hybridized to 2.5-kb message on the NRF2+/+ RNA, to 2.5- and 4.0-kb messages in the NRF2+/− RNA, and only to the 4.0-kb message in the NRF2−/− RNA (Fig. 6). A lacZ probe only hybridized to this 4.0-kb message (data not shown). This larger 4.0-kb transcript produced from the mutated allele appears to be unstable, as it hybridized much less intensely than the 2.5 kb RNA, and was barely discernible in the RNA from some tissues.
Figure 6.
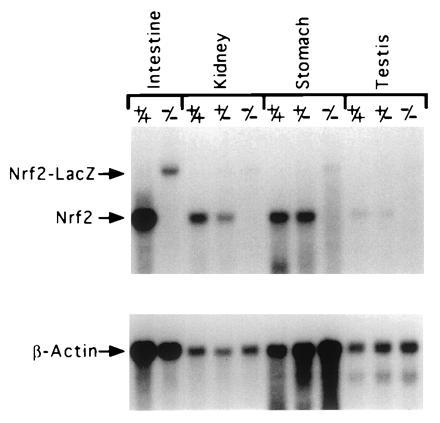
Northern blot analysis of RNA from wild-type, NRF2+/−, and NRF2−/− mice. The NRF2 message is 2.5 kb, and the NRF2-LacZ chimeric message is 4 kb.
DISCUSSION
Our analysis on DNA from the targeted ES cells as well as the DNA from the ES cells derived mice shows that we have disrupted the NRF2 gene. Several facts indicate that the modification in the NRF2 gene results in a null mutation. The LacZ cassette linked to the neomycin cassette in the targeting construct replaced part of exon 4 and the whole of exon 5, which contain the CNC, DNA binding, and leucine zipper domain, leaving about 125 amino acids of the amino end of the NRF2 protein linked in-frame to the LacZ reporter gene. This extensive deletion is borne out by Northern blot analysis, reverse transcription–PCR, and in situ hybridization. From these observations, we conclude that no functional NRF2 protein could possibly be made in the Nrf2 null mice.
It was unfortunate that the lacZ gene fused to the NRF2 gene failed to generate any detectable β-galactosidase activity for it could have provided a means of monitoring the expression of NRF2 in the mice throughout their development. A test construct using a cDNA that contained exon 2 to part of exon 4 fused to the same LacZ cassette driven by the cytomegalovirus promoter successfully showed β-galactosidase activity upon X-Gal staining when transiently transfected in 293 kidney cells. It is possible that the 16 extra amino acids coded by exon 1 in addition to the one coded by exon 2 to exon 4 created enough steric bulk to abolish the activity of the chimeric lacZ molecule. Alternately, as shown by the Northern blot data, chimeric NRF2-LacZ transcript is much less abundant than the wild type and is unstable.
As NRF2 is expressed widely, and even in ES cells, one might expect that the homozygous null mice would exhibit defect in embryogenesis. Yet, interbreeding of heterozygous animals resulted in a normal Mendelian ratio of homozygous null offspring, which are indistinguishable from their heterozygous and normal littermates. They are fertile and produce normal litter size. These results show that NRF2 gene function is not necessary for mouse development, growth, and fertility.
It is interesting that using a p45-NFE2 probe to screen a chicken cDNA library, Itoh and coworkers (33) isolated a cDNA, termed Ech, that encodes a bZIP protein more homologous to NRF2 than p45-NFE2. Of the 40 of 42 ECH clones Itoh and coworkers (33) identified, all were homologous to NRF2 and none to p45-NFE2. They also showed that this chicken NRF2-like gene is highly expressed in hematopoietic cells as well as some other tissues. It heterodimerizes with MAF protein and transactivates through the NFE2 motifs. Perhaps NRF2 serves important hematopoietic functions in the avian system, but is replaced by p45-NFE2 or NRF1 in mammals through evolution.
Although the NRF2 null mice are viable and exhibit an outwardly normal phenotype, we cannot rule out that lack of NRF2 results in subtle changes that do not produce a recognizable phenotype under laboratory conditions, but will only reveal changes that can be detected when subjected to specific challenges or when the mice age. NRF2 absence may also have consequences in compound knock-out animals involving other CNC-bZIP genes. Experiments are in progress to address these issues.
Acknowledgments
We thank Dr. Jefferson Chan for reviewing the manuscript, Dr. T. S. B. Yen for reviewing the sections from the mice, Mei-Chi Cheung for her help in the art work, and Lisa Woldin for editorial assistance. The work is supported in part by National Institutes of Health Grants DK16666 and DK50267. Y.W.K. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Tuan D, Solomon W, Li Q, London I M. Proc Natl Acad Sci USA. 1985;82:6384–6388. doi: 10.1073/pnas.82.19.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forrester W C, Thompson C, Elder J T, Groudine M. Proc Natl Acad Sci USA. 1986;83:1359–1363. doi: 10.1073/pnas.83.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forrester W C, Takegawa S, Papayannopoulou T, Stamatoyannopoulos G, Groudine M. Nucleic Acids Res. 1987;15:10159–10177. doi: 10.1093/nar/15.24.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosveld F, van Assendelft G B, Greaves D R, Kollias G. Cell. 1987;51:975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 5.Curtin P T, Liu D P, Liu W, Chang J C, Kan Y W. Proc Natl Acad Sci USA. 1989;86:7082–7086. doi: 10.1073/pnas.86.18.7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enver T, Ebens A J, Forrester W C, Stamatoyannopoulos G. Proc Natl Acad Sci USA. 1989;86:7033–7037. doi: 10.1073/pnas.86.18.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan T M, Behringer R R, Martin N C, Townes T M, Palmiter R D, Brinster R L. Genes Dev. 1989;3:314–323. doi: 10.1101/gad.3.3.314. [DOI] [PubMed] [Google Scholar]
- 8.Enver T, Raich N, Ebens A J, Papayannopoulou T, Costantini F, Stamatoyannopoulos G. Nature (London) 1990;344:309–313. doi: 10.1038/344309a0. [DOI] [PubMed] [Google Scholar]
- 9.Liu D, Chang J C, Moi P, Liu W, Kan Y W, Curtin P T. Proc Natl Acad Sci USA. 1992;89:3899–3903. doi: 10.1073/pnas.89.9.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moi P, Kan Y W. Proc Natl Acad Sci USA. 1990;87:9000–9004. doi: 10.1073/pnas.87.22.9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikuta T, Kan Y W. Proc Natl Acad Sci USA. 1991;88:10188–10192. doi: 10.1073/pnas.88.22.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ney P A, Sorrentino B P, McDonagh K T, Nienhuis A W. Genes Dev. 1990;4:993–1006. doi: 10.1101/gad.4.6.993. [DOI] [PubMed] [Google Scholar]
- 13.Collis P, Antoniou M, Grosveld F. EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang J C, Liu D, Kan Y W. Proc Natl Acad Sci USA. 1992;89:3107–3110. doi: 10.1073/pnas.89.7.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohler J, Vani K, Leung S, Epstein A. Mech Dev. 1991;34:3–9. doi: 10.1016/0925-4773(91)90086-l. [DOI] [PubMed] [Google Scholar]
- 16.Andrews N C, Erdjument B H, Davidson M B, Tempst P, Orkin S H. Nature (London) 1993;362:722–728. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 17.Andrews N C, Kotkow K J, Ney P A, Erdjument-Bromage H, Tempst P, Orkin S H. Proc Natl Acad Sci USA. 1993;90:11488–11492. doi: 10.1073/pnas.90.24.11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan J Y, Han X L, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11371–11375. doi: 10.1073/pnas.90.23.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moi P, Chan K, Asunis I, Cao A, Kan Y W. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caterina J J, Donze D, Sun C W, Ciavatta D J, Townes T M. Nucleic Acids Res. 1994;22:2383–2391. doi: 10.1093/nar/22.12.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan J Y, Cheung M C, Moi P, Chan K, Kan Y W. Hum Genet. 1995;95:265–269. doi: 10.1007/BF00225191. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M, Yamamoto M. Nature (London) 1994;367:568–572. doi: 10.1038/367568a0. [DOI] [PubMed] [Google Scholar]
- 23.Shivdasani R A, Rosenblatt M F, Zucker-Franklin D, Jackson C W, Hunt P, Saris C J, Orkin S H. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 24.Shivdasani R A, Orkin S H. Proc Natl Acad Sci USA. 1995;92:8690–8694. doi: 10.1073/pnas.92.19.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotkow K J, Orkin S H. Proc Natl Acad Sci USA. 1996;93:3514–3518. doi: 10.1073/pnas.93.8.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas K R, Capecchi M R. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 27.Angerer L M, Angerer R C. In: In Situ Hybridization: A Practical Approach. Wilkinson D G, editor. Oxford: IRL; 1992. pp. 15–32. [Google Scholar]
- 28.Tybulewicz V L, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 29.Peschon J J, Behringer R R, Brinster R L, Palmiter R D. Proc Natl Acad Sci USA. 1987;84:5316–5319. doi: 10.1073/pnas.84.15.5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsh E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Chui D H, Tang W, Orkin S H. Biochem Biophys Res Commun. 1995;209:40–46. doi: 10.1006/bbrc.1995.1467. [DOI] [PubMed] [Google Scholar]
- 32.Lu S J, Rowan S, Bani M R, Ben-David Y. Proc Natl Acad Sci USA. 1994;91:8398–8402. doi: 10.1073/pnas.91.18.8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Mol Cell Biol. 1995;15:4184–4193. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]