

Abstract
Various studies suggest that eukaryotic chromosomes may occupy distinct territories within the nucleus and that chromosomes are tethered to a nuclear matrix. These constraints might limit interchromosomal interactions. We have used a molecular genetic test to investigate whether the chromosomes of Saccharomyces cerevisiae exhibit such territoriality. A chromosomal double-strand break (DSB) can be efficiently repaired by recombination between flanking homologous repeated sequences. We have constructed a strain in which DSBs are delivered simultaneously to both chromosome III and chromosome V by induction of the HO endonuclease. The arrangement of partially duplicated HIS4 and URA3 sequences around each HO recognition site allows the repair of the two DSBs in two alternative ways: (i) the creation of two intrachromosomal deletions or (ii) the formation of a pair of reciprocal translocations. We show that reciprocal translocations are formed approximately as often as the pair of intrachromosomal deletions. Similar results were obtained when one of the target regions was moved from chromosome V to any of three different locations on chromosome XI. These results argue that the broken ends of mitotic chromosomes are free to search the entire genome for appropriate partners; thus, mitotic chromosomes are not functionally confined to isolated domains of the nucleus, at least when chromosomes are broken.
Mitotic recombination in Saccharomyces cerevisiae can be initiated by site-specific double-strand breaks (DSBs) created by the HO endonuclease (1, 2). One efficient mechanism of DNA repair, termed single-strand annealing (SSA), occurs intrachromosomally and results in deletions between homologous DNA sequences flanking the DSB (3). This process involves extensive 5′ to 3′ degradation of DNA from the site of a DSB until flanking regions of homology are exposed and the complementary sequences on either side of break can anneal. SSA occurs efficiently even when the flanking homologous regions are separated by as much as 15 kb or when the extent of shared homology of the flanking regions is only a few hundred bp (4, 5). It is important to note that SSA is not a minor pathway in yeast, used only when cells cannot repair a break by gene conversion. When a DSB is created in a region that can be repaired either by intrachromosomal gene conversion or by SSA, more than two-thirds of the events occur by SSA (5). Very similar results are observed when a different site-specific endonuclease, I-SceI, is expressed in S. cerevisiae (6).
But is SSA inherently an intrachromosomal pathway? Given a choice, would the two ends of one DSB reanneal more readily than the ends of two independent DSBs on different chromosomes? There is abundant evidence from many organisms that chromosomes exist in association with a nuclear matrix that may significantly restrict the ability of different chromosomes or chromosome regions to interact with each other (7, 8, 9). These studies suggest that chromosomal DNA is arranged in loops of 50–100 kb. Other studies imply that each chromosome may occupy a distinct territory within the nucleus (10, 11).
In S. cerevisiae, specific chromosomal regions such as origins of DNA replication, centromeres, and telomeres appear to be preferentially associated with the nuclear envelope, at least at some stages of the cell cycle (8, 12, 13, 14). Such tethering might restrict how broken chromosome ends can behave. For example, if each chromosome were restricted to a domain of the nucleus one might expect that intrachromosomal SSA would be considerably favored over interchromosomal interactions. This might be especially true if a DSB and its flanking homologous regions were all initially tethered within one chromosomal loop.
To test this idea, we have adopted a strategy illustrated in Fig. 1. Two simultaneous DSBs were introduced into two nonhomologous chromosomes of a haploid strain. DNA sequences flanking the DSBs were arranged in such a way that these breaks could be repaired by SSA, but in two alternative ways. First, the two breaks could be repaired by a pair of intrachromosomal deletions (Fig. 1A). Alternatively, the breaks could be healed by a pair of interchromosomal events, creating a pair of reciprocal translocations (Fig. 1B). If there were no constraints on how each broken chromosome end would locate and anneal with another end, we would expect that inter- and intrachromosomal repair should be approximately equivalent; however, if each chromosome was confined to a local territory, we would predict that intrachromosomal repair should be much more frequent than interchromosomal translocations. Surprisingly, we find that intrachromosomal deletions are not favored over interchromosomal joinings.
Figure 1.
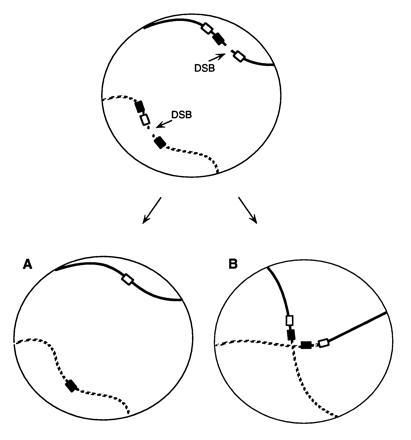
Alternative ways of repairing two DSBs. In the nucleus of a haploid strain illustrated here, repair of two DSBs by SSA can occur in two different ways. There may be two intrachromosomal deletion events (A); alternatively, there may be a pair of reciprocal translocations by annealing between equivalently sized homologous regions on two different chromosomes (B).
MATERIALS AND METHODS
Strains and Plasmids.
Plasmid pJH825, a derivative of plasmid pJH186 (15, 16), was constructed by the insertion of a BamHI fragment carrying two inverted copies of the 117-bp MATa HO recognition/cleavage site (cs) flanking the 2.0-kb HpaI–SalI fragment of the LEU2 gene. This cs::LEU2::cs insert was in turn derived from plasmid pVR196, kindly provided by Victoria Lundblad (Baylor College of Medicine), that carried the two cut sites flanking a URA3 gene with HindIII sites at either end. Plasmid pJH825 was then inserted into the HIS4 region of strain XW161 as described (15, 16) to generate the chromosome III structure shown in Fig. 2A. This MATα strain was then crossed with its congenic strain, NR238-7A, to obtain a MATa-inc segregant carrying the his4::(URA3–cs::LEU2::cs–Δhis4Δ) region (strain G304). Similarly, plasmid pJH1113 was constructed by inserting the cs::LEU2::cs BamHI fragment into the BamHI site of plasmid pJH816. This plasmid was then targeted into the ura3-52 locus of XW161, yielding strain G372, which has the insertion on chromosome V that is illustrated in Fig. 2A. Strains G304 and G372 were then crossed and meiotic segregants were screened to obtain strain G378, carrying MATa-inc his4:: (URA3–cs::LEU2::cs–Δhis4Δ)ura3-52::(Δhis4Δ-cs::LEU2:: cs::URA3) and the GAL::HO plasmid pFH800 (17). The MATa-inc locus cannot be cleaved by HO so that only the cs::LEU2::cs regions are cleaved.
Figure 2.
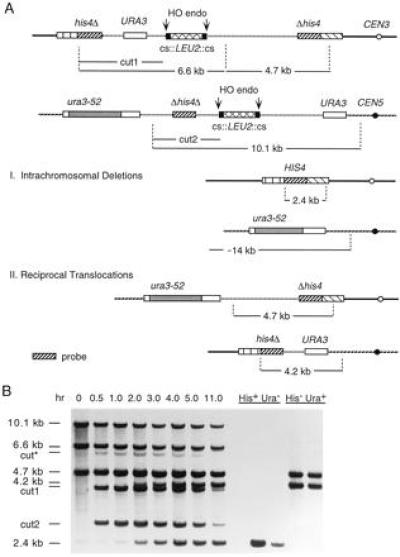
Repair of two broken chromosomes by single-stand annealing between URA3 and HIS4 sequences on chromosomes III and V. (A) pBR322 plasmids carrying URA3 and Δhis4Δ sequences and the LEU2 gene flanking by a pair of HO cs were integrated into chromosome III (at HIS4) and chromosome V (at ura3-52), as shown. The ura3-52 allele contains a large insertion of a Ty element. After induction of HO endonuclease, the broken chromosomes may be repaired by two intrachromosomal deletion events, leading to the loss of all plasmid sequences (vertical hatched lines). Alternatively, there may be a pair of reciprocal translocations by annealing between equivalently sized homologous regions on two different chromosomes. The vertical dotted lines indicate the positions of PvuII restriction endonuclease sites. (B) Southern blot analysis of the kinetics of appearance of deletions and translocations. DNA was extracted from cells at the intervals indicated after HO induction and digested with PvuII. The Southern blot was probed with Δhis4Δ fragment indicated by the dark diagonally hatched lines in A. The fragments characteristic of a pair of intrachromosomal deletions (His+ Ura−) and of a pair of reciprocal translocations (His− Ura+) are shown on the right side of the figure. Two examples of each type are shown. cut* indicates the expected size of a PvuII restriction fragment if HO cleaves only the centromere-proximal site on chromosome III, thus replacing fragment “cut1” with a 6.4-kb fragment (see A, above). The 4.7-kb fragment is present in both the parental strain and in derivatives containing reciprocal translocations. The 4.2-kb fragment is diagnostic of reciprocal translocations while the 2.4-kb fragment is indicative of an intrachromosomal deletion.
In a second set of experiments, plasmid pJH1113 was inserted by homologous recombination between URA3 sequences on the plasmid and a URA3 gene that had been introduced into each of three different sites on chromosome XI: at YKL008C::URA3, an open reading frame located on the left arm of chromosome XI, 11 kb from its centromere; at apn1::URA3, 215 kb from the CEN11 and at tif1::URA3, 115 kb from the centromere on the opposite arm. Strains containing each of these gene disruptions were in the same S288c background as the other strains used in these experiments, and were provided by C. Fairhead and B. Dujon (Institut Pasteur). The URA3 genes of the disruptions on chromosome XI are each oriented in the same direction (5′ to 3′ toward the centromere) as ura3-52 on chromosome V and do not create duplicated sequences surrounding the URA3 insertions. Each of these strains were transformed with plasmid pJH1113 and shown to have an insertion of the plasmid at the chromosome XI URA3 locus. Meiotic segregants, designated G542, G549, and G552, were obtained after crossing G304 or a MATα-inc derivative with strains containing the pJH1113 insertion at one of the three chromosome XI genes (YKL008C, TIF1, and APN1, respectively). These MATa-inc haploid segregants contain the his4::(URA3–cs::LEU2::cs–Δhis4Δ) region on chromosome III and the URA3::(Δhis4Δ-cs::LEU2::cs::URA3) insert on chromosome XI, plus plasmid pFH800.
Growth and Induction of HO Endonuclease.
Cells were grown to stationary phase in synthetic dextrose medium lacking tryptophan (18) (to retain plasmid pFH800) and then transferred to yeast extract/peptone-lactate medium (1% yeast extract/2% peptone/2% lactic acid) overnight. A logarithmically growing culture (doubling time = 5 hr) was then induced for expression of the HO endonuclease gene by adding galactose to a final concentration of 2% (19). Cells were plated either on yeast extract/peptone-dextrose medium or synthetic dextrose medium lacking tryptophan medium and colonies were tested for nutritional requirements by replica plating to appropriate drop-out media.
DNA Analysis.
DNA extraction and Southern blot analysis was carried out as previously described by White and Haber (19).
Statistical Analysis.
A matrix G-test (20), written for Macintosh Hypercard by E. Louis (John Radcliffe Hospital, Oxford), was used to evaluate the statistical significance of the results.
RESULTS AND DISCUSSION
Interchromosomal End-Joining Is as Efficient as Intrachromosomal Repair.
To explore how broken chromosome ends are repaired in yeast we have created haploid strain G378 in which two pairs of DSBs are generated on different chromosomes (Fig. 2A). The HO cs were inserted ≈35 kb from the centromeres on chromosomes III and V, and 67 and 152 kb from their respective telomeres. Induction of HO endonuclease from a galactose-inducible promoter is very efficient, so that HO cleavage of the cut sites results in the liberation of a LEU2 fragment from each chromosome. Thus, the chromosomal ends of the DSBs are separated by about 2 kb after HO cleavage. The DSB on chromosome III is surrounded by partially duplicated copies of the HIS4 gene, so that an intrachromosomal deletion repair event produces a His+ recombinant. The interval between his4Δ and Δhis4 also contains the URA3 gene, which would also be lost when the flanking his4 segments recombine. On chromosome V, the pair of HO cut sites are flanked by ura3-52 and URA3; nearly always, SSA produces a deletion containing ura3-52 (4); in 1% of the cases, SSA yields a URA3 cell. This chromosome V interval also contains the internally duplicated piece of his4 (Δhis4Δ) that is found on chromosome III; this segment will also be lost after annealing between the ura3 sequences on chromosome V. The pair of intrachromosomal deletions nearly always yield a His+ Ura− cell, that is also Leu− (Fig. 2A). Rare Ura+ His+ Leu− colonies were presumed to have undergone a similar process, but where SSA produced URA3 and not ura3-52 on chromosome V. these were not counted in subsequent analysis.
The additional URA3 sequence on chromosome III and the Δhis4Δ piece on chromosome V are both oriented so that an alternative repair event can occur: the joining of URA3 on chromosome III with URA3 on chromosome V and the annealing of Δhis4Δ on chromosome V with the Δhis4 segment on chromosome III. This produces a His− Ura+ (and Leu−) cell that contains a pair of reciprocal translocations (Fig. 2A). The sizes of the homologous regions that lead to the pair of intrachromosomal deletions are identical to those that yield the pair of interchromosomal joinings.
Single cells of strain G378 were plated on synthetic galactose medium plates lacking tryptophan and grown into colonies. This medium retains the TRP1 GAL::HO plasmid pFH800 and thus the HO endonuclease is expressed. Among 79 colonies, 23 were His+ Ura− and Leu−, the phenotype expected for the pair of intrachromosomal deletions; the remaining 56 were His− Ura+ and Leu−, the phenotype expected for the pair of interchromosomal joinings to produce the reciprocal translocations. As expected, all of the cells had lost both LEU2 copies originally situated in between pairs of HO cut sites. We used Southern blots of four randomly chosen colonies of each phenotype to confirm that the His+ Ura− cells were indeed produced by two intrachromosomal SSA events and that the His− Ura+ cells had the restriction fragments expected for reciprocal translocations (Fig. 2B).
In a second experiment, strain G378 was grown in liquid YEP-lactate medium and then HO endonuclease was induced by the addition of galactose. DNA was collected at intervals and the time course of DNA breakage and repair was analyzed by the Southern blot shown in Fig. 2B. Induction of HO is sufficient to cleave nearly all of the four HO cs on chromosomes III and V, as the intensities of higher molecular weight fragments indicative of cutting only one of the two sites flanking LEU2 are very faint relative to the fragments created by cleaving both sites (compare bands cut* and cut1 in Fig. 2B). Approximately 2 hr after HO induction, one sees the appearance of the 2.4- and 4.2-kb PvuII restriction fragments indicative of the intrachromosomal deletion and the interchromosomal translocation, respectively. A very faint amount of deletion product is seen at time 0, most likely because of the leaky expression of HO during growth of the culture under noninducing conditions (4, 5). The time of appearance of both types of events is approximately the same.
In this and similar experiments, cells were spread on yeast extract/peptone-dextrose medium plates after 2 hr of galactose induction and grown into colonies. Despite having to repair two DSBs, cell viability after galactose induction was 65% (413/634) as measured for all cells and 64% (354/556) for those that retained the TRP1-marked GAL::HO plasmid pFH800. Much of the inviability is apparently the consequence of adding galactose to cells, independent of the expression of the GAL::HO gene, as the proportion of cells lacking the TRP1 plasmid did not decrease among cells plated after galactose induction. Approximately 85% of the Trp+ cells had become Leu−, indicating that HO had cleaved both chromosomes III and V. In this experiment, too, the reciprocal translocations predominated (57%) over internal deletions, by a ratio of 170 to 129. The genetic results substantiate the physical analysis of HO-cut DNA in showing that the level of HO endonuclease is high enough to ensure that both chromosomal breaks occurred at roughly the same time. If one chromosome had been cut well before the other, we would expect that the first chromosome would be repaired by intrachromosomal deletion, forcing the second to be subsequently repaired in the same way. The fact that many of the events were reciprocal translocations argues that one of the HO cleavage events did not precede the other by a significant period of time.
The fact that we recovered more reciprocal translocations than internal deletions seems to agree with what is understood about the mechanism of SSA. In a situation where a DSB was flanked by three copies of the URA3 gene: ura3-52—URA3—HO cut site—URA3 (4), SSA strongly favored the formation of a deletion between the two URA3 genes closest to the DSB. This suggests that the closer URA3 gene to the left of the DSB became single-stranded earlier than the ura3-52 gene and thus could anneal preferentially with the URA3 partner on the right. This result is supported by previous studies showing that the appearance of SSA deletion products could be delayed by one hr simply by inserting 4.4 kb of DNA in between the HO cut site and one of the flanking homologous regions, thus extending the time before one region became single-stranded (5). In the experiment described here, the Δhis4Δ on chromosome V and the URA3 segment on chromosome III should become single-stranded earlier than the more distal ura3-52 and his4Δ sequences; hence annealing between Δhis4Δ and Δhis4 and between the two URA3 regions should be slightly favored.
These results demonstrate that the broken ends of a chromosome are free to search for a homologous partner anywhere in the genome. There is no constraint imposed by the way chromosomes are placed in the nucleus that favors intrachromosomal repair events between sequences that are initially less than 10 kb apart, compared with sequences that might be located across the nucleus.
This conclusion appears to be true for cells both in the G1 and G2 phases of the cell cycle. First, unbudded (G1) cells that had been induced for 2 hr were micromanipulated onto a rich medium (yeast extract/peptone-dextrose medium) plate and these single cells were grown into colonies and then analyzed genetically to ascertain what type of repair event had occurred. Because the doubling time of cells in lactate medium is greater than 5 hr, the unbudded cells experienced HO cleavage in G1 and not in a previous cell cycle. Among 15 G1-induced cells, five contained intrachromosomal deletions and 10 carried reciprocal translocations. Thus, the G1 population of cells did not differ from the spectrum of results found for cells induced at all phases of the cell cycle.
A Test of the Generality of Frequent Interchromosomal Repair.
One concern about the generality of these results is that the construction on chromosome III and chromosome V were each located about 35 kb from their respective centromeres. If chromosomes had a tendency to line up from their centromeres toward their telomeres (22), the two locations might fortuitously lie near each other, thus enhancing interchromosomal interactions. To rule out this explanation, we inserted the (Δhis4Δ-cs::LEU2::cs::URA3) sequences into a URA3 gene that had been previously introduced as transplacements at each of three different sites on chromosome XI (Fig. 3B), as a disruption of the APN1, YKL008C, and TIF1 open reading frames, located 215, 11, and 115 kb from CEN11, respectively (Fig. 3B). In all three cases, URA3 was oriented to allow viable translocations to be recovered.
Figure 3.
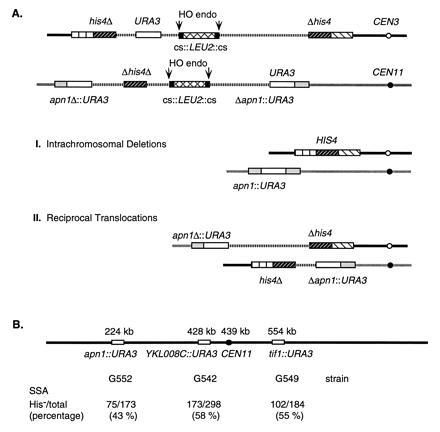
Repair of two broken chromosomes by single-stand annealing between URA3 and HIS4 sequences on chromosomes III and XI. (A) pBR322 plasmids pJH825 and pJH1113 carrying URA3 and Δhis4Δ sequences and the LEU2 gene flanking by a pair of HO cs were integrated into chromosome III (at HIS4) and chromosome XI (at YKL0086::URA3), respectively, as shown. After induction of HO endonuclease, the broken chromosomes may be repaired by two intrachromosomal deletion events (A), leading a His+ cell in which there has been a loss of all plasmid sequences (vertical hatched lines). Alternatively, there may be a pair of reciprocal translocations (B), producing a His− cell. Similar analyses were carried out where the chromosome XI target was at apn1::URA3 or tif1::URA3 (see below). (B) Locations of three sites where the URA3-pBR322-cs::LEU2::cs-URA3 target was inserted. The distances (in kb) indicate the position of each target relative to the left telomere. The frequencies of reciprocal translocations between the target and the site on chromosome III were scored as His− Ura+ cells (see above) and are indicated for each construct.
After HO induction of strains G542, G549, and G552 (Fig. 3B), Leu− colonies, which had excised both LEU2 genes from their surrounding HO cut sites, were scored for the type of SSA that had repaired both broken chromosomes. In each case, between 43% and 58% of the repair events were His− [there were no Ura− colonies as both types of repair yield Ura+ colonies (Fig. 3A)]. Southern blots of five His− and five His+ colonies from strain G542 confirmed that the His+ colonies arose from two intrachromosomal repair events while the His− colonies resulted from the formation of reciprocal translocations (data not shown). In these chromosome III to chromosome XI interactions, reciprocal translocations appeared at approximately the same frequency as when the recombining sequences were on chromosomes III and V (0.60). The results for apn1::URA3 are statistically different from the other three cases (P < 0.05) and could indicate that there is a minor constraint on interchromosomal interactions in region near APN1. However, even in this case, reciprocal translocations were formed almost as often as intrachromosomal deletions.
Relation of These Studies to Previous Analysis of Inter- and Intrachromosomal Interactions.
The conclusions we have drawn from these studies of recombination, induced by DSBs and repaired by SSA, are noticeably different than those from previous studies of spontaneous mitotic recombination between auxotrophic heteroalleles of a 2-kb gene inserted in a variety of different chromosomal locations (16, 23). In those studies there was a significant increase in mitotic recombination when the two alleles were situated on the same chromosome arm, compared with interchromosomal events (16). This would suggest that regions of DNA that were originally part of the same DNA molecule had a notable advantage in recombination, whereas the present study does not show this. One difficulty in interpreting the earlier studies is that there are probably several pathways of recombination contributing to these results (24, 25, 26) and some of these mechanisms may strongly favor intrachromosomal interactions before or coincident with the initiation of recombination. SSA apparently does not labor under this constraint. One possibility is that the broken ends are all recruited to a specific DNA repair site within the nucleus where SSA can occur.
The joining of broken chromosome ends was first described in maize by McClintock (27), who demonstrated fusions between nonhomologous chromosomes in cells where there had been two chromosome breaks. The mechanism by which these ends become joined in maize is not known at the molecular level, but it is well known that maize has many interspersed repeated sequences that could serve as the end-points of joinings by SSA (28). SSA has been shown to be an important repair pathway in both Xenopus (29) and in mammalian cells (3, 30). A major source of reciprocal translocations could be similar annealing events between cells in which damage had produced more than one broken chromosome.
Our results suggest that interphase yeast chromosomes, at least when they have suffered a DSB, are not significantly constrained in the ways in which the single-strand regions at broken ends can anneal with complementary single-stranded regions on other chromosomes. Whether this apparent absence of chromosome territoriality is a general phenomenon or is a consequence of chromosome breakage awaits further study. It will also be interesting to determine if interchromosomal interactions are greatly constrained if this type of DSB repair occurs in metaphase arrested cells, where chromosomes are more condensed and where territoriality may be more evident.
Acknowledgments
We gratefully acknowledge the gifts of the URA3 insertions on chromosome XI from Cecile Fairhead and Bernard Dujon and plasmid pVR196 from Victoria Lundblad. We thank members of the Haber lab and Susan Lovett for their comments on the manuscript. This work was supported by National Institutes of Health Grant GM20056 and Department of Education Grant DE-FG02-91ER61235.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: DSB, double-strand break; SSA, single-strand annealing; cs, cleavage site(s).
References
- 1.Haber J. BioEssays. 1995;17:609–620. doi: 10.1002/bies.950170707. [DOI] [PubMed] [Google Scholar]
- 2.Jensen R, Herskowitz I. Cold Spring Harbor Symp Quant Biol. 1984;49:97–104. doi: 10.1101/sqb.1984.049.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Lin F L, Sperle K, Sternberg N. Mol Cell Biol. 1990;10:113–119. doi: 10.1128/mcb.10.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugawara N, Haber J E. Mol Cell Biol. 1992;12:563–575. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fishman-Lobell J, Rudin N, Haber J E. Mol Cell Biol. 1992;12:1292–1303. doi: 10.1128/mcb.12.3.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plessis A, Perrin A, Haber J E, Dujon B. Genetics. 1992;130:451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laemmli U K, Kas E, Poljak L, Adachi Y. Curr Opin Genet Dev. 1992;2:275–285. doi: 10.1016/s0959-437x(05)80285-0. [DOI] [PubMed] [Google Scholar]
- 8.Roberge M, Gasser S M. Mol Microbiol. 1992;6:419–423. doi: 10.1111/j.1365-2958.1992.tb01485.x. [DOI] [PubMed] [Google Scholar]
- 9.Hiraoka Y, Dernburg A F, Parmelee S J, Rykowski M C, Agard D A, Sedat J W. J Cell Biol. 1993;120:591–600. doi: 10.1083/jcb.120.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher M R, Mathieu U, Jauch A, Emmerich P, Scherthan H, Ried T, Cremer C, Lichter P. Cold Spring Harbor Symp Quant Biol. 1993;5884:777–792. doi: 10.1101/sqb.1993.058.01.085. [DOI] [PubMed] [Google Scholar]
- 11.Manuelidis L. Science. 1990;250:1533–1540. doi: 10.1126/science.2274784. [DOI] [PubMed] [Google Scholar]
- 12.Gasser S M. Curr Opin Cell Biol. 1991;3:407–413. doi: 10.1016/0955-0674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 13.Amati B B, Gasser S M. Cell. 1988;54:967–578. doi: 10.1016/0092-8674(88)90111-0. [DOI] [PubMed] [Google Scholar]
- 14.Palladino F, Laroche T, Gilson E, Axelrod A, Pillus L, Gasser S M. Cell. 1993;75:543–555. doi: 10.1016/0092-8674(93)90388-7. [DOI] [PubMed] [Google Scholar]
- 15.Lichten M, Borts R H, Haber J E. Genetics. 1987;115:233–246. doi: 10.1093/genetics/115.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lichten M, Haber J E. Genetics. 1989;115:261–268. doi: 10.1093/genetics/123.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nickoloff J A, Singer J D, Hoekstra M F, Heffron F. J Mol Biol. 1989;207:527–541. doi: 10.1016/0022-2836(89)90462-2. [DOI] [PubMed] [Google Scholar]
- 18.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. [Google Scholar]
- 19.White C I, Haber J E. EMBO J. 1990;9:663–674. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guacci V, Hogan E, Koshland D J. Cell Biol. 1994;125:517–530. doi: 10.1083/jcb.125.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokol R R, Rolhf F J. Biometrics. San Francisco: Freeman; 1969. [Google Scholar]
- 22.Rabl C. Morphol Jahrb. 1885;10:214–330. [Google Scholar]
- 23.Jinks-Robertson S, Michelitch M, Ramcharan S. Mol Cell Biol. 1993;13:3937–3950. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas B J, Rothstein R. Genetics. 1989;123:725–738. doi: 10.1093/genetics/123.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein H L. Genetics. 1988;120:367–377. doi: 10.1093/genetics/120.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haber J E, Hearn M. Genetics. 1985;111:7–22. doi: 10.1093/genetics/111.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McClintock B. Genetics. 1942;28:458–463. doi: 10.1073/pnas.28.11.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hake S, Walbot V. Chromosoma. 1980;79:251–270. [Google Scholar]
- 29.Carroll D, Lehman C W. Methods Cell Biol. 1991;36:467–486. doi: 10.1016/s0091-679x(08)60292-7. [DOI] [PubMed] [Google Scholar]
- 30.Lin F L, Sperle K, Sternberg N. Mol Cell Biol. 1984;4:1020–1034. doi: 10.1128/mcb.4.6.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]