

Abstract
The multisubunit (α1S, α2/δ, β1, and γ) skeletal muscle dihydropyridine receptor transduces transverse tubule membrane depolarization into release of Ca2+ from the sarcoplasmic reticulum, and also acts as an L-type Ca2+ channel. The α1S subunit contains the voltage sensor and channel pore, the kinetics of which are modified by the other subunits. To determine the role of the β1 subunit in channel activity and excitation-contraction coupling we have used gene targeting to inactivate the β1 gene. β1-null mice die at birth from asphyxia. Electrical stimulation of β1-null muscle fails to induce twitches, however, contractures are induced by caffeine. In isolated β1-null myotubes, action potentials are normal, but fail to elicit a Ca2+ transient. L-type Ca2+ current is decreased 10- to 20-fold in the β1-null cells compared with littermate controls. Immunohistochemistry of cultured myotubes shows that not only is the β1 subunit absent, but the amount of α1S in the membrane also is undetectable. In contrast, the β1 subunit is localized appropriately in dysgenic, mdg/mdg, (α1S-null) cells. Therefore, the β1 subunit may not only play an important role in the transport/insertion of the α1S subunit into the membrane, but may be vital for the targeting of the muscle dihydropyridine receptor complex to the transverse tubule/sarcoplasmic reticulum junction.
Voltage-dependent Ca2+ channels are key factors in the control of Ca2+-linked cellular functions including muscle contraction (1). Mammalian skeletal muscle contains a dihydropyridine sensitive voltage-dependent Ca2+ channel, that is called the dihydropyridine receptor (DHPR), and it is localized to the transverse tubules (T tubules) in adult muscle. The DHPR, in addition to acting as an L-type Ca2+ channel, also couples T tubule depolarization to release of Ca2+ from the sarcoplasmic reticulum (SR), by triggering the opening of the SR Ca2+ release channel also called the ryanodine receptor (RyR-1). This process is referred to as excitation-contraction (E–C) coupling. The DHPRs are present in clusters that are juxtaposed to the junctions between the T tubules and SR, so-called diads, triads, and, in immature cells, peripheral couplings (2). Freeze-fracture analyses have shown that the DHPR complex is present in tetrads (groups of four DHPRs) that are present as arrays, that are arranged opposite an ordered array of ryanodine receptors (2).
The skeletal muscle DHPR is comprised of four subunits, α1S, α2/δ, β1, and γ (3). The α1S subunit is able to direct the formation of a functional channel when introduced into heterologous expression (4, 5, 6). However, the gating kinetics of the channel are considerably different from the native channel. These differences appear to be due to the absence of the other (α2/δ, β, and γ) subunits that make up the normal DHPR. Coexpression of the α1 subunits with combinations of the other subunits in heterologous expression systems can return the activity and kinetics of the channel to values more representative of those seen for the native channel. However, the particular effect observed is dependent on both the expression system used and the specific subunits under study. The skeletal muscle β subunit has a large effect on the functional expression of the α1S subunit from skeletal muscle (7). Coexpression of the β subunit with α1 subunits increases the Ca2+ current and accelerates activation and inactivation more than tenfold (8, 9, 10). Features of the primary structure of the rabbit skeletal β1 polypeptide (11) and biochemical data suggested that it is a peripheral membrane protein that interacts with an intracellular domain of the α1 subunit (12). Subsequently, a β binding site on the α1 subunits (13), and an α1 binding site on the β subunits (14) have been identified. The β subunit also has been shown to facilitate insertion of the α1 subunit into the membrane of HEK293 cells (15).
The β1 subunit is expressed at high levels in skeletal muscle and brain and at lower levels in spleen and heart (11, 16, 17). Alternate splicing produces three isoforms, one of which is expressed exclusively in skeletal muscle (17). To determine directly the role of the β1 subunit in DHPR kinetics and E–C coupling we used gene targeting to inactivate the β1 gene. The absence of the β1 subunit eliminates E–C coupling and modifies the Ca2+ currents in skeletal muscle cells. In addition, the absence of the β1 subunit results in a failure of the α1 subunit to be transported to and/or inserted appropriately into the T tubule membrane.
MATERIALS AND METHODS
Gene Targeting.
A bacteriophage clone containing a portion of the β1 gene was isolated from a mouse 129Sv genomic library (Stratagene no. 946305). A 6.5-kb genomic DNA fragment was subcloned into a pBluescript-derived vector that contains a herpes simplex virus thymidine kinase expression cassette. To inactivate the β1 gene, an internal 1.5-kb fragment, containing exons 6 (used in all transcripts), 7a (used in muscle), and 7b (used in brain), was replaced by a 1.6-kb fragment containing the neo gene linked to a thymidine kinase promoter and a polyadenylylation signal (provided by R. Behringer, M. D. Anderson Cancer Center, Houston). The targeting vector also contains the herpes simplex virus thymidine kinase gene to allow for negative selection of nonrecombinants. The targeting vector retains 2.5 kb of identity with the native β1 gene upstream and 2.1 kb of identity downstream of the neo cassette. A unique BamHI restriction site was used to linearize the plasmid prior to its introduction into mouse embryonic stem (ES) cells by electroporation. Colony selection and identification of targeted clones using probe 2 was as described (18). 5 μg of the targeting vector was electroporated into 5 × 106 AB1 ES cells. Ninety-two G418 and 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-iodouracil- resistant clones were analyzed and 19 showed specific targeting with probe 2. Three were expanded and analyzed with probe 1, and used to produce germ-line chimeras.
Histology.
For light microscopy, tissues were fixed in formalin in saline, embedded in paraffin and 7-μm sections prepared and stained with hematoxylin and eosin. For electron microscopy, the hindlimbs were immersed in Karnovsky’s fixative, post fixed in osmium tetroxide, dehydrated, and embedded in resin. Sections were cut and stained with uranyl acetate and lead citrate.
Contraction Measurements.
Experiments were done essentially as described (19). Embryonic day 16 (E16) fetuses were used for these experiments because bone calcification in E18 fetuses prevented forelimb contraction. The skinned forelimb was attached by a silk filament at the wrist and the humerus, to a force transducer, and the other end to a fixed wire. The preparation was immersed in Krebs–Ringer solution (136 mM NaCl/5 mM KCl/2 mM CaCl2/1 mM MgCl2/10 mM Hepes, pH 7.2) at 23°C. Electrical field stimulation was provided by platinum electrodes placed on each side of the preparation. Caffeine was added by changing the bath solution.
Action Potentials, Ca2+ Currents and Ca2+ Transients.
Ribcages of E18 fetuses were dissected in Krebs–Ringer solution. The tissue was incubated at 37°C for 10 min in PBS containing 3 mg/ml collagenase (Sigma, type I) and 1 mg/ml trypsin (Sigma, type III) to release myotubes. Cells were held under either current or voltage clamp with pipettes that had a tip resistance of 2–7 MΩ when filled with the internal solution. Action potentials were recorded in Krebs–Ringer. The pipette solution was 140 mM KCl, 5 mM MgCl2, 10 mM Hepes·Tris, pH 7.3. For Ca2+ current recordings the external solution was 130 mM tetraethylammonium-methanesulfonate, 10 mM CaCl2, 1 mM MgCl2, 2 μM tetrodotoxin, and 10 mM Hepes, pH 7.2, and the pipette solution was 140 mM Cs-aspartate, 5 mM MgCl2, 5 mM EGTA, 10 mM Mops, pH 7.2. Intracellular Ca2+ was measured in a photomultiplier-based microfluorimeter. Cells were loaded with 0.1 μg/ml fura-2AM (Molecular Probes) for 20–30 min. A 30 × 50-μm slit was centered on the cell held under current clamp. Fluorescence intensity ratios, F340/F380, were taken at a rate of 200 ratios/sec. Fluorescence intensity ratios during stimulation were divided by the ratio at rest.
Immunofluorescence Labeling of Cultured Cells.
Primary muscle cultures were derived from myoblasts isolated from E18 fetuses. Cultures were grown and fixed on gelatin-coated primaria culture dishes (Becton Dickinson) and processed for immunohistochemistry as described (20). Monoclonal antibodies to DHPR subunits α1S (Chemicon) and β1 (Upstate Biotechnology, Lake Placid, NY) were used at dilutions of 1:200 and 1:10 respectively. Antibodies to RyR-1 (Upstate Biotechnology) were used at 1:100. Primary antibodies have been shown to be specific to the various proteins by Western blot analysis. Secondary antibodies used were fluorescein conjugated polyclonal goat anti mouse IgG (Cappel) at a dilution of 1:200.
RESULTS
Targeted Disruption of the β1 Subunit Gene (cchb1) Produces Perinatal Lethality.
Gene targeting was done as described in Materials and Methods (Fig. 1). Three independent targeted ES cell clones were injected into C57BL/6 blastocysts and chimeras obtained. All three ES clones were transmitted through the germ line, and all showed the same phenotype when the mutation was homozygous. Heterozygous mutant mice (+/cchb1tm1) were indistinguishable from wild type (+/+) animals. Mice homozygous for the targeted mutation (cchb1tm1/cchb1tm1), hereafter called β1-null, failed to survive the perinatal period. However, analyses of E18 fetuses revealed that ≈25% were homozygous for the modified β1 allele. The β1-null fetuses fail to show any movement, have flexed necks and extremities, curvature of the spine, and thin limbs. Compared with control littermates the β1-null fetuses show a drastic reduction in muscle mass (Fig. 2 a and b). Muscle from β1-null fetuses contains myofibrils with well-defined Z-bands, but the thick and thin filaments are disorganized compared with controls (either +/+ or +/cchb1tm1, Fig. 2 d and e). T tubule and SR junctional complexes were identified in β1-null fetal muscle (Fig. 2c). Both the gross morphology and the histology of the β1-null fetuses are similar to the muscular dysgenesis (21) and RyR-1 knockout (skrrm1) mutants (19).
Figure 1.
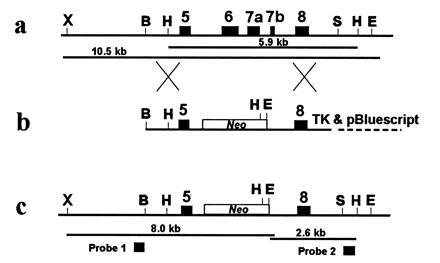
Gene targeting of the β1 subunit gene. (a) Portion of the β1 gene showing relevant restriction enzyme sites and gene structure. (b) Targeting vector. (c) Modified β1 allele expected after homologous recombination between the endogenous locus (a) and the targeting vector (b). Exons are represented by solid boxes. The neo gene is shown by an open box. X, XmnI; B, BamHI; H, HindIII; S, SpeI; E, EcoRV.
Figure 2.
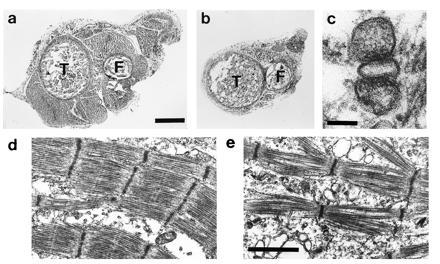
Morphology of E18 β1-null fetuses shows a reduction in muscle mass and disorganization of the myofibrils. (a and b) Transverse sections through the hindlimbs of a control (a) and β1-null (b) fetus. (Bar = 0.3 mm.) Sections were cut at approximately the same position and the tibia (T) and fibula (F) can be seen. The amount of muscle in the β1-null fetus is markedly decreased. (c) Electron micrograph of a triad from a β1-null fetus. (Bar = 0.1 μm.) (d and e) Electron micrograph of muscle from control (d) and β1-null (e) fetus. (Bar = 1.2 μm.)
β1-Null Fetuses Lack E–C Coupling.
The absence of any movement by the β1-null fetuses indicated that E–C coupling may be impaired. This was examined by measuring the twitch contractions of the forelimb of E16 day control and β1-null fetuses. Electrical field stimulation-induced twitches in forelimbs from control fetuses (Fig. 3a), but failed to induce twitches in forelimbs from β1-null fetuses (Fig. 3b). Exposure of both preparations to caffeine produced a slow increase in tension (Fig. 3 c and d), although the force produced by the β1-null forelimb is reduced 20–30-fold compared with controls. This is probably due to the decrease in muscle mass, because Ca2+ transients in single cells, while highly variable in magnitude, are similar to those in control cells (data not shown). Tetrodotoxin-sensitive action potentials can be elicited in both control and β1-null myotubes (Fig. 3 e and f) indicating membrane excitability is normal. In control myotubes, the induced action potential produced a Ca2+ transient (Fig. 3g). However, in β1-null myotubes the action potential failed to elicit a Ca2+ transient (Fig. 3h). These data show that membrane excitability and the ability of the SR to accumulate Ca2+ are normal in β1-null myotubes. The lack of E–C coupling indicates that the β1 subunit has an important role in either transport and/or assembly or function of the DHPR.
Figure 3.

Contractile properties, action potentials and Ca2+ transients in control and β1-null tissues. (a) Electrical field stimulation induced contractions in forelimb from control fetus. Contraction is seen in response to each stimulus. (b) as in a except from β1-null fetus. Stimulus fails to produce contraction. (c and d) Caffeine (25 mM) addition to the bath solution induced contraction in control (c) and β1-null (d) tissue. The response in the β1-null tissue is 20-fold lower than the control tissue. (e and f) Action potentials can be induced in both control (e) and β1-null (f) myotubes and are tetrodotoxin sensitive. (g and h) Action potentials (Inset) induce a Ca2+ transient in control myotubes (g), but fail to induce a Ca2+ transient in myotubes from β1-null (h) fetuses.
L-Type Ca2+ Currents are Decreased in the β1-Null Myotubes.
Whole cell patch-clamp was used to characterize the Ca2+ currents in freshly dissociated intercostal myotubes. Both control and β1-null myotubes contain a transient, rapidly inactivating (T-type) current that can be observed using a test potential of −20 mV (Fig. 4 a and b). T-type currents in control and mutant myotubes had similar peak amplitudes (legend to Fig. 4). At positive test potentials a large sustained slowly inactivating (L-type) inward Ca2+ current is observed in control cells (Fig. 4a). In contrast, the L-type current present in the β1-null myotubes at positive test potentials, while present, is decreased ≈10- to 20-fold (Fig. 4b). Current-voltage relationships of the L-type currents were determined 1 ms before the end of a 300-ms test pulse, and are shown in Fig. 4c. The L-type current density in the β1-null cells is 16-fold lower and the peak current is shifted 10 mV to more positive potentials when compared with control myotubes. In the current-voltage curves of β1-null cells the peak at −20 mV represents residual, approximately 10% of maximum, T-type current that is only detectable in the records at negative test potentials. A similar amount of T-type current is present in control cells at negative test potentials, however this does not appear on the curves because of the scale difference for the two curves. The peak at +25 mV represents the L-type current and contains no T-type current.
Figure 4.
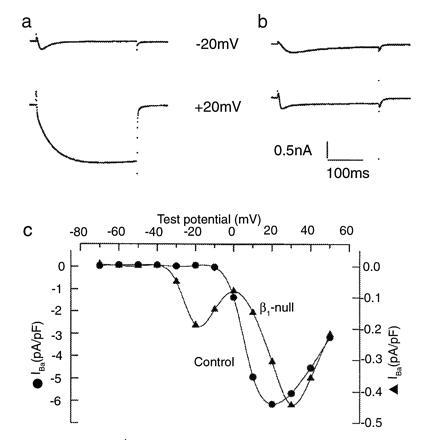
Ca2+ currents and current-voltage curves for control and β1-null myotubes. (a and b) Ca2+ currents from control (a) and β1-null (b) myotubes. A holding potential of −80 mV was used. Test potentials are indicated next to each trace and lasted 300 ms. The control myotubes show a T-type current activated by weak depolarizations [I(peak) = −1.62 ± 0.19 pA/pF at −20 mV; n = 14] and larger L-type currents activated by the strong depolarizations [I(end of pulse) = −5.20 ± 0.38 pA/pF at +20 mV; n = 14]. In the β1-null myotube, the T-type current is unaltered [I(peak) = −1.59 ± 0.17 pA/pF at −20 mV; n = 19], however the L-type current is reduced 10-fold [I(end of pulse) = −0.40 ± 0.04 pA/pF at +20 mV; n = 19]. c, Current-voltage relationships for control and β1-null myotubes. Current amplitude was determined at the end of a 300-ms test pulse. Note difference in ICa scales for the control (•) and β1-null (▴) myotubes.
The β1 Subunit Alters Expression of α1 Subunit.
Our observation that the absence of the β1 subunit drastically reduces the L-type current could arise from two alternative mechanisms. First, the β1 subunit may be required to produce a functional channel. Second, the β1 subunit may be required for correct insertion and/or localization of the α1 subunit into the T tubule membrane. To examine this latter possibility, we used immunohistochemistry to determine the distribution of the α1 and β1 subunits in control, β1-null, and dysgenic myotubes. In normal myotubes there is a punctate expression pattern for both the β1 (Fig. 5a) and α1S (Fig. 5b) subunits. The punctate staining pattern is sometimes found in a random array or in curled patterns (arrows in Fig. 5 a and b) indicative of early T tubule/SR junctions. The pattern of staining for α1S and β1 are equivalent in control myotubes, however the staining intensity is not always the same because of the different affinities of the two antibodies. Double labeling of the α1S subunit and the RyR-1, reveals that they colocalize in control mouse myotubes (22). In the β1-null myotubes, there is no β1 subunit (Fig. 5c) (as would be expected), and also no detectable α1S subunit (Fig. 5d). In addition, there is no evidence that the α1S subunit is present elsewhere in the cell. Because the absence of the α1S subunit was unexpected, we routinely stained dysgenic myotubes as negative controls and normal cultures as a positive controls. In all experiments the staining for the α1S subunit in the β1-null and dysgenic cells were essentially identical. Because the α1S subunit is absent in the dysgenic myotubes (20) these data indicate that the α1S subunit is in fact either absent, or present at undetectable levels, in the β1-null myotubes.
Figure 5.

Immunofluorescence of control, β1-null and dysgenic, α1S-null, myotubes with antibodies to β1 and α1S DHPR subunits and RyR-1. (a and b) Control myotubes labeled with either anti-β1 (a) or anti-α1S (b). Both show a punctate pattern (arrows) reflecting aggregates of the respective DHPR subunits at putative coupling junctions (triads, diads, pentads, and/or peripheral couplings) (20). (c and d) β1-Null myotubes labeled with either anti-β1 (c) or anti-α1S (d). (e) Dysgenic myotubes labeled with anti-β show a weak, but punctate pattern (arrows). (f) β1-Null myotubes labeled with anti-RyR-1 in a punctate pattern (arrows). The diffuse background staining in the myotube may represent RyR-1 that is not membrane localized. (Bar = 20 μM.)
The β subunit is present in dysgenic myotubes and shows a punctate and diffuse expression pattern (Fig. 5e). Similarly, RyR-1 is present in β1-null myotubes as both punctate and diffuse staining (Fig. 5f), a pattern reminiscent of the staining for RyR-1 in dysgenic cells (22). The punctate staining is most likely from T tubule/SR junctions and the diffuse staining from mistargeted or nonclustered proteins. Staining of dysgenic myotubes for the α2 subunit produces a diffuse pattern and no punctate staining, indicating it is not clustered at T tubule/SR junctions (20, 23).
The absence of either diffuse or punctate staining for the α1S subunit in the β1-null myotubes suggests that the β1 subunit is required for either correct transport, insertion, or localization of the α1S subunit into the T tubules. In contrast, the α1 subunit is not required for the appropriate localization of the β1 subunit as indicated by punctate staining for the β1 subunit in dysgenic myotubes.
DISCUSSION
We used gene targeting to inactivate the β1 subunit of the multisubunit skeletal muscle L-type Ca2+ channel in mice. Homozygous mutant fetuses have a phenotype that is very similar to that seen in mice with mutations in either the α1S subunit, muscular dysgenic (21), or RyR-1, skrr (19). All three mutants lack E–C coupling, and so it is likely that the gross phenotype is due to the effects of the absence of E–C coupling rather than the absence of the respective subunit. Based on heterologous expression experiments, the β1 subunit was thought to modulate channel kinetics and possibly increase the expression of the α1 subunit and so the complete lack of E–C coupling in the β1-null mice was somewhat unexpected. Analyses of skeletal muscle myotubes from the β1-null mice demonstrates that the L-type Ca2+ current is reduced 10-20-fold. Given our data show that the α1S subunit is absent or extremely low, the residual L-type current in the β1-null cells could be the same as Idys, present in dysgenic cells (24). However, confirmation that this is true will require careful analyses of the residual currents in both dysgenic and β1-null myotubes.
There is considerable evidence that β subunits modulate channel kinetics (25). In addition, evidence is accumulating that β subunits play an important role in increasing the expression of α1 subunits (26). Most recently, Chien et al. (15) demonstrated that the β subunit was involved in the transport and/or insertion of the α1C subunit into the membrane of HEK293 cells. In contrast, other studies using charge movement as a measure of the amount of α1 subunit present in the membrane, showed that coexpression of a β2A subunit had little effect on the expression of the α1C subunit (27). These differing results could reflect the differences in the expression system used. We demonstrate, using immunohistochemistry, that the β subunit is important for clustering of the α1 subunit at the triad in vitro. It is possible that the α1 subunit is dispersed throughout the cell membrane and therefore difficult to detect, however, the 60% decrease in charge movement and 70% reduction in PN200-110 binding in the β1-null cells (28) would not support this as the only explanation.
The mechanism by which expression of the α1 subunit is controlled by the β1 subunit is unknown, but it is possible that the failure of the formation of the α1/β complex results in the degradation of the α1 subunit. Many channel complexes are assembled at the endoplasmic reticulum and the interaction between newly synthesized subunits may be important to prevent rapid intracellular degradation (29). A recent study of potassium channel assembly showed that the KVβ2 subunit binds to the KV1.2 α subunit in the endoplasmic reticulum. This binding facilitates the posttranslational modification of KV1.2, and its subsequent transport, probably as a KVβ2-KV1.2 complex, to the plasma membrane (30). Chien et al. (15) proposed a similar role for the β subunits of the voltage-dependent Ca2+ channels.
Our studies indicate that the transport and/or insertion of the α1S subunit into the membrane is dependent on the presence of the β1 subunit, but that the converse is not true. We show that in skeletal muscle cells, the β1 subunit is clustered, most likely at T tubule/SR junctions in the absence of the α1S subunit. The insertion of the α2 subunit into the membrane also is independent of the expression of the α1 subunit, although it is not localized appropriately as indicated by diffuse staining in dysgenic myotubes (20). Whether the α2 subunit is inserted into and clustered in the membrane of the β1-null myotubes is currently under investigation. At this time the role of the γ subunit in any of these processes is unknown.
The skeletal muscle DHPR occupies a unique niche among Ca2+ channels in that its primary function, to transduce T tubule membrane depolarization into opening of the SR Ca2+ release channel or RyR-1, is independent of its role as a Ca2+ channel. The biogenesis of the T tubule/SR junctional complex containing the DHPRs and RyR-1 has received considerable attention. Franzini-Armstrong and colleagues (31) have shown that the junctional regions in skeletal muscle myotubes are formed in the absence of the RyR-1 and DHPR, but that the insertion of RyR-1 and DHPR into the SR and T tubules, respectively, serves to expand and stabilize the tertiary structure. The insertion of RyR-1 is independent of the insertion of the DHPR α1 and β1 subunits because in both dysgenic (20, 23) and β1-null myotubes (Fig. 5f), the immunofluorescence staining shows a punctate pattern for RyR-1 similar to that seen in control myotubes. Studies on myotubes from mice that lack RyR-1 (skrr1) indicate that the normal tetrad arrays of DHPRs in the T tubule are not formed (31), even though biophysical data indicate that the DHPRs are present in the membrane, however they do not act as Ca2+ channels presumably because they fail to form tetrads (32). Therefore, unlike RyR-1 that is positioned appropriately in the T tubule/SR junctional region in the absence of DHPRs, the localization and/or organization of the DHPRs in this region appears dependent on interactions with RyR-1. The punctate distribution of the β subunit in the absence of the α1 subunit in dysgenic myotubes (Fig. 5e), indicates that the β subunit can be transported to the T tubule/SR junction in the absence of the α1 subunit. Presumably the β subunit binds to an as yet unidentified component of the T tubule/SR junction. The availability of β1-null mice will provide valuable tools with which to dissect the multifunctional nature of the skeletal muscle β1 subunit. These functions include, modulation of channel kinetics, transport, and/or insertion of the α1 subunit into the membrane and possibly the targeting of the DHPRs to the T tubule/SR junction. Whether β subunits in other tissues are involved in appropriate cellular or subcellular localization of voltage-dependent Ca2+ channels remains to be determined. However, this possibility is intriguing given that many tissues express more than one of the four β subunit genes and that each gene produces splice variants. The availability of β1-null mutant mice will provide a valuable resource for the complete dissection of this multifunctional protein.
Acknowledgments
We would like to thank A. Bradley for AB-1 and SNL 76/7 cells, R. Behringer for advice and providing us with the herpes simplex virus thymidine kinase containing plasmid, H. Peickert for technical assistance, and S. Hunsaker and R. Fish for photographic work. This work was supported by grants from the National Science Foundation (R.G.G. and P.A.P.); the National Institutes of Health (R.G.G., P.A.P., and R.C.), Philippe Foundation (C.S. and M.B.), and the MDA and Blakeslee Endowment Fund (J.A.P.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: T tubule, transverse tubule; SR, sarcoplasmic reticulum; DHPR, dihydropyridine receptor; RyR-1, ryanodine receptor; E–C, excitation–contraction; ES cells, embryonic stem cells; En, embryonic day n.
References
- 1.Melzer W, Herrman-Frank A, Luttagii H C. Biochim Biophys Acta. 1995;1241:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- 2.Franzini-Armstrong C, Jorgensen A O. Ann Rev Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- 3.Catterall W A, Seager M J, Takahashi M. J Biol Chem. 1988;263:3535–3538. [PubMed] [Google Scholar]
- 4.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Nature (London) 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 5.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Nature (London) 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Reyes E, Kim H S, Lacerda A E, Horne W, Wei X, Rampe D, Campbell K, Brown A M, Birnbaumer L. Nature (London) 1989;340:233–236. doi: 10.1038/340233a0. [DOI] [PubMed] [Google Scholar]
- 7.Varadi G, Lory P, Schultz D, Varadi M, Schwartz A. Nature (London) 1991;352:159–162. doi: 10.1038/352159a0. [DOI] [PubMed] [Google Scholar]
- 8.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 9.Mori Y, Friedrich T, Kim M-S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T, Numa S. Nature (London) 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 10.Lacerda A E, Kim H S, Ruth P, Perez-Reyes E, Flockerzi V, Hofmann F, Birnbaumer L, Brown A M. Nature (London) 1991;352:527–530. doi: 10.1038/352527a0. [DOI] [PubMed] [Google Scholar]
- 11.Ruth P, Rohrkasten A, Biel M, Bosse E, Regulla S, Meyer H E, Flockerzi V, Hofmann F. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640. [DOI] [PubMed] [Google Scholar]
- 12.Catterall W A. Science. 1988;242:50–61. doi: 10.1126/science.2459775. [DOI] [PubMed] [Google Scholar]
- 13.Pragnell M, DeWaard M, Mori Y, Tanabe T, Snutch T P, Campbell K P. Nature (London) 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 14.De Waard M, Witcher D R, Pragnell M, Liu H, Campbell K P. J Biol Chem. 1995;270:12056–12064. doi: 10.1074/jbc.270.20.12056. [DOI] [PubMed] [Google Scholar]
- 15.Chien A J, Zhao X, Shirokov R E, Puri T S, Chang C F, Sun D, Rios E, Hosey M M. J Biol Chem. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- 16.Pragnell M, Sakamoto J, Jay S D, Campbell K P. FEBS Lett. 1991;291:253–258. doi: 10.1016/0014-5793(91)81296-k. [DOI] [PubMed] [Google Scholar]
- 17.Powers P A, Lui S, Hogan K, Gregg R G. J Biol Chem. 1992;267:22967–22972. [PubMed] [Google Scholar]
- 18.Ramírez-Solis R, Davis A C, Bradley A. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- 19.Takeshima H, Masamitsu I, Takekura H, Nishi M, Kuno J, Minowa O, Takano H, Noda T. Nature (London) 1994;369:556–559. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- 20.Flucher B E, Phillips J L, Powell J A. J Cell Biol. 1991;269:21770–21777. doi: 10.1083/jcb.115.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell J A. FASEB J. 1990;4:2798–2808. doi: 10.1096/fasebj.4.10.2197156. [DOI] [PubMed] [Google Scholar]
- 22.Flucher B E, Andrews S B, Fleisher S, Marks A R, Caswell A, Powell J A. J Cell Biol. 1993;123:1161–1174. doi: 10.1083/jcb.123.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powell J A, Petherbridge L, Flucher B E. J Cell Biol. 1996;134:375–387. doi: 10.1083/jcb.134.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams B A, Beam K G. J Gen Physiol. 1989;94:429–444. doi: 10.1085/jgp.94.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei X, Perez-Reyes E, Lacerda A E, Schuster G, Brown A M, Birnbaumer L. J Biol Chem. 1991;266:21943–21947. [PubMed] [Google Scholar]
- 26.Perez-Reyes E, Schneider T. Drug Dev Res. 1994;33:295–318. [Google Scholar]
- 27.Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- 28.Strube, C., Beurg, M., Powers, P. A., Gregg, R. G. & Coronado, R. (1996) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 29.Green W N, Millar N S. Trends Neurosci. 1995;18:280–287. [PubMed] [Google Scholar]
- 30.Shi G, Nakahira K, Hammond S, Rhodes K J, Schechter L E, Trimmer J S. Neuron. 1996;16:843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 31.Takehura H, Nishi M, Noda T, Takeshima H, Franzini-Armstrong C. Proc Natl Acad Sci USA. 1995;92:3381–3385. doi: 10.1073/pnas.92.8.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakai J, Dirksen R T, Nguyen H T, Pessah I N, Beam K G, Allen P D. Nature (London) 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]