

Abstract
Recent data implicates a role for the CD40–CD40 ligand (CD40L) pathway in graft rejection. One potential mechanism is direct costimulation of T cells through CD40L. Alternatively, the ability of CD40 stimulation to induce CD80 (B7-1) and CD86 (B7-2) expression on antigen-presenting cells (APCs) has led to the hypothesis that the role of CD40–CD40L interactions in transplant rejection might be indirect, i.e., to promote the costimulatory capacity of APCs. Here, we have used a murine vascularized cardiac allograft model to test this hypothesis. Treatment of the recipients with donor splenocytes and a single dose of anti-CD40L mAb induces long-term graft survival (>100 days) in all animals. This is associated with marked inhibition of intragraft Th1 cytokine [interferon γ and interleukin (IL) 2] and IL-12 expression with reciprocal up-regulation of Th2 cytokines (IL-4 and IL-10). In untreated allograft recipients, CD86 is strongly expressed on endothelial cells and infiltrating mononuclear cells of the graft within 24 hr. In contrast, CD80 expression is not seen until 72 hr after engraftment. Anti-CD40L mAb has no detectable effect on CD86 up-regulation, but almost completely abolishes induction of CD80. However, animals treated with anti-CD80 mAb or with a mutated form of CTLA4Ig (which does not bind to CD86) rejected their cardiac allografts, indicating that blockade of CD80 alone does not mediate the graft-prolonging effects of anti-CD40L mAb. These data support the notion that the role of CD40–CD40L in transplant rejection is not solely to promote CD80 or CD86 expression, but rather that this pathway can directly and independently costimulate T cells. These data also suggest that long-term graft survival can be achieved without blockade of either T cell receptor-mediated signals or CD28–CD86 engagement.
It is well-accepted that T cells require costimulatory signals for optimal activation (1, 2). At present, CD28 is the best-characterized costimulatory receptor on T cells (3, 4). Its known ligands, CD80 (B7-1) and CD86 (B7-2), are expressed on activated antigen-presenting cells (APCs) (3, 4). Blockade of CD28–B7 interactions has been shown to inhibit a number of immune responses in vitro and in vivo, including transplant rejection, induction of graft versus host disease, and autoimmune syndromes (for review, see ref. 5).
Recently, other costimulatory pathways have been characterized (1). One which has been the subject of intense study is that of CD40–CD40 ligand (CD40L) (6, 7). CD40, a member of the tumor necrosis factor (TNF)-receptor family is expressed on the surface of B cells. T cell activation induces expression of a molecule of the TNF family known as CD40L, and binding of CD40L to CD40 during cognate T–B interactions provides B cell help (for review, see ref. 8). CD40 is expressed on other APCs (macrophages and dendritic cells) as well (9). Recent studies showing that CD40 engagement induces the expression of CD80 and CD86 (10, 11) suggest an indirect function of this pathway, i.e., to promote CD28-mediated costimulation. However, it also has been reported that engagement of CD40L provides a direct costimulatory signal to the T cell (12). Here, we have addressed this question by examining the ability of anti-CD40L mAb to prevent murine cardiac allograft rejection and modulate the expression of B7 molecules in vivo.
MATERIALS AND METHODS
Murine Cardiac Allografts.
C57BL/6 (H-2b) and BALB/c (H-2d) mice aged 6–8 weeks were purchased from The Jackson Laboratory and housed in pathogen-free conditions. Cardiac allografts from BALB/c donors into C57BL/6 recipients were placed in an intraabdominal location (13). Graft function was assessed daily by palpation. Animals received mAbs or fusion proteins (at a dose of 200 μg unless otherwise stated) by intravenous injection at the time of transplantation or 2 days after engraftment. In some instances, animals also received an intravenous injection of 5 × 106 donor splenocytes at the time of transplantation. Rejection was defined as the day of cessation of palpable heartbeat, and was verified by autopsy and selective pathological examination. Loss of graft function within 48 hr of transplant was considered a technical failure (<10% on average), and these animals were omitted from further analysis.
Monoclonal Antibodies and Fusion Proteins.
The anti-CD40L mAb MR-1 and a control hamster Ig were gifts of Randy Noelle (Dartmouth Medical School, Hanover, NH). The anti-CD80 mAb 16-10A1 was a gift of Gary Gray (Repligen, Boston). The anti-CD86 mAb GL1 was obtained from American Type Culture Collection. The fusion protein CTLA4Ig has been described (14). The fusion protein CTLA4IgY100F was produced by introducing a phenylalanine residue at position 100 in place of tyrosine using PCR primer-directed mutagenesis. In brief, in vitro and in vivo studies indicate that CTLA4IgY100F binds CD80 with similar avidity as does CTLA4Ig, but has at least a 200-fold lower avidity for CD86. In vitro, CTLA4Ig has detectable binding (by fluorescence-activated cell sorter) to CD86-transfected Chinese hamster ovary cells at concentrations as low as 10-30 ng/ml, whereas CTLA4IgY100F fails to bind to CD86-transfected Chinese hamster ovary cells at concentrations as high as 100 μg/ml. Both CTLA4Ig and CTLA4IgY100F bind equivalently to CD80-transfected Chinese hamster ovary cells (R.P. and P.S.L., unpublished work). The half-life of CTLA4IgY100F in mice is similar to that of CTLA4Ig. CTLA4IgY100F effectively blocks CD80-dependent responses (32).
Immunopathology.
Portions of each graft were snap-frozen and stored at −70°C until sectioning, or formalin-fixed and paraffin-embedded for standard light microscopy. Isotype-matched control mAbs and rat and hamster mAbs against mouse proteins were purchased from PharMingen, unless specified. These consisted of mAbs for CD80 (16-10A1) and CD86 (GL1); cell surface markers expressed by all leukocytes (CD45, 30F11.1), T cells (CD5, 53-7.3), B cells (CD45R/B220, RA3-6B2), monocytes (CD11b, M1/70), natural killer cells (NK1.1, PK136), and granulocytes (Gr-1, RB6-8C5); the activation antigens VCAM-1 (CD106, 429) and interleukin (IL)-2R (CD25, 3C7); and the cytokines IL-2 (S4B6), IL-4 (11B11), interferon (IFN) γ (R4-6A2), IL-10 (JES5-2A5), and TNF-α (MP6-XT22), plus a polyclonal antibody to IL-12 (R & D Systems). Cryostat sections were fixed either in paraformaldehyde-lysine-periodate for demonstration of cell surface antigens or in acetone for localization of cytokines, and were stained by a four-layer PAP method as described (15, 16). Each graft was analyzed at three or more levels, with counts of CD45+ leukocytes in 10-20 fields per section (expressed as mean ± SD of cells per high power field). The specificity of labeling was assessed using isotype-matched mAbs or purified Ig. In addition, the specificity of cytokine staining was confirmed by overnight mAb absorption with recombinant cytokines (IL-2, IL-4, IL-10, and IFN-γ, obtained from PharMingen) prior to immunohistologic labelling of selected cytokine-rich day 3 allografts (15, 16).
RESULTS
Effect of anti-CD40L mAb on Cardiac Allograft Survival.
We first studied the effect of a blocking hamster anti-murine CD40L mAb on cardiac allograft rejection. As shown in Fig. 1, administration of a single dose of anti-CD40L mAb on the day of transplantation significantly delayed rejection in all recipients, and led to indefinite graft survival in the majority (five of seven recipients). Previously, using a rat model of cardiac allografts, we found that the use of donor-specific transfusion (DST) consisting of donor-type splenocytes, was synergistic with CTLA4Ig in preventing graft rejection (17). Similar results were reported recently with murine islet allografts, in which a 7-week course of treatment with anti-CD40L mAb (1 week pre- and 6 weeks posttransplant) alone prevented rejection in 40% of animals, whereas the addition of a single DST led to indefinite graft survival in 96% of the recipients (17). To determine if DST similarly improved graft survival in animals treated with anti-CD40L mAb, mice received a transfusion of 5 × 106 donor-type splenocytes at the time of transplantation (Fig. 1). DST alone had a slight graft-prolonging effect, although most grafts were rejected within 2 weeks, and all were lost by 3 weeks. The combination of DST and anti-CD40L mAb was clearly synergistic, with all animals maintaining allograft function beyond 150 days.
Figure 1.
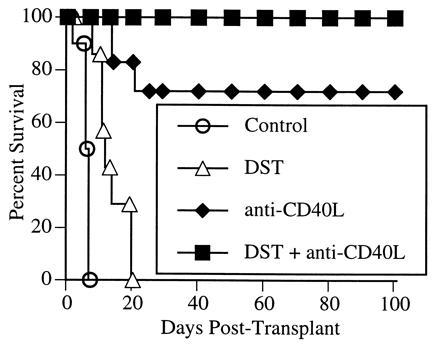
Effect of anti-CD40L mAb on cardiac allograft survival. BALB/c hearts were transplanted into C57BL/6 recipients. Allograft recipients were treated with either a control Ig or anti-CD40L mAb (200 μg by intravenous injection) at the time of transplantation. Selected recipients also received a transfusion of 5 × 106 donor lymphocytes (DST) at the time of transplantation.
Effect of CD40L mAb on Intragraft Cytokine Expression.
We next examined the effect of anti-CD40L mAb on the pattern of intragraft cytokine gene expression (Fig. 2 and Table 1). At day 3 after transplantation, control grafts (treated with DST plus control Ig) contained mononuclear cells expressing IL-2 and IFN-γ, but essentially no cells producing IL-4 or IL-10 were detectable. In contrast, while there was an equivalent mononuclear infiltrate in the grafts of DST plus anti-CD40L mAb-treated animals (Table 1), these cells lacked detectable IL-2 or IFN-γ, but showed a striking induction of both IL-4 and IL-10 expression. In additional experiments, we found that the intensity and pattern of expression of IL-2, IL-4, IL-10, and IFN-γ, as well as the extent of the mononuclear cell infiltrate, were identical in animals treated with a control Ig alone compared with animals treated with DST plus control Ig (data not shown). Furthermore, animals treated with anti-CD40L mAb alone (without DST) had identical findings as those treated with anti-CD40L mAb plus DST (data not shown). Thus, the effects of DST plus anti-CD40L mAb on cytokine expression are a specific result of blockade of CD40L. They are neither the result of DST alone nor require the concomitant use of DST. Animals treated with anti-CD40L mAb also had reduced intragraft expression of the proinflammatory cytokines IL-12 and TNF-α (Table 1). Interestingly, anti-CD40L mAb had no effect on the expression of vascular cell adhesion molecule-1, a ligand for Very Late Antigen-4 (Table 1).
Figure 2.

Effect of anti-CD40L mAb on intragraft cytokine gene expression. C57BL/6 recipients of BALB/c cardiac allografts were treated with either DST plus 200 μg of control Ig (Upper) or DST plus 200 μg of anti-CD40L mAb (Lower) at the time of transplantation. The animals were killed after 3 days and the hearts were stained for expression of IL-2, IFN-γ, IL-4, and IL-10 as described. Arrowheads (a and c) indicate the typical mononuclear cell labeling for IL-2 and IFN-γ confined to control grafts, whereas grafts from anti-CD40L mAb-treated recipients showed dense mononuclear and some adjacent endothelial cell labeling for IL-4 (f) and IL-10 (h). Cryostat sections, hematoxylin counterstain. (Bar = 50 microns.)
Table 1.
Immunopathology of day 3 cardiac allografts
| Feature | Control Ig plus DST | Anti-CD40L mAb plus DST |
|---|---|---|
| Leukocyte infiltration* | Moderate, multifocal infiltrate of T cells (<25%) and monocytes (>75%) | Same |
| IL-2R+ cells | 10–20% of MNCs | <1% of MNCs |
| IL-2, IFN-γ | 5–10% of MNCs | Negative |
| IL-4, IL-10 | <1% of MNCs | >50% of MNCs and ECs |
| IL-12, TNF-α | 20–50% of MHC and focal ECs | <1% of MNCs |
| VCAM-1 | Most ECs | Same |
Comparable data were seen in mice given control Ig alone and mice given DST plus control Ig. Data reflect evaluation of 10-20 fields per graft and three grafts per group. Normal hearts lacked any of these features apart from the presence of small numbers of CD45+ resident dendritic cells and a rare VCAM-1+ endothelial cell. MNC, mononuclear cell; EC, endothelial cell; VCAM-1, vascular cell adhesion molecule-1.
Control Ig plus DST grafts contained 43 ± 11 CD45+ leukocytes per high power field, and anti-CD40L mAb plus DST grafts contained 39 ± 12 leukocytes per high power field (P = not significant).
Expression of CD80 and CD86 During the in Vivo Response to Alloantigen.
Given the role of the CD40L-CD40 pathway in modulating the expression of CD80 and CD86 in vitro (10, 11), and the known importance of B7 molecules in graft rejection (3, 5), it was important to characterize the normal pattern of CD80 and CD86 expression during an alloimmune response in vivo, and to determine the effects of anti-CD40L mAb on these parameters. For these experiments, cardiac allografts were examined by immunohistologic labeling at serial time points posttransplant (Fig. 3). Neither CD80 nor CD86 molecules were detected in significant amounts in the cardiac tissue before transplantation, with only rare and scattered interstitial dendritic cells and an occasional endothelial cell being labeled (Fig. 1, day 0). By 24 hr after transplantation of cardiac allografts, CD86 was densely expressed on virtually all endothelial cells and interstitial dendritic cells. CD86 expression was not increased in control cardiac isografts (Fig. 3) within the limits of detection, suggesting that up-regulation of this molecule is occurring due to specific immune recognition of foreign major histocompatibility complex proteins and the ensuing immune response, and not merely as a result of a nonspecific inflammation secondary to the “trauma” of transplantation (tissue manipulation, ischemia, etc.). In contrast to the prompt up-regulation of CD86 expression, CD80 expression remained unchanged (i.e., weak dendritic cell labeling) within the graft both 24 and 48 hr after transplantation. A striking difference was seen 3 days after transplantation however, at which point CD80 (as well as CD86) was densely expressed on most dendritic cells and infiltrating macrophages, i.e., a population of cells that expressed CD86 at 24 hr. CD80 expression on endothelial cells was more focal and never appeared to be as strong as CD86 expression on those cells, which remained high even at 3 days.
Figure 3.
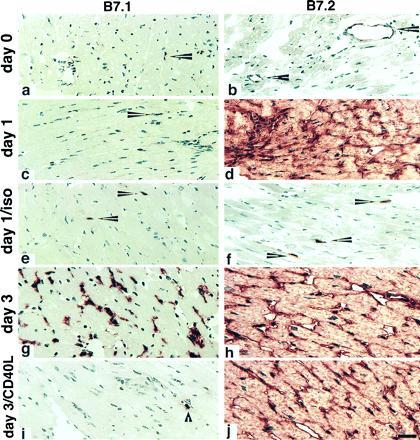
Serial changes in B7-1 and B7-2 expression within murine cardiac allografts. BALB/c hearts transplanted into C57BL/6 recipients were harvested after 1 or 3 days and sections stained for the expression of CD80 (B7-1) or CD86 (B7-2). Day 0 panels represent a harvested donor heart immediately before transplantation. An isograft control (BALB/c donor into BALB/c recipient) was harvested on day 1. Arrowheads in a and c indicate the weak labeling for B7-1 that is restricted to interstitial dendritic cells in the first day posttransplantation, whereas by day 3, dense mononuclear and some capillary endothelial cell labeling is seen (g). In contrast, B7-2 is expressed by occasional graft endothelial cells (arrowheads in b) but is rapidly and densely up-regulated in allografts within 24 hr (d) but not in a day 1 isograft (f). Where indicated, the recipients were treated with DST plus 200 μg of anti-CD40L mAb at the time of transplantation (i and j). DST plus CD40L mAb administration blocked up-regulation of B7-1, without affecting graft cellularity (compare g and i). The dense endothelial expression of B7-2 was not modulated by DST plus anti-CD40L mAb (compare h and j). DST plus control Ig did not affect the marked increase in intragraft B7-1 seen at day 3 in control allografts (data not shown). Cryostat sections, hematoxylin counterstain. (Bar = 50 microns.)
Effect of CD40L mAb on the Expression of B7 Molecules.
To determine whether anti-CD40L mAb affected CD80 or CD86 expression, the animals were killed at day 3 (a time when control animals express both CD80 and CD86 in their grafts; see Fig. 3) for immunopathologic assessment (Fig. 3). By comparison with untreated animals, it can be seen that DST plus control Ig by itself had no observable effect on the pattern or intensity of CD80 or CD86 expression. Interestingly, however, there was a differential effect of anti-CD40L mAb treatment on the expression of CD80 and CD86. While anti-CD40L mAb treatment had no detectable effect on the expression of CD86, it almost completely abrogated the induction of CD80 expression, with treated animals having only a small number of residual CD80+ cells in their grafts, similar to what was seen before transplantation or in control isografts. This was not due to alterations in the populations of cells available for examination, as both control-Ig and anti-CD40L-treated animals had equivalent mononuclear infiltrates as assessed by cell morphology and immunohistochemical staining for T cells, macrophages, natural killer cells, and B cells (Table 1, and data not shown). The lack of effect of anti-CD40L mAb on CD86 expression also serves as a control for cells that are potentially capable of expressing CD80. Therefore, as assessed by immunohistochemical staining of the grafts, we find that the ability of anti-CD40L treatment to prevent acute cardiac allograft rejection, and to promote long-term survival, is associated with selective blockade of CD80 induction. Furthermore, the staining patterns for CD80 and CD86 were not altered by DST alone, and were identical in animals treated with anti-CD40L mAb alone, as in animals receiving anti-CD40L mAb plus DST (data not shown). Thus, as with the findings regarding intragraft cytokine staining (Fig. 2), the effects of anti-CD40L mAb on intragraft staining of CD80 and CD86 are not dependent upon the use of DST.
Effect of Selective CD80- and CD86-Blockade on Allograft Survival.
The data in Fig. 3 suggested the hypothesis that the mechanism by which anti-CD40L mAb prevented graft rejection was through inhibiting the induction of CD80 on APCs. Therefore, we next tested the ability of selective blockade of CD80 or CD86 to prevent rejection. As seen in Table 2, and consistent with our previous studies in rats (5, 17), a single dose of CTLA4Ig (given on day 2) plus DST induced indefinite graft survival in all cardiac allograft recipients. In contrast, DST plus anti-CD80 mAb minimally delayed rejection and was unable to induce long-term survival. Blockade of CD86 alone with mAb induced long-term survival in a minority of recipients.
Table 2.
Effects of CD80 and CD86 blockade on graft survival
| Treatment | Cardiac allograft survival |
|---|---|
| Control Ig | 6, 7, 7, 7, 7, 7, 8, 8, 8, 8, 8 |
| DST | 11, 12, 12, 13, 19, 20, 20 |
| CTLA4Ig + DST | >150 (n = 12) |
| Anti-B7-1 + DST | 20, 22, 23, 42, 42 |
| CTLA4IgY100F + DST | 5, 5, 7, 11, 16 |
| Anti-CD86 + DST | 7, 25, 28, 100+, 100+ |
| CTLA4IgY100F + anti-CD86 + DST | >150 (n = 4) |
BALB/c hearts were transplanted into C57BL/6 recipients. Allograft recipients were treated with a control Ig, anti-CD80 mAb, or anti-CD86 mAb (200 μg by intravenous injection), CTLA4Ig (200 μg), or CTLA4IgY100F (600 μg) 2 days after transplantation. Selected recipients also received a transfusion of 5 × 106 donor lymphocytes (DST) at the time of transplantation.
Although the anti-CD80 mAb used in the studies above (16-10A1) clearly blocks CD28 binding (18), it has been suggested that intact anti-CD80 mAbs might have uncharacterized positive signaling effects on CD80-expressing cells (19). Therefore, in additional experiments, we used a mutated form of CTLA4Ig, called CTLA4IgY100F, in which the tyrosine at position 100 was replaced by a phenylalanine. This mutation causes CTLA4IgY100F to retain binding activity for CD80 but abolishes binding to CD86. An advantage of this reagent over Fab fragments of anti-CD80 mAb are its relatively longer half-life (53 hr) compared with expected half-life of Fab fragments in vivo. CTLA4IgY100F had no effect, either at the same dose as CTLA4Ig (200 μg, data not shown) or at a 3-fold higher dose (600 μg, Table 2). The inability of CTLA4IgY100F to prevent rejection was not due to failure to adequately block CD80. Table 2 shows that CTLA4IgY100F and anti-CD86 mAb synergize in preventing allograft rejection, indicating that CTLA4IgY100F is an effective competitive inhibitor for CD80 binding. Thus, selective blockade of CD80 in vivo is unable to replicate the immunosuppressive effects of anti-CD40L mAb.
DISCUSSION
To our knowledge, this is the first report describing the pattern of expression of the costimulatory ligands CD80 and CD86 in vivo during the response to a vascularized organ allograft. We find that both molecules are up-regulated as part of a specific immune response (rather than as a result of the transplant procedure itself), and that CD86 is expressed significantly earlier than CD80. The expression pattern of the two molecules is distinct, in that CD86 appears to be quite a bit more prominent on endothelial cells than does CD80. The endothelium is the first site of contact of host T cells with recipient tissue in the case of vascularized grafts. This, coupled with the known ability of endothelium to express major histocompatibility complex class II molecules, makes it likely that endothelial cells play a very prominent role in the early stages of alloactivation in vivo.
Many previous studies attest to the importance of CD80 and CD86 in transplant rejection (5, 17, 20, 21, 22). More recent evidence suggests a role for the CD40-CD40L pathway in these responses as well (6, 7, 8, 23). This might be as a direct T cell costimulator (12, 24) or via induction of CD80 and CD86 on B cells (10, 11). In addition, CD40 is expressed on macrophages, and ligation of CD40 has been shown to potentiate macrophage production of nitric oxide and selective monokines such as IL-12, and to enhance macrophage cytotoxicity (25).
Our data indicate that blockade of CD40L by administration of a single dose of mAb at the time of transplantation is able to induce long-term survival of vascularized cardiac allografts in ≈70% of murine recipients. The addition of DST leads to indefinite graft survival in all animals, although the mechanisms by which DST augment the effects of anti-CD40L mAb are not known. Parker et al. (23) have shown that anti-CD40L antibody combined with DST blocked rejection of murine islet allografts, although in that system prolonged administration (2–7 weeks) of the antibody was required. Our model, one of vascularized organ transplantation, introduces an additional level of complexity.
Recently, Larsen et al. (26) have shown that anti-CD40L mAb alone could prevent murine cardiac allograft rejection in the majority of recipients. In their studies, CTLA4Ig, given at the time of transplantation, did not prevent ultimate graft rejection, consistent with our own previous reports regarding the need to delay administration of CTLA4Ig (17). However, combination of anti-CD40L mAb and CTLA4Ig initiated at the time of transplantation was synergistic, leading to long-term survival in all animals (27). While anti-CD40L mAb alone did not effect the expression of T cell cytokines or of B7 molecules, the combination of CTLA4Ig plus anti-CD40L inhibited the expression of IL-2, IL-4, IL-10, and IFN-γ, as determined by reverse transcription–PCR. Transcripts for CD80 and CD86 were only minimally affected. This is in contrast to our own data, where blockade of CD40L alone induced a Th2 immune deviation in association with a loss of CD80 expression. The reasons for this discrepancy are not immediately apparent, but may relate to their use of multiple doses of anti-CD40L mAb to prevent rejection, or may be due to the use of different detection methods [protein detection by immunohistochemistry in the present study versus mRNA detection by RT-PCR in Larsen et al. (26)].
It is important to note that our results of costimulatory molecule and cytokine expression were all obtained using immunohistochemistry. An advantage of this technique is the ability to detect protein itself, and the preservation of tissue architecture, allowing for spatial localization of the relevant gene products. While we cannot exclude low residual CD86 expression or a small affect on CD80 expression in the grafts of anti-CD40L-treated animals, the large alterations in CD80, CD86, and cytokine gene expression seen in the present study are real, and seem likely to be physiologically meaningful.
Endothelial cells, when activated, are immunogenic, expressing major histocompatibility complex class I and II molecules, adhesion receptors, and costimulatory molecules. Recently, three groups have shown that CD40 is expressed on endothelial cells and that ligation of CD40 on endothelial cells in vitro up-regulates intercellular adhesion molecule-1, E-selectin, and vascular cell adhesion molecule-1 (28, 29, 30). In that system, neither CD80 nor CD86 was induced by CD40 ligation of endothelium (28). While that signal may not by itself induce these molecules, our data clearly indicate that, in the case of alloimmune responses, CD40-mediated signals are required for CD80 induction in vivo.
In is also interesting to note that treatment with anti-CD40L mAb was able to prevent rejection in the face of abundant CD86 expression throughout the graft endothelium and graft infiltrating APCs. This result implies that long-term graft survival can be achieved without blockade of either T-cell antigen receptor-mediated signals or the CD28 pathway. Thus, while CD28-mediated costimulation may be necessary for graft rejection, our data suggest that it is not sufficient (at least not when CD86 is the ligand), and that signals mediated through CD40L-CD40 interactions are required as well. Although extremely unlikely, it should be noted that we cannot exclude a direct effect of anti-CD40L mAb on TCR or CD28 signaling.
In our studies, prolongation of graft survival by anti-CD40L mAb was accompanied by immune deviation toward Th2 cytokines, and by specific down-regulation of CD80 expression without an effect on CD86 expression. Recently, Stüber et al. (31) reported that anti-CD40L Ab prevented Th1-mediated inflammatory colitis by blocking the secretion of IL-12, an effect observed in our study as well (Table 1). Whether or not the loss of Th1 and induction of Th2 cytokines we observed is responsible for the graft prolonging effects of anti-CD40L mAb is not known. This pattern of selective cytokine sparing is associated with enhanced graft survival in a variety of models; however, a causal role for Th2 cells in transplantation tolerance has yet to be established.
Our studies define the effects of CD40L-blockade on the expression of the T cell costimulators known to be required for transplant rejection (i.e., CD80 and CD86), and suggest that the effects of anti-CD40L mAb on these molecules alone cannot account for its tolerogenic effects in organ transplantation. We cannot, however, completely exclude the possibility that anti-CD80 mAb and CTLA4IgY100F provided only incomplete blockade of CD80, relative to the inhibition seen with anti-CD40L mAb. Further experiments using CD80-knockout mice will be required to answers this question as well as to address the potentially distinct roles of CD80 on donor compared with recipient cells, and on T cells versus APCs.
Acknowledgments
We thank Julie Rogers for expert assistance in preparing CTLA4IgY100F and Charles B. Carpenter for critical review of the manuscript. This work was supported by National Institutes of Health Grants AI-37691 (L.A.T.) and AI-34965 (M.H.S.). L.A.T. is an Established Investigator of the American Heart Association.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: APC, antigen-presenting cell; CD40L, CD40 ligand; DST, donor-specific transfusion; TNF, tumor necrosis factor; IL, interleukin; IFN, interferon.
References
- 1.Janeway C H, Bottomly K. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz R H. Cell. 1989;57:1073–1081. doi: 10.1016/0092-8674(89)90044-5. [DOI] [PubMed] [Google Scholar]
- 3.Guinan E C, Gribben J G, Boussiotis V A, Freeman G J, Nadler L M. Blood. 1994;84:3261–3282. [PubMed] [Google Scholar]
- 4.June C H, Bluestone J A, Nadler L M, Thompson C B. Immunol Today. 1994;15:321–331. doi: 10.1016/0167-5699(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 5.Sayegh M H, Turka L A. J Am Soc Nephrol. 1995;6:1143–1150. doi: 10.1681/ASN.V641143. [DOI] [PubMed] [Google Scholar]
- 6.Durie F H, Aruffo A, Ledbetter J, Crassi K M, Green W R, Fast L D, Noelle R J. J Clin Invest. 1994;94:1333–1338. doi: 10.1172/JCI117453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noelle R J, Roy M, Shephard D M, Stamekovic I, Ledbetter J A, Aruffo A. Proc Natl Acad Sci USA. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durie F H, Foy T M, Masters S R, Laman J D, Noelle R J. Immunol Today. 1994;15:406–411. doi: 10.1016/0167-5699(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 9.Alderson M R, Armitage R J, Tough T W, Stockbine L, Fanslow W C, Spriggs M K. J Exp Med. 1993;178:669–674. doi: 10.1084/jem.178.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranheim E A, Kipps T J. J Exp Med. 1993;177:925–935. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy M, Aruffo A, Ledbetter J, Linsley P, Kehry M, Noelle R. Eur J Immunol. 1995;25:596–603. doi: 10.1002/eji.1830250243. [DOI] [PubMed] [Google Scholar]
- 12.Cayabyab M, Phillips J H, Lanier L L. J Immunol. 1994;152:1523–1531. [PubMed] [Google Scholar]
- 13.Corry R J, Winn H S, Russell P S. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Linsley P S, Brady W, Urnes M, Grosmaire L S, Damle N K, Ledbetter J A. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hancock W W, Polanski M, Zhang J, Blogg N, Weiner H L. Am J Pathol. 1995;155:1193–1199. [PMC free article] [PubMed] [Google Scholar]
- 16.Mottram P L, Han W R, Purcell L J, McKenzie I F C, Hancock W W. Transplantation. 1995;59:559–565. [PubMed] [Google Scholar]
- 17.Lin H, Bolling S F, Linsley P S, Wei R, Gordon D, Thompson C B, Turka L A. J Exp Med. 1993;178:1801–1806. doi: 10.1084/jem.178.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Razi-Wolf Z, Freeman G J, Galvin F, Benacerraf B, Nadler L, Reiser H. Proc Natl Acad Sci USA. 1992;89:4210–4214. doi: 10.1073/pnas.89.9.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller S D, Vanderlugt C L, Lenschow D J, Pope J G, Karandikar N J, Dal Canto M C, Bluestone J A. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 20.Pearson T C, Alexander D Z, Winn K J, Linsley P S, Lowry R P, Larsen C P. Transplantation. 1994;57:1701–1706. [PubMed] [Google Scholar]
- 21.Sayegh M H, Akalin E, Hancock W W, Russell M E, Carpenter C B, Linsley P S, Turka L A. J Exp Med. 1995;181:1869–1874. doi: 10.1084/jem.181.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenschow D J, Zeng Y, Hathcock K S, Zuckerman L A, Freeman G, Thistlethwaite J R, Gray G S, Hodes R J, Bluestone J A. Transplantation. 1995;60:1171–1178. doi: 10.1097/00007890-199511270-00019. [DOI] [PubMed] [Google Scholar]
- 23.Parker D C, Greiner D L, Phillps N E, Appel M C, Steele A W, Durie F H, Noelle R J, Mordes J P, Rossini A A. Proc Natl Acad Sci USA. 1995;92:9560–9564. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Essen D, Kikutani H, Gray D. Nature (London) 1995;378:621–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- 25.Tian L, Noelle R J, Lawrence D A. Eur J Immunol. 1995;25:306–309. doi: 10.1002/eji.1830250152. [DOI] [PubMed] [Google Scholar]
- 26.Larsen C P, Alexander D Z, Hollenbaugh D, Elwood E T, Ritchie S C, Aruffo A, Hendrix R, Pearson T C. Transplantation. 1996;61:4–9. doi: 10.1097/00007890-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 27.Larsen C P, Elwood E T, Alexander D Z, Ritchie S C, Hendrix R, Tucker-Burden C, Cho H R, Aruffo A, Hollenbaugh D, Linsley P S, Winn K J, Pearson C T. Nature (London) 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 28.Yellin M J, Brett J, Baum D, Matsushima A, Szabolcs M, Stern D, Chess L. J Exp Med. 1995;182:1857–1864. doi: 10.1084/jem.182.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karmann K W, Hughes C C, Schechner J, Fanslow W C, Pober J S. Proc Natl Acad Sci USA. 1995;92:4342–4346. doi: 10.1073/pnas.92.10.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollenbaugh D, Mischel-Petty N, Edwards C P, Simons J C, Denfeld R W, Kiener P A, Aruffo A. J Exp Med. 1995;182:33–40. doi: 10.1084/jem.182.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stüber E, Strober W, Neurath M. J Exp Med. 1996;183:693–698. doi: 10.1084/jem.183.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khoury, S. J., Gallon, L., Verburg, R. R., Chandraker, A., Peach, R., Linsley, P. S., Turka, L. A., Hancock, W. W. & Sayegh, M. S. (1996) J. Immunol., in press. [PubMed]