

Abstract
The two cell surface receptors for tumor necrosis factor (TNF) interact with a number of intracellular signal transducing proteins. The association of TRADD, a 34-kDa cytoplasmic protein containing a C-terminal death domain, with aggregated TNF receptor 1 (TNF-R1) through their respective death domains leads to NF-κB activation and programmed cell death. In contrast, TNF receptor 2 (TNF-R2) interacts with the TNF receptor associated factors 2/1 (TRAF2/TRAF1) heterocomplex, which mediates the recruitment of two cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) to TNF-R2. Here we show that the TNF-R2 signal transducers TRAF2 and c-IAP1 are a part of the TNF-R1 signaling complex. The recruitment of TRAF2 and c-IAP1 to TNF-R1 is TNF-dependent, is mediated by TRADD, and is independent of TNF-R2. These data establish the physiological involvement of TRAF2 and c-IAP1 in TNF-R1 signaling and help provide a molecular explanation for both the overlapping and distinct signals generated by the two TNF receptors.
Keywords: tumor necrosis factor signaling
Tumor necrosis factor (TNF) is a potent cytokine produced primarily by activated macrophages and monocytes. TNF elicits a broad range of biological effects (1, 2, 3) through two distinct membrane receptors, TNF-R1 (≈55 kDa) and TNF-R2 (≈75 kDa), which are expressed at low levels on most cell types (3, 4, 5, 6, 7). The extracellular domains of the two TNF receptors share a similar architecture with characteristic cysteine-rich motifs that define them as members of a receptor superfamily that also includes Fas, CD27, CD30, CD40, and the 75-kDa nerve growth factor receptor (8).
Binding of TNF to the extracellular domains of the two TNF receptors initiates many similar cellular responses, notably the activation of the proinflammatory transcription factor NF-κB (9, 10, 11, 12, 13, 14). In contrast to the extracellular domains, the primary amino acid sequences of the cytoplasmic domains of TNF-R1 and TNF-R2 are unrelated. This led to the prediction that the two receptors would be found to initiate distinct signal transduction pathways by interacting with different signaling proteins (15, 16, 17). Indeed, gene knockout experiments (18, 19, 20) and studies with receptor-specific agonistic antibodies (21, 22, 23, 24, 25) have confirmed that in many instances the two TNF receptors mediate divergent biological responses.
Efforts to understand the molecular mechanisms of TNF signaling have been aided by the discoveries of several proteins that interact with the intracellular domains of TNF-R1 and TNF-R2. Rothe et al. (12) identified two proteins, TNF-R2 associated factors 1 and 2 (TRAF1 and TRAF2, respectively), that exist in a heterodimeric complex that associates directly with the intracellular domain of TNF-R2. Overexpression of TRAF2 causes activation of NF-κB, whereas a dominant negative mutant of TRAF2 blocks TNF-R2 mediated NF-κB activation, suggesting a central role for TRAF2 in TNF-R2 signaling (26). Recently, two novel proteins, c-IAP1 and c-IAP2 (for cellular inhibitor of apoptosis protein 1 and 2, respectively), were identified that are recruited to TNF-R2 by the TRAF2/TRAF1 heterocomplex (27). Although the physical interactions among these molecules have been well-established in experimental systems, the signaling mechanisms and, in the cases of TRAF1 and c-IAPs, the physiological functions of these individual molecules are not clear.
The intracellular regions of both TNF-R1 and Fas contain a so-called “death domain” of ≈80 amino acids that is responsible for signaling cell death by the respective receptors (28, 29). In contrast, the intracellular domain of TNF-R2 does not contain a death domain and activation of TNF-R2 does not cause apoptosis in most cell types (8). In a yeast two-hybrid screening using the intracellular domain of TNF-R1 as bait, Hsu et al. (13) identified TRADD, a 34-kDa cytoplasmic protein containing a C-terminal death domain. TRADD and TNF-R1 interact through their death domains and TRADD is recruited to TNF-R1 in a ligand-dependent process (13, 14). As observed for TNF-R1, overexpression of TRADD causes both programmed cell death and activation of NF-κB (13). These studies suggest that TRADD transduces TNF-R1 signals.
We recently found that the N-terminal domain of TRADD interacts directly with TRAF2, and that overexpression of a dominant negative TRAF2 mutant blocks TNF-R1-induced NF-κB activation (14). These data argue that TRAF2 may play a role in NF-κB activation mediated by both TNF receptors. The death domain of TRADD also interacts with other two death domain proteins, fas-associated death domain protein and receptor interacting protein (14, 30). Since a dominant negative mutant of FADD blocks TNF-R1- and TRADD-mediated cell death (14), the TRADD–FADD interaction may be part of the TNF-R1 pathway that signals apoptosis. Thus, TRADD–TRAF2 and TRADD–FADD interactions appear to define two distinct TNF-R1 signal transduction pathways.
Here we show that TRAF2 and c-IAP1 are rapidly recruited to TNF-R1 signaling complex in a TNF-dependent manner in untransfected mammalian cells. The recruitment of TRAF2 and c-IAP1 to TNF-R1 is mediated by TRADD and is independent of TNF-R2. These data provide direct evidence for the physiological involvement of TRAF2 and c-IAP1, proteins first identified as part of the TNF-R2 signaling complex, in TNF-R1 signaling.
MATERIALS AND METHODS
Reagents.
Recombinant human TNF was provided by Genentech. The 985 mAb to TNF-R1, the 1040 mAb to TNF-R2, and the rabbit polyclonal antibodies against TNF-R2, TRADD, c-IAP1, and c-IAP2 were described previously (12, 13, 23, 24, 27). Rabbit polyclonal antibodies against human TRAF1 and human TRAF2 were raised against a 30-mer peptide, RGEDLQSISPGSRLRTQEKAHPEVAEAGIG, and a 34-mer peptide, VHEGIYEEGISILESSSAFPDNAARREVESLPAV, respectively (Babco, Richmond, CA).
Mammalian cell expression vectors encoding TNF-R1, TRADD, TRAF2, and c-IAP1 have been described (13, 26, 27).
Cell Culture and Transfection.
Human U937 histiocytic lymphoma cells (provided by C. Ware, University of California, Riverside) were grown in RPMI medium 1640 containing 10% fetal calf serum and 100 μg/ml each of penicillin G and streptomycin. Human HeLa-S3 cells (provided by J. Anzola, Tularik) were maintained in MEM for suspension cells (GIBCO) containing 10% fetal calf serum, 100 μg/ml each of penicillin G and streptomycin, and 1% Pluronic F-68 (GIBCO). Human 293 embryonic kidney cells were maintained in high glucose DMEM containing 10% fetal calf serum.
For transient transfection, ≈2 × 106 cells per well were seeded on 100-mm plates. Cells were transfected the following day by the calcium phosphate precipitation method (31). Coimmunoprecipitation following transient transfection has been described (13).
Immunoprecipitation and Immunoblotting.
For endogenous coimmunoprecipitation experiments, U937 or HeLa cells were washed in warm PBS and incubated in the presence or absence of TNF (100 ng/ml) for variable time periods as indicated in the text. Cells were lysed in 5 ml lysis buffer (20 mM Tris, pH 7.5/150 mM NaCl/1% Triton/1 mM EDTA/30 mM NaF/2 mM sodium pyrophosphate/10 μg/ml aprotinin/10 μg/ml leupeptin). Lysates were incubated with 25 μg of mAb or mouse IgG control, or 10 μl polyclonal antibody or preimmune serum control, and 50 μl of a 1:1 slurry of protein GammaBind G Plus-Sepharose (Pharmacia) overnight at 4°C. The Sepharose beads were washed four times with 5 ml of lysis buffer containing 0.5 M NaCl, and once more with lysis buffer. The precipitates were fractionated on 10% SDS/PAGE and transferred to a nitrocellulose membrane. Immunoblotting analyses were performed with various polyclonal antibodies and visualized with horseradish peroxidase-coupled goat antirabbit IgG (Amersham) or horseradish peroxidase-coupled protein A (Bio-Rad) using the Enhanced Chemiluminescence Detection system (Amersham).
Gel Filtration.
HeLa cells (≈1 × 108) were collected and washed twice with PBS containing 5 mM EDTA. All the following procedures were performed at 4°C. Washed cells were lysed in 5 ml lysis buffer 400 (50 mM Tris, pH 7.4/400 mM NaCl/0.1% Nonidet P-40/10% glycerol/50 mM NaF/1 mM Na orthovanadate/1 mM DTT/1 mM phenylmethylsulfonyl fluoride/10 μg/ml aprotinin/10 μg/ml leupeptin/10 μg/ml pepstatin A). The lysate was centrifuged at 3500 rpm for 10 min. The supernatant was further cleared by centrifugation at 35,000 rpm for 1 hr, and then separated by size exclusion chromatography on a Superdex 200 column in lysis buffer 400. Samples were collected in fractions of 1.0 ml. For immunoblotting analysis, 100 μl samples were precipitated with trichloroacetic acid and dissolved in SDS/PAGE loading buffer. Immunoblotting was performed as described above.
RESULTS
TNF-Dependent Recruitment of TRAF2 to TNF-R1.
We have previously demonstrated in mammalian cell overexpression systems and yeast two-hybrid assays that TRADD can simultaneously interact with TNF-R1 and TRAF2 (14). However, the presence of TRAF2 in the TNF-R1 signaling complex has not been established under physiological conditions. Since TRADD is recruited to TNF-R1 in a ligand dependent process (14), we attempted to determine whether TRAF2 is also recruited to TNF-R1 following TNF treatment of untransfected human cells. U937 cells were left untreated or treated with TNF for variable time periods. Cell lysates were immunoprecipitated with a nonagonistic TNF-R1 mAb and the immunoprecipitates were analyzed by immunoblotting with various antisera. We found that TRADD was recruited to TNF-R1 within 2 min of TNF treatment, and remained associated with TNF-R1 for at least 40 min after TNF treatment (Fig. 1A). Similarly, TRAF2 was rapidly recruited to TNF-R1 following TNF treatment (Fig. 1B). In similar experiments, we could not detect TRAF1 in the TNF-R1 complex (data not shown). These data suggest that TRAF2 is involved in TNF-R1 signaling under physiological conditions.
Figure 1.
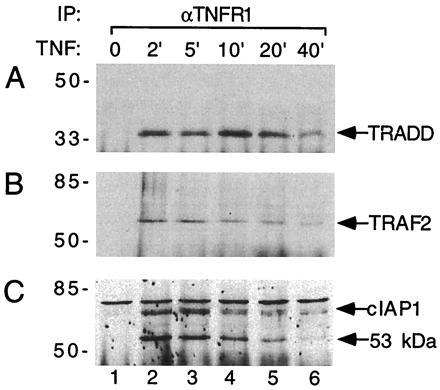
TNF-dependent recruitment of TRADD, TRAF2, and c-IAP1 to TNF-R1 in U937 cells. TNF (100 ng/ml) was used to treat U937 cells (≈2 × 108) for the indicated times (lanes 2–6) or left the U937 cells were left untreated (lane 1). Immunoprecipitations were carried out with the 985 mAb to TNF-R1. Immunoprecipitates were analyzed by immunoblotting with rabbit polyclonal antibodies against TRADD (A), TRAF2 (B), or c-IAP1 (C). Positions of molecular mass standards (in kDa) are shown on the left.
TNF-Dependent Recruitment of c-IAP1 to TNF-R1.
In mammalian cell expression systems, TRAF2 interacts with c-IAP1 and c-IAP2 and helps recruit them to the TNF-R2 signaling complex (27). Furthermore, different domains of TRAF2 are used for their interactions with c-IAP and TRADD (14, 27). Therefore, we examined whether c-IAP1 and/or c-IAP2 could be found in the TNF-R1 complex. Remarkably, the 68-kDa c-IAP1 was recruited to TNF-R1 in a pattern similar to the recruitment of TRAF2 (Fig. 1C). The c-IAP1 polyclonal antibody also recognized a protein of ≈53 kDa whose presence in the TNF-R1 complex was likewise ligand-dependent (Fig. 1C). This ≈53 kDa protein appears to be either a truncated form of c-IAP1 or a closely related homolog. In similar experiments, c-IAP2 could not be detected in the TNF-R1 signaling complex in U937 cells (data not shown). These data provide the first evidence for c-IAP1 playing a physiological role in TNF-R1 signaling.
Recruitment of TRAF2 and c-IAP1 to TNF-R1 Is Independent of TNF-R2.
Since TRAF2 and c-IAP1 were first identified as signal transducers in the TNF-R2 signaling pathway, we considered the possibility that the presence of TRAF2 and c-IAP1 in the TNF-R1 complex might be due to TNF-induced formation of complexes containing both TNF-R1 and TNF-R2. To examine this possibility, we compared U937 cells, which express both receptors, with HeLa cells, which do not express detectable TNF-R2 (Fig. 2A). We found that both TRAF2 and c-IAP1 were recruited to TNF-R1 in a TNF-dependent manner in HeLa cells (Fig. 2 B and C). The amount of TRAF2 and c-IAP1 recruited to TNF-R1 in HeLa cells is equal to, or greater than, that in U937 cells. The ≈53 kDa protein identified with c-IAP1 antibody in U937 cells (Figs. 1C and 2C) was not observed in the TNF-R1 complex in HeLa cells (Fig. 2C). In fact, although this ≈53 kDa protein was easily detectable in U937 cells by immunoprecipitation with the c-IAP1 antibody, it could not be detected by this approach in HeLa cells (data not shown). Consistent with our earlier experiments, c-IAP2 could not be detected in the TNF-R1 complex in either U937 cells or HeLa cells (data not shown). These data demonstrate that TNF-dependent recruitment of TRAF2 and c-IAP1 to TNF-R1 occurs independently of TNF-R2.
Figure 2.
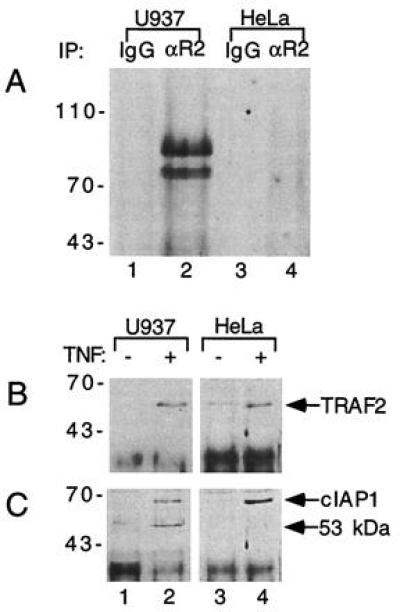
TNF-dependent recruitment of TRAF2 and c-IAP1 to TNF-R1 is TNF-R2-independent. (A) Comparison of TNF-R2 expression in U937 and HeLa cells. Lysates of U937 cells (≈1 × 108) or HeLa cells (≈1 × 108) were immunoprecipitated with the 1040 mAb to TNF-R2 (lanes 2 and 4) or mouse IgG control (lanes 1 and 3). The immunoprecipitates were subsequently analyzed by immunoblotting with a polyclonal TNF-R2 antibody. Positions of molecular mass standards (in kDa) are shown on the left. (B and C) Recruitment of TRAF2 and c-IAP1 to the TNF-R1 complex in U937 and HeLa cells. TNF (100 ng/ml) was used to treat 2 × 108 U937 cells (lanes 1 and 2) or HeLa cells (lanes 3 and 4) for 5 min (lanes 2 and 4). Lanes 1 and 3 were left untreated. Cell lysates were immunoprecipitated with the 985 mAb to TNF-R1 and immunoprecipitates were analyzed by immunoblotting with polyclonal antibodies against TRAF2 (B) or c-IAP1 (C). Positions of molecular mass standards (in kDa) are shown on the left.
Recruitment of c-IAP1 to TNF-R1 Is Mediated by TRADD and TRAF2.
Previously, different domains of TRAF2 have been shown to interact with TRADD (14) and c-IAP1 (27) in mammalian cells overexpressing these proteins. Therefore, the TNF-dependent recruitment of c-IAP1 to TNF-R1 might be mediated by TRAF2 and TRADD. To test this possibility, 293 cells were transfected with various combinations of expression vectors for TNF-R1, TRADD, TRAF2, and myc-epitope-tagged c-IAP1. Transfected cell lysates were immunoprecipitated with polyclonal antibody against TNF-R1, followed by immunoblotting analysis with anti-myc antibody. We found that c-IAP1 did not directly interact with TNF-R1, and neither TRADD nor TRAF2 alone could mediate the recruitment of c-IAP1 to TNF-R1. However, in the presence of both TRADD and TRAF2, c-IAP1 was recruited to TNF-R1 complex (Fig. 3). These data suggest that the recruitment of endogenous c-IAP1 to TNF-R1 occurs through TRADD and TRAF2.
Figure 3.
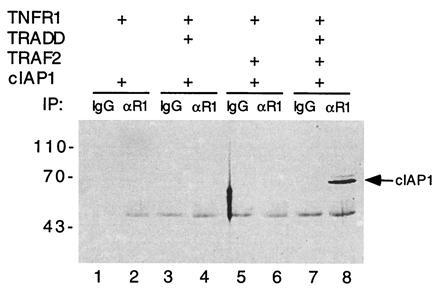
TRADD and TRAF2 mediate recruitment of c-IAP1 to TNF-R1. Combinations of expression vectors for TNF-R1, myc tagged c-IAP1, TRADD, and TRAF2 (as indicated) were used to transfect 293 cells (≈2 × 106). Transfected cell lysates were immunoprecipitated with either mouse IgG control (lanes 1, 3, 5, and 7) or a goat anti human TNF-R1 antibody (lanes 2, 4, 6, and 8). Immunoprecipitates were analyzed for c-IAP1 by immunoblotting with a myc epitope antibody. Positions of molecular mass standards (in kDa) are shown on the left.
Sequential Assembly of the TNF-R1 Signaling Complex.
Since TRADD, TRAF2, and c-IAP1 are all recruited to TNF-R1 rapidly following TNF treatment, the time course experiments described above were not able to determine whether these signaling proteins are recruited to TNF-R1 sequentially or in the form of a preexisting complex. To address this question, we examined the biochemical nature of these signaling proteins in normal cells. HeLa cell lysates were separated by gel filtration, and individual fractions were analyzed by immunoblotting with antibodies against TRADD, TRAF2, and c-IAP1. We found that TRADD was present in fractions that eluted in the 35–80 kDa range (Fig. 4), suggesting that TRADD exists as a monomer and/or dimer in normal cells. However, TRAF2 and c-IAP1 were found in overlapping fractions eluting at ≈350 kDa (Fig. 4). These data are consistent with the hypothesis that TRADD is first recruited to TNF-R1 following TNF treatment and then acts as an adaptor for the subsequent recruitment of TRAF2 and c-IAP1.
Figure 4.
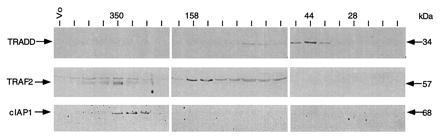
Biochemical properties of TRADD, TRAF2, and c-IAP1 in HeLa cells. HeLa cell lysates (1 × 108 cells) were separated by size exclusion chromatography on Superdex 200 column. Individual fractions were analyzed by immunoblotting with polyclonal antibodies against TRADD, TRAF2, or c-IAP1 as indicated. Positions of molecular mass standards (in kDa) are shown on the left.
To further determine how TRAF2 and c-IAP1 are recruited to TNF-R1, we asked whether these signal transducers exist in the same complex under physiological conditions. HeLa cells were treated with TNF or left untreated and cell extracts were immunoprecipitated with antisera directed against TRADD, TRAF1, TRAF2, or c-IAP1. Each immunoprecipitate was analyzed by immunoblotting with the c-IAP1 antibody. We found large amounts of c-IAP1 coimmunoprecipitated with TRAF2 and significantly lower amounts associated with TRAF1 (Fig. 5). No c-IAP1 was found in association with TRADD in untreated cells, and barely detectable amounts were seen in TNF treated cells (Fig. 5). In these experiments, TNF treatment had no significant effect on the association of TRAF2 with c-IAP1 (Fig. 5), suggesting that the presence of physiological TRAF2-c-IAP1 complexes is not TNF-dependent.
Figure 5.
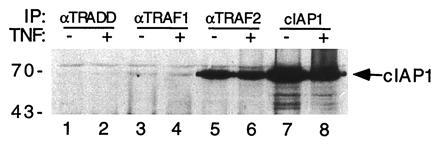
Association of TRAF2 and c-IAP1 in HeLa cells. HeLa cells (≈2 × 108) were treated with TNF (100 ng/ml) for 5 min (lanes 2, 4, 6, and 8) or left untreated (lanes 1, 3, 5, and 7). Cell lysates were immunoprecipitated with rabbit polyclonal antisera against TRADD (lanes 1 and 2), TRAF1 (lanes 3 and 4), TRAF2 (lanes 5 and 6), or c-IAP1 (lanes 7 and 8). Immunoprecipitates were analyzed by immunoblotting with a polyclonal antibody against c-IAP1. Positions of molecular mass standards (in kDa) are shown on the left.
DISCUSSION
The Pleiotropic Activities of TNF are Mediated by Two Distinct TNF Receptors, TNF-R1 and TNF-R2.
Recently, several novel proteins have been identified that interact with the intracellular domains of TNF-R1 and TNF-R2 to initiate signal transduction events (12, 13, 14, 30). TRADD is a 34-kDa protein containing a C-terminal death domain that interacts with the death domain of TNF-R1 (13). Overexpression of TRADD in mammalian cells causes both apoptosis and NF-κB activation, mimicking the effects of TNF treatment and TNF-R1 activation (13). Under physiological conditions, TRADD is recruited to TNF-R1 in a ligand-dependent process (14), suggesting that TRADD is critically involved in TNF-R1 signaling.
The intracellular domain of TNF-R2 does not contain a death domain and does not interact with TRADD (13), suggesting that TRADD is not involved in TNF-R2 signaling. This is consistent with the observations that TNF-R2 does not mediate programmed cell death in most cell types (8). Instead, the intracellular domain of TNF-R2 interacts with a TRAF2/TRAF1 heterocomplex (12), which can then recruit c-IAP1 and c-IAP2 to the TNF-R2 complex (27). Whereas the functional roles of TRAF1, c-IAP1, and c-IAP2 in TNF-R2 signaling are not yet known, TRAF2 appears to be an essential component in the TNF-R2-mediated NF-κB activation pathway (26).
The initial identification of these distinct TNF receptor-associated proteins seemed to confirm earlier predictions (15, 16, 17) that TNF-R1 and TNF-R2 would be found to activate independent and distinct signal transduction pathways. However, a recent study (14) suggested that functional crosstalk between these pathways may account for certain overlapping functions of the two TNF receptors, such as NF-κB activation. In this study, we found that TRAF2 interacts strongly with the N-terminal half of TRADD and that overexpression of a dominant negative mutant of TRAF2 inhibited TNF-R1-mediated NF-κB activation (14). Thus, TRAF2 may be a common player in both TNF-R1- and TNF-R2-mediated NF-κB activation pathways.
In this study, we show that TRAF2 is rapidly recruited to TNF-R1 in a ligand-dependent manner in normal mammalian cells. These data provide the strongest evidence yet for an essential role of TRAF2 in TNF-R1-mediated NF-κB activation. Additionally, c-IAP1, a mammalian homolog of baculoviral inhibitor of apoptosis proteins, which was first identified in the TNF-R2 complex, is also recruited to the TNF-R1 complex in a similar ligand-dependent manner. Interestingly, in U937 cells a ≈53-kDa protein recognized by c-IAP1 antibody is also recruited to TNF-R1 following TNF treatment. This ≈53 kDa protein might be a closely related homolog of c-IAP1. In fact, several members of the human IAP family have been recently identified (26, 32). Alternatively, the ≈53 kDa protein might be a proteolytically derived form of c-IAP1. Consistent with this latter possibility, we observed that a fraction of overexpressed full-length c-IAP1 and c-IAP2 is degraded to a size of ≈53 kDa in 293 cells (data not shown).
Since TRAF2 interacts with c-IAP1 in mammalian cells, recruitment of c-IAP1 to TNF-R1 may itself not be as surprising as the notion that activation of TNF-R1 can cause cell death while members of IAP family are proposed to be involved in antagonizing apoptosis (27, 33, 34, 35). In fact, TNF-R1-induced cell death is generally weaker and slower than that induced by Fas (36), another member of the TNF receptor superfamily. In this context, c-IAP1 may be recruited to TNF-R1 to protect or delay cells from TNF induced cell death. However, the exact function of c-IAP1 in TNF-R1 signaling is still unresolved. Nonetheless, our data suggest that TRAF2 and c-IAP1 are involved in both TNF-R1 and TNF-R2 mediated signal transduction pathways. Utilization of these common proteins may help explain certain shared functions of the two TNF receptors, such as NF-κB activation. Our finding that TRAF2 and c-IAP1 are recruited to TNF-R1 in HeLa cells, which have no detectable TNF-R2, shows that the ligand dependent recruitment of TRAF2 and c-IAP1 to TNF-R1 is not dependent on TNF-R2. Thus, it seems that the two TNF receptors can independently utilize common downstream signal transducers following TNF stimulation.
In mammalian cells, TRADD and c-IAP1 do not interact under physiological conditions or when overexpressed (data not shown). Since TRAF2 can interact with TRADD through its C-terminal TRAF-C domain (14) and with c-IAP1 through its TRAF-N domain (27) in mammalian overexpression systems, recruitment of c-IAP1 to TNF-R1 seems to be mediated through TRADD and TRAF2. Since TRADD is present in either monomer or dimer form and is not associated with the TRAF2-cIAP1 complex in normal cells, the TRADD–TRAF2 interaction possibly occurs only after aggregation of TRADD, either by TNF stimulation or overexpression in mammalian cells. These data suggest that TRADD may be recruited to TNF-R1 in a monomeric or dimeric form where it then functions as an adapter for recruitment of TRAF2-cIAP1 complex.
In mammalian overexpression systems, recruitment of c-IAP1 to TNF-R2 complex requires a TRAF2/TRAF1 heterocomplex (27). In contrast, our current study suggests that TRADD and TRAF2, but not TRAF1, are necessary for the recruitment of c-IAP1 to TNF-R1. Thus, it is possible that the presence or absence of TRAF1 may determine whether c-IAP1 associates with TNF-R1 or both TNF receptors.
Although TRADD, TRAF2, and c-IAP1 are recruited to the TNF-R1 complex after TNF treatment, the association of TRADD with c-IAP1 following TNF treatment in untransfected cells was barely detectable (Fig. 5). Although we do not exclude other possibilities, the simplest explanation for this observation is that TRADD associated with the TNF-R1 signaling complex is not accessible to antibody in this experiment or that anti-TRADD antibody disrupts the complex. To date, we have been unable to detect either TRAF1 or c-IAP2 in the TNF-R1 signaling complex in either U937 or HeLa cells. This may suggest that TRAF1 and c-IAP2 are not components of the TNF-R1 signaling complex, or that they are not expressed in these cells. The third possibility is that our TRAF1 and c-IAP2 antibodies are not potent enough to detect low levels of TRAF1 and c-IAP2 associated with TNF-R1.
Based on the current data and our earlier studies, we propose the following model for TNF-R1 signaling (Fig. 6). In this model, the death domains of TRADD and TNF-R1 do not interact with each other as monomers. Instead, the trimeric TNF is likely to induce trimeric aggregates of TNF-R1 (37) that are stabilized by its self-associating death domain (38, 39). These aggregated TNF-R1 death domains would then provide a high affinity binding site for TRADD monomers or dimers. The aggregated TNF-R1-TRADD complex may recruit RIP, another death domain protein to the TNF-R1 complex through its C-terminal death domain (30). Following the association of TRADD with TNF-R1, the N-terminal domain of TRADD may become accessible to TRAF2, thereby permitting recruitment of the TRAF2/cIAP1 heterocomplex. This recruitment of the TNF-R2 signal transducers TRAF2 and cIAP1 to TNF-R1 provides a molecular explanation for the functional overlap of the two TNF receptors. It is now of great interest to decipher how signals are transduced from the TNF receptor signaling complexes to downstream components, such as the inducible kinase(s) for IκB.
Figure 6.
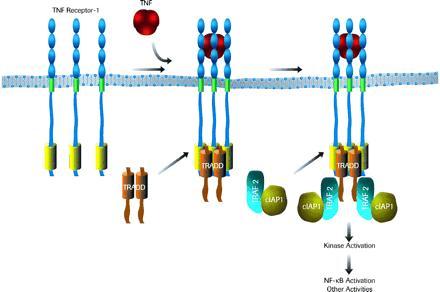
A model for the assembly of TNF-R1 signaling complex. See text for details.
Acknowledgments
We thank Drs. Carl Ware and Todd VanArsdale for advice on the U937 cell immunoprecipitations, Drs. Jianing Huang and Ho Yeong Song for critical reading of the manuscript, and Ms. Laura Medin for help in preparing the manuscript. H.-B.S. would like to thank Dr. Harish C. Joshi (Emory University, Atlanta) for his continued support and encouragement.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: TNF, tumor necrosis factor; TNF-R1 and TNF-R2, TNF receptor type 1 and type 2, respectively; c-IAP, cellular inhibitor of apoptosis protein; TRAF1 and TRAF2, TNF receptor associated factors 1 and 2, respectively; TRADD, TNF-R1-associated death domain protein.
References
- 1.Goeddel D V, Aggarwal B B, Gray P W, Leung D W, Nedwin G E, Palladino M A, Patton J S, Pennica D, Shepard H M, Sugarman B J, Wong G H W. Cold Spring Harbor Symp Quant Biol. 1986;51:597–609. doi: 10.1101/sqb.1986.051.01.072. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B, Cerami A. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 3.Fiers W. FEBS Lett. 1991;285:199–212. doi: 10.1016/0014-5793(91)80803-b. [DOI] [PubMed] [Google Scholar]
- 4.Loetscher H, Pan Y-C E, Lahm H W, Gentz R, Brockhaus M, Tabuch H, Lesslauer W. Cell. 1990;61:351–359. doi: 10.1016/0092-8674(90)90815-v. [DOI] [PubMed] [Google Scholar]
- 5.Schall T J, Lewis M, Koller K J, Lee A, Rice G C, Wong G H W, Gatanaga T, Granger G A, Lentz R, Raab H, Kohr W J, Goeddel D V. Cell. 1990;61:361–370. doi: 10.1016/0092-8674(90)90816-w. [DOI] [PubMed] [Google Scholar]
- 6.Smith C A, Davis T, Anderson D, Solam L, Beckmann M P, Jerzy R, Dower S K, Cosman D, Goodwin R G. Science. 1990;248:1019–1023. doi: 10.1126/science.2160731. [DOI] [PubMed] [Google Scholar]
- 7.Tartaglia L A, Goeddel D V. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 8.Smith C A, Farrah T, Goodwin R G. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 9.Osborn L, Kunkel S, Nabel G J. Proc Natl Acad Sci USA. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiegmann K, Schütze S, Kampen E, Himmler A, Machleidt T, Krönke M. J Biol Chem. 1992;267:17997–18001. [PubMed] [Google Scholar]
- 11.Lægreid A, Medvedev A, Nonstad U, Bombara M P, Ranges G, Sundan A, Espevik T. J Biol Chem. 1994;269:7785–7791. [PubMed] [Google Scholar]
- 12.Rothe M, Wong S C, Henzel W J, Goeddel D V. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 13.Hsu H, Xiong J, Goeddel D V. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 14.Hsu H, Shu H B, Pan M G, Goeddel D V. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 15.Dembic Z, Loetscher H, Gubler U, Pan Y-C E, Lahm H W, Genz R, Brockhaus M, Lesslauer W. Cytokine. 1990;2:231–237. doi: 10.1016/1043-4666(90)90022-l. [DOI] [PubMed] [Google Scholar]
- 16.Lewis M, Tartaglia L A, Lee A, Bennett G L, Rice G C, Wong G H W, Chen E Y, Goeddel D V. Proc Natl Acad Sci USA. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin R G, Anderson D, Jerzy R, Davis T, Brannan C I, Copeland N G, Jenkins N A, Smith C A. Mol Cell Biol. 1991;11:3020–3026. doi: 10.1128/mcb.11.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeffer K, Matsuyama T, Kundig T M, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi P S, Kronke M, Mak T W. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 19.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Nature (London) 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 20.Erickson S L, de Sauvage F J, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan K C F, Schreiber R D, Goeddel D V, Moore M W. Nature (London) 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 21.Englemann H, Holtmann H, Brakebush C, Avni Y S, Sarov I, Nophar Y, Hadas E, Leitner O, Wallach D. J Biol Chem. 1990;265:14497–14504. [PubMed] [Google Scholar]
- 22.Espevik T, Brockhaus M, Loetscher H, Nonstad U, Shalaby R. J Exp Med. 1990;171:415–426. doi: 10.1084/jem.171.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tartaglia L A, Weber R F, Figari I S, Reynolds C, Palladino M A, Goeddel D V. Proc Natl Acad Sci USA. 1991;88:9292–9296. doi: 10.1073/pnas.88.20.9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong G H W, Tartaglia L A, Lee M S, Goeddel D V. J Immunol. 1992;149:3550–3553. [PubMed] [Google Scholar]
- 25.Gehr F, Gentz R, Brockhaus M, Loetscher H, Lesslauer W. J Immunol. 1992;149:911–917. [PubMed] [Google Scholar]
- 26.Rothe M, Sarma V, Dixit V M, Goeddel D V. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 27.Rothe M, Pan M G, Henzel W J, Ayres T M, Goeddel D V. Cell. 1995;83:1243–1253. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 28.Tartaglia L A, Ayres T M, Wong G H W, Goeddel D V. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 29.Itoh N, Nagata S. J Biol Chem. 1993;268:10932–10937. [PubMed] [Google Scholar]
- 30.Hsu H, Huang J, Shu H B, Baichwal V, Goeddel D V. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 32.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda J, MacKenzie A, Korneluk R G. Nature (London) 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 33.Birnbaum M J, Clem R J, Miller L K. J Virol. 1994;68:2531–2538. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clem R J, Miller L K. In: Apoptosis II: The Molecular Basis of Apoptosis in Disease. Tomei D L, Cope F O, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. pp. 89–110. [Google Scholar]
- 35.Hay B A, Wassarman D A, Rubin G M. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 36.Nagato S, Goldstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 37.Banner D W, D’Arcy A, Janes W, Gentz R, Schoenfeld H-J, Broger C, Loetscher H, Lesslauer W. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 38.Song H Y, Dunbar J D, Donner D B. J Biol Chem. 1994;269:22492–22495. [PubMed] [Google Scholar]
- 39.Boldin M P, Mett I L, Varfolomeev E E, Chumakov I, Shemer-Avni Y, Camonis J H, Wallach D. J Biol Chem. 1995;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]