

Abstract
Affinity maturation of antibodies requires localized hypermutation and antigen selection. Hypermutation is particularly active in certain regions (notably the CDRs of light and heavy chains) due to the local accumulation of hot spots. We have now analyzed the role of individual nucleotides in the origin of hot spots and show that mutability is largely defined by the nucleotide sequence. We compared the mutability profile of wild-type and modified κ transgenes that contain silent mutations in the CDR1 segment. We found a new hot spot created at the third base of Ser-31 when its wild-type AGT codon was substituted by AGC. Two major hot spots associated with this AGC vanished when Ser-31 was encoded by the synonymous TCA. In addition to these, which were the most prominent changes, there were compensatory alterations in mutability of residues not directly related to the introduced silent mutations, so that the average hypermutation remained constant. Thus, mutations arising early in the immune response, even silent ones, could affect the mutability of critical residues and alter the pattern of affinity maturation. When analyzing hybridomas, we detected such alterations, but they seemed to better correlate with changes in average rather than local mutation rates. Overall, this paper shows how evolution could have optimized the mutability of individual residues to minimize deleterious mutations. Thus, the optimal strategy for affinity maturation may involve the incorporation of multiple point mutations before antigen selection of the relevant cells.
During the course of immunization, the affinity of the antibodies directed against the immunogen usually increases. This process of affinity maturation relies on hypermutation of the immunoglobulin genes expressed by antigen-selected cells and is at the root of the gradual improvement of antigen-binding properties of antibodies (1). Hypermutation is restricted in time and space to antigen-stimulated B-cells and occurs during their growth and differentiation in the germinal centers (2, 3, 4, 5, 6). It is highly localized to ≈2 kb of DNA, which includes the heavy and light chain V-gene segments. There is a sharp upstream hypermutation boundary in the middle of the intron leader and a gradual decrease of mutation frequencies well into the J-C intron (7, 8, 9, 10, 11, 12). The hypermutation rate has been estimated to be between 10−3 and 0.4 × 10−4 mutations/cell/generation (13, 14), a value orders of magnitude higher than the rate of spontaneous mutation of antibody genes in tissue-cultured myelomas and transfected cells (15, 16). This hypermutation rate was estimated to be the optimum to produce maximal diversity without catastrophic degeneration (17, 18). Such calculations assumed random mutation and gradual accumulation of mutants at a rate not higher than one per cycle. However, point mutations are not evenly distributed along the V-gene segments, and this is only partly due to antigen selection. Indeed antigen-independent, hence intrinsic, hot spots make a major contribution to the general pattern of hypermutation.
Although long suspected (19), the existence of intrinsic hot spots was first deduced from a statistical analysis of independent repeats of silent mutations of a single antibody gene (20). The analysis of the hypermutation of transgenes provided not only definitive evidence for the presence of intrinsic hot spots but also a deeper understanding of their origin, as well as their potential functional importance. Suitable hybridomas and transgenic animals permitted researchers to dissociate intrinsic mutations from the skewing effects of antigen selection (21, 22). The analysis of Peyer’s patches germinal center B cells facilitated the derivation of sufficiently large data bases, from which significant conclusions could be drawn (23, 24, 25).
It is of considerable interest that intrinsic hot spots are concentrated in CDRs, a point which has obvious evolutionary as well as functional implications (26, 27, 28). The probable importance of evolutionary pressure to define an optimal nucleotide sequence directed to produce potentially useful mutants is also manifested by the high ratio of replacement/silent mutations imprinted in CDR segments (29).
The origin of hot spots has been associated with certain sequence motifs (e.g., G/A-G-T/C-A/T and TAA; ref. 30), and also with secondary structure features of the DNA, like palindromic sequences, direct and inverted repeats, etc. (31, 32, 33, 34). In this paper we show that key silent changes in the nucleotide sequence of hot spots have highly significant effects on the frequency at which individual bases hypermutate. In particular, the most prominent hot spots of VκOx1, AGT, or AGC at Ser-31 are no longer hot spots when the serine residue is encoded by a TCA triplet.
MATERIALS AND METHODS
Transgenes and Transgenic Mice.
Lκ contains a mouse VκOx1-Jκ5 linked to the LOU allotype rat Cκ (21), LκΔB is derived from Lκ by deletion of the DNA between the BamHI sites 3′ of the Cκ (25). Modified versions of the Lκ and LκΔB (M7 and ΔBM7) were made by replacing the PstI/KpnI fragment in the wild-type (WT) VκOx1 region (from Cys-23 to Tyr-36) by a synthetic analogue containing seven silent mutations (see Fig. 1a). ΔBTCAM7 was made taking advantage of the XhoI site present in ΔBM7 but not in LκΔB, by replacing the XhoI/KpnI fragment of ΔBM7, from Ser-27 to Tyr-36, by a synthetic analogue in which the AGC codon of Ser-31 was substituted for TCA.
Figure 1.
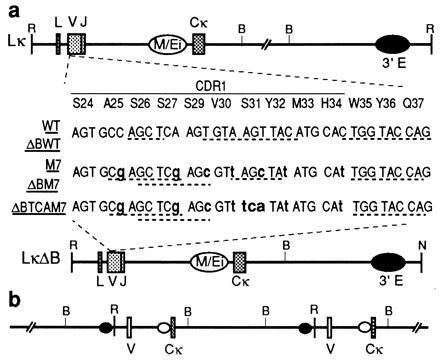
Variations of the Lκ transgene construct. (a) The portion of the VκOx1 where WT and ΔBWT and mutants differ has been expanded. WT and ΔBWT are identical to Lκ and LκΔB (19, 23). Modified bases shown in boldface lowercase type. Potential hairpins and palindromic sequences are underlined. (b) Genetic map of LκWTM7 mouse. One of the V segments is WT and the other M7. Both hypermutate implying that the 3′ enhancer drives the process when placed upstream of the VJ segment. The restriction endonuclease cleavage sites are abbreviated as follows: R, EcoRI; B, BamHI; N, NotI.
The LκNG transgenic line, containing LκΔB and ΔB7M, as well as Lκ-Vgpt* and Lκ-Vneo*[XS]i, has been described (25). The LκWTM7 line was prepared by injecting an equimolar mixture of EcoRI fragments of Lκ and M7. The LκΔBTCAM7/Δ[Li] line was prepared by injecting an equimolar mixture of EcoRI/NotI fragments of ΔB7M and LκΔ[Li], which carries a deletion in the leader intron. For the purpose of this paper, Lκ and LκΔB are usually referred to as WT and ΔBWT, respectively.
Founder animals were identified by expression of rat κ chain in serum, as revealed by ELISA (35), and by Southern blot analysis of tail DNA digested with BamHI. The XhoI/NotI fragment of the 3′ enhancer and the XbaI/SacII fragment of the J-C intron were used as probes. The number of transgenic copies was estimated by Southern blots and by sequence analysis of hybridomas. LκNG mice contained two ΔB7M and three copies of ΔBWT, one of them (ΔBWTm) carrying a fortuitously introduced silent mutation (G for A) in Lys-18. LκWTM7 mice contained one intact and two truncated copies in a head-to-tail tandem arrangement (see Fig. 1b). LκΔBTCAM7/Δ[Li] mice contained one copy of ΔBTCAM7 and four copies of LκΔ[Li]. There were two head-to-tail and one head-to-head transgene junctions in the tandem rearrangement. Founders were bred with (C57BL/6 × CBA)F1 animals.
Derivation of Hybridomas.
Adult mice were immunized with alum-precipitated phOx14-CSA and 109 heat-killed Bortedella pertussis, and 6 weeks later boosted with an i.v. injection of soluble phOx14-CSA (1). Hybridomas were derived 3 days later as described (36).
Sequence Analysis.
Mouse Peyer’s patches B220+PNAhi cells were collected as described (23) from 4- to 5-month-old mice. Two mice were analyzed independently from the LκNG line, four from the LκWTM7 line and three from the LκΔBTCAM7/Δ[Li] line.
Preparation of Peyer’s patches and hybridoma DNA and further PCR amplification were as described (23, 24). The ΔBTCAM7 transgene was selectively amplified using primers LKFOR (37) and BEALEADER (5′-GGGAATTCGAGATCAGAATACAACCAA-3′, containing an EcoRI site), which hybridizes in the leader intron absent from the LκΔ[Li] construct.
The PCR product was cloned into M13 mp18, and positive clones were identified by hybridization and sequenced as described (23). The PCR error rate was determined by sequencing transgenes from characterized hybridomas.
Statistical Analysis.
The statistical significance of differences, estimated by a Pearson’s χ2 test (38) with 1 degree of freedom, was performed with the mutants versus the WT* data base and the M7* versus ΔBTCAM7 data base, using the formula:
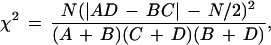 |
where A and B belong to one data base and C and D to the other. A and C are the number of clones carrying mutations at a specific position. B and D are the number of clones carrying no mutations at the same position. N = A + B + C + D. The formula was applied for each of the 282 nt of the V-segment. For significance at the 95% level or better, χ2 must be >3.8.
RESULTS
Hypermutation of Mutant Transgenes.
We have previously shown that two transgene versions (Lκ and LκΔB) encoding the dominant light chain (VκOx1) of the antibody response to 2-phenyloxazolone include the necessary controlling elements to target hypermutation (21, 24). In this paper we refer to them as WT or ΔBWT, and we refer to the pooled data derived from them both as WT*. The most mutable segment of Lκ is CDR1, and the most prominent hot spot is the second base of the AGT triplet encoding Ser-31. We have now derived mouse lines containing two alternative transgene versions. One of them (M7 or ΔBM7) differs from the wild type by seven silent substitutions in the CDR1, including C for T in the Ser-31 codon (Fig. 1). In the other (ΔBTCAM7), the Ser-31 codon of ΔBM7 was substituted by the synonymous TCA.
Below we describe the results obtained with three independently derived transgenic lines. One contained M7 and WT, the second contained ΔBM7 and ΔBWT, and the third contained ΔBTCAM7. The pattern of hypermutation was analyzed by isolating germinal center B-cells from the Peyer’s patches of 4- to 5-month-old mice. DNA was extracted, and the VJ-gene segment was amplified, cloned into M13, and sequenced. At least two mice from each transgenic line were separately analyzed. In all cases, the results were comparable within each line and therefore the data have been pooled.
All the transgenes were highly mutated in some of the B cells (Fig. 2). The frequency at which mutations accumulated in ΔBTCAM7 when all clones (mutated and unmutated) were computed was, however, lower than in the others (Table 1). This is due to the higher proportion of unmutated clones. Indeed, when these were ignored, the average number of mutations accumulated in the modified and WT transgenes were similar. From Table 1, it can be calculated that in WT*, M7*, and ΔBTCAM7, the clones with more than one mutation accumulated on average 5.3, 4.2, and 5.2 mutations/clone, respectively, in the 282 bases sequenced. As expected, the vast majority were point mutations, transitions were more common than transversions, and T in the coding strand (and to a lesser degree C) mutated less frequently than A or G (results not shown).
Figure 2.
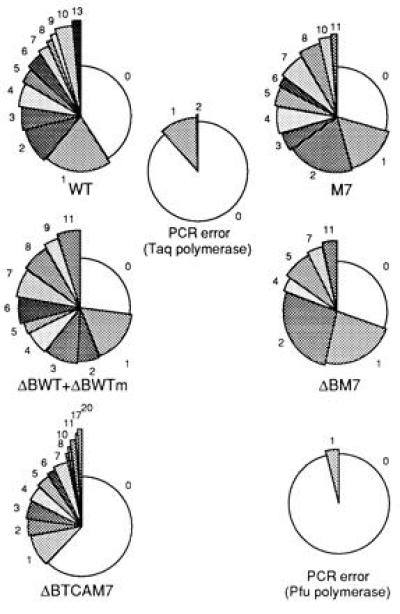
Frequency distribution of clones with respect to the number of mutations they carry. Each part depicts the proportion of sequences with mutations as specified. The controls for PCR errors for each set of experiments are shown at the center or to the right of the relevant results. PCR errors were measured by sequencing transgenes from characterized hybridomas. ΔBWTm refers to one of the ΔBWT copies that carries an accidental G for A silent substitution in the Lys-18 codon.
Table 1.
Transgene mutations in PCR clones derived from Peyer’s patches germinal center B cells
| Mouse line and transgene | No. of clones*
|
Total PM† | PMs per 103 base pairs‡
|
||
|---|---|---|---|---|---|
| Total | >1 PM | Total | >1 PM | ||
| LκWTM7 | |||||
| WT | 88 | 34 | 184 | 7.4 | 17.3 |
| M7 | 81 | 44 | 211 | 9.2 | 16.0 |
| LκNG | |||||
| ΔBWT + ΔBWTm | 59 | 33 | 197 | 11.8 | 20.1 |
| ΔBM7 | 26 | 12 | 52 | 7.1 | 13.6 |
| PCR error | 245 | 1 | 29 | 0.4 | |
| LκΔBTCAM7/ΔLi | |||||
| ΔBTCAM7 | 172 | 48 | 267 | 5.5 | 18.5 |
| PCR error | 52 | 0 | 2 | 0.1 | |
PM, point mutation.
Clones sequenced, and number of those with more than one point mutation (>1 PM).
Sum of point mutations in all clones.
Frequency of PMs per 103 base pairs computed for all clones or for those with two or more mutations.
Mutability of Ser 31 (AGT) Is Increased or Decreased When Encoded by AGC or TCA, Respectively.
The overall distribution of hot spots in wild type and mutants showed very few differences, but some of them were remarkable (Fig. 3). There was an additional hot spot in M7* at Ser-31(III), where T was replaced by C. The emergence of this hot spot was not at the expense of the one at Ser-31(II), which was unaffected. When the Ser-31 codon (AGC), containing the two most prominent hot spots of M7*, was substituted for TCA, no mutants were found in either the second or third position (Fig. 3, ΔBTCAM7). In addition to these prominent changes, there were other differences scattered not only within or near the segment containing the mutations but also elsewhere.
Figure 3.
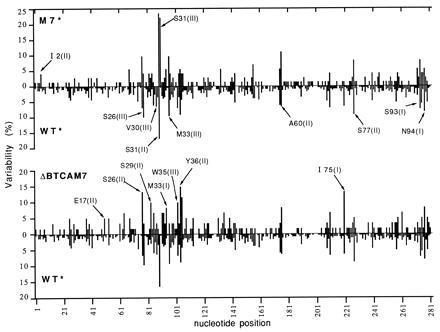
Comparison of hot spots of WT*(WT+ΔBWT), M7*(M7+ΔBM7), and ΔBTCAM7. Variability is the number of times a given nucleotide was mutated divided by the number of clones sequenced (in %). The number of clones used to compile this figure was 239 WT* (data from refs. 23 and 37), 75 M7*, and 61 ΔBTCAM7.
To assess the statistical significance of all these differences, we performed a χ2 test (Fig. 4). The new hot spot at Ser-31(III) in M7* was highly significant. Equally, the conclusion that Ser-31 is not a hot spot when encoded by TCA is highly reliable, with a confidence level in the region of 0.001.
Figure 4.
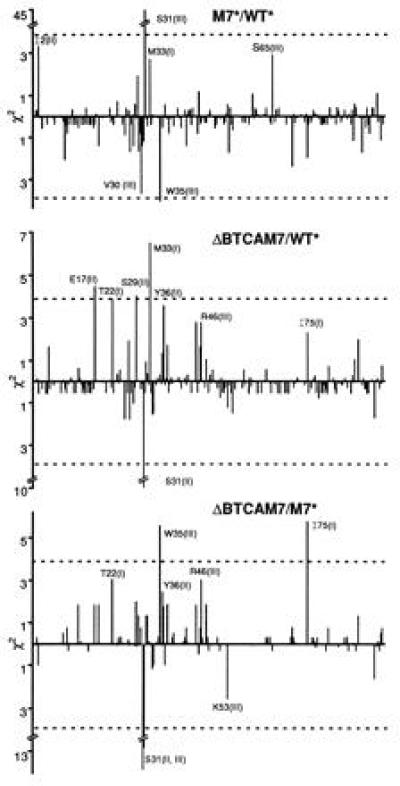
χ2 Pearson test of statistical significance of the mutability differences between WT*, M7*, and ΔBTCAM7. Vertical lines above and below 0 indicate, respectively, increased or decreased mutation frequency of individual nucleotides. Comparisons are for M7* relative to WT*, ΔBTCAM7 relative to WT*, and ΔBTCAM7 relative to M7*. Broken horizontal line indicates the 95% confidence level.
Compensatory Changes of Mutation Frequency.
Variations of Ser-31 hot spots induced by codon changes were accompanied by variations of mutation rates at a variety of other positions scattered along the V-segment. Those that were statistically significant were opposite to the ones observed at Ser-31 (Fig. 4). Thus, while the prominent Ser-31 hot spot(s) disappeared in ΔBTCAM7, the mutation rates of Met-33(I), Trp-35(III), Ile-75(I), and others increased relative to WT* or M7*. On the other hand, the new hot spot in M7* was counterbalanced by a decreased mutability of Trp-35(III) and probably Val-30(III). We conclude that when new hot spots are created or eliminated, compensatory changes elsewhere occur, so that the average number of mutations accumulated in each mutated clone is not affected. This generalization is also illustrated in Fig. 2.
TA, a Hot-Spot Motif.
The increased mutation frequency of Met-33(I) of ΔBTCAM7 relative to WT* was statistically significant at a confidence level of 99.9%, well above the average (Fig. 4). The Met-33(I) in M7* was also more mutated than in WT* but only marginally, possibly due to the compensatory effect induced by the new Ser-31 hot spot. Since the Met codon is invariant, higher mutability must have been induced by other changes. The most obvious one was the substitution of C for T encoding Tyr-32(III). This and the following A of Met introduced a TA sequence. This observation supports the view that TA is part of an alternative motif involved in the origin of hot spots (30, 34).
Other changes of mutation frequencies could not be directly correlated with alterations of primary sequence motifs, for example Trp-35(III) and Ile-75(I). Thus, a statistically significant change in mutation frequency occurred 12 and 127 residues downstream of the only difference between ΔBTCAM7 and M7*. A similar comment is applicable to other changes that were at or near the 95% confidence level.
Hypermutation of Mutant Transgenes in Hybridomas.
Do changes of silent codons affect the pattern of affinity maturation? The hallmark of affinity maturation to 2-phenyloxazolone is the substitution of His-34 for Asn or Gln, usually associated with a Tyr-36 for Phe substitution. This increases the affinity of the antibody by a factor of 10 and 8, respectively (20). Transgenic lines expressing WT and M7 or ΔBWT and ΔBM7 simultaneously gave us an opportunity to investigate if one of the copies was preferentially targeted to include the diagnostic mutations. Thus, we derived hybridomas from secondary response to 2-phenyloxazolone and sequenced the relevant segment of each transgene. At least three M13 clones of each transgene copy were sequenced to correct for PCR errors.
Table 2 shows the summary of the results. LκNG contained five VκOx1 copies, three ΔBWT (one identifiable by a fortuitous silent substitution of G for A at the Lys-18), and two ΔBM7. Of 10 hybridomas, five (identified by an asterisk) included a copy with the His-34 for Asn or Gln, accompanied by Tyr-36 for Phe substitutions. All of them were in the WT copies. At first sight, this may suggest that the WT is more efficient than the M7 copies in generating the higher affinity mutants. However, the opposite was found in the hybridomas derived from the LκWTM7 line, which contains one copy of each transgene. Here, out of five mutated hybridomas, two include the diagnostic substitution pair, and both are in the M7 copy.
Table 2.
Transgene mutations in PCR clones derived from anti-phOx hybridomas
| Hybridoma | Antibody
|
No. of PMs per
copy*
|
||||||
|---|---|---|---|---|---|---|---|---|
| Cκ, rat | Cκ, mouse | ΔBWT | ΔBWTm† | ΔBM7 | WT | M7 | ΔBTCAM7 | |
| LκNG | ||||||||
| C15.2.7 | + | − | 8‡ and 6 | 7 | 3 and 3 | |||
| C10.5.4 | + | − | 5‡ and 8 | 6 | 1 and 1 | |||
| A6.8 | − | + | 3 and 3 | 1 | 0 | |||
| C23.1.5 | + | − | 7‡ and nf | NF | 2 and 2 | |||
| D1.1.5 | + | − | 10‡ and 0 | NF | 10 and 0 | |||
| B20.6 | + | − | 4 and 0 | 2 | 4 and 0 | |||
| C6.3.6 | + | − | 8‡ and nf | 4 | 5 and 2 | |||
| D12.3.8 | + | − | 0 | 2 | 0 | |||
| C4.6.8 | + | − | 0 | 2 | 0 | |||
| D23.4 | + | − | 1 and 0 | 1 | 1 and 0 | |||
| LκWTM7 | ||||||||
| D3.3.3 | + | − | 3§ | 7‡ | ||||
| C12.1.3 | + | − | 5§ | 7‡ | ||||
| EH2.5 | ± | + | 0 | 4 | ||||
| RB3.6 | ± | + | 0 | 3 | ||||
| EA8.7 | ± | + | 0 | 1 | ||||
| C3.23.2 | ± | + | 0 | 0 | ||||
| C11.4.8.8 | + | − | 0 | 0 | ||||
| LκΔBTCAM7/ΔLi | ||||||||
| B2.4.4 | + | − | 3 | |||||
| C14.1.9 | + | − | 6‡ | |||||
| A20.5.2 | + | − | 4‡ | |||||
PM, point mutation; NF, not found.
Number of PMs found in each transgenic copy.
This copy was identified by a silent substitution (G for A) in codon Lys-18.
Copies containing the critical mutation of His-34 to Asn or Gln. In addition, all of them included the mutation Tyr-36 to Phe.
These copies also carried deletions that result in a coding sequence out of frame.
The transgenic copy maturation bias in the two lines did not correlate with the nucleotide differences. It did, however, correlate with the average hypermutation rates of the copies. The ratio M7/WT of all mutations in hybridomas were 22/8 for LκWTM7 and 34/63 for ΔBM7/ΔBWT in LκNG. A similar (but less marked) overall accumulation of mutants was found in Peyer’s patches (Table 1, penultimate column). Thus the higher mutation frequencies in both hybridomas and Peyer’s patches correlate with the preferential maturation of transgene copies.
DISCUSSION
The Role of Primary Sequence Motifs and Higher Order Structure in Determining Hot Spots.
The results of this paper show that primary sequence motifs are of prime importance in generating hot spots and provides an experimental base to the view that the AGC/T codon for serine has been selected through evolution to target hot-spot positions (26, 27). However, not all AGC/T give rise to hot spots, and not all hot spots include such a sequence. The CDR1 of VκOx1 includes this motif as a prominent hot spot, as well as extensive palindromes and hairpin loops (Fig. 1). Using synthetic mutants, we were able to test the relative importance of primary sequence and secondary structure motifs in the origin of hot spots.
Changing the nucleotide sequence without altering the encoded amino acids, we distorted the local secondary structure (Fig. 1) and went on to show that it was the motifs rather than the distortions the ones that correlated with changes in targeting efficiency. Thus, the M7* nucleotide substitutions removed a potential hairpin loop encompassing the major hot spot G in Ser-31 codon. This did not affect the mutation frequency of the G. However, the third base T of Ser-31, a cold spot in WT*, became a major hot spot when replaced by C. The importance of local sequence motifs is also illustrated by the loss of the two hot spots following replacement of AGC with the synonymous TCA.
The G in the motif AGC/T has been found associated with hot spots in a variety of examples. Surprisingly, when the G is followed by a C (but not by a T), this residue also becomes a hot spot. This explains why Ser-26 AGC contains two hot spots and similarly why both G and C are hot spots in the sequence gcgaGC encoding Glu-Leu in the gpt gene (25). The reason for this peculiarity seems to be that G and C hot spots have overlapping motifs (unpublished results). In any event, the fact that it is found in independent examples supports the view that it is due to the primary sequence difference.
In addition to G and C, hot spots involve A residues, but in such cases the neighboring sequences are quite different. In our studies, a mutation leading to a TA sequence increases the mutability of A, thus adding experimental evidence to speculations implicating TA in an alternative hot-spot motif. Indeed, A hot spots are generally preceded by T (unpublished results), and it is not rare to find the second base of tyrosine (TAT/C) highly mutated. TA hot spots are, of course, also associated with other residues and present in untranslated segments (30, 34).
At least two sequence motifs seem necessary to define C, G, and A hot spots, but this is unlikely to be the whole story. Motifs could involve a respectable number of as yet undefined residues, but, even so, the change in mutation frequency in residues well removed from the mutated CDR1 segment [e.g., Ile-75(I)] provide clues about other features controlling the variation in mutability of individual nucleotides. We suggest that higher order structural features are also involved in determining hot spots. This is simpler to imagine if only one strand, capable of adopting a complex higher order structure(s), is the hypermutating unit. It has been proposed that hypermutation occurs as a consequence of an error prone repair process that operates during transcription, along the lines of the excision repair mechanism (39, 40, 41, 42, 43). It is therefore not unreasonable to speculate that the strand of DNA being transcribed, adopts a folded structure involving long-range interactions. These (and/or DNA–protein interactions) may also control the efficiency of the mutability of different motifs. Thus, mutations anywhere in the gene could have unpredictable consequences regarding mutation frequencies of residues elsewhere. This conclusion has important implications in terms of affinity maturation or antibodies.
Preferential Affinity Maturation of Transgene Copies.
Since minor sequence alterations of transgenes have a direct effect on the mutability of both neighboring and distant bases, early mutations during the process of affinity maturation could be important in skewing the pattern of mutations during subsequent cycles of hypermutation. Thus, even silent mutations could increase (or decrease) the mutation rate of nucleotides involved in affinity maturation.
In the experiments described here, the comparison of mutation rates between WT and mutant transgenes did not disclose a change in mutability of the diagnostic His-34. Thus, it was surprising to find preferential expression of this maturation hallmark in either the mutant or the WT copy. However, the preference was opposite in two transgenic lines, suggesting that the advantage was not due to the sequences but to a peculiarity of the line. Different transgene copy numbers did not explain our results, since there were the same number of relevant copies in both lines. A better explanation arose by correlating overall targeting efficiency and maturation of individual copies. The overall mutation rate of the “preferred” transgene was higher, both in hybridomas and in Peyer’s patches, supporting the hypothesis that the preference was a consequence of marginal changes in the total rate of accumulation of mutants in the individual transgene copies. It is possible that overall mutability is correlated with differences in expression, which in turn could be related to the configuration of the individual copies and/or the site of integration.
Hot Spots and Hypermutation Rate.
The localization of intrinsic hot spots played an important role in the evolution of antibody genes, but this and the primary amino acid sequence requirements may have been in conflict. Even so, natural selection seems to have shaped the pattern of intrinsic hypermutation to allow critical diversity with minimum degeneracy. The corollary is that the frequency of deleterious mutations is much lower than the average. A second corollary is that as hypermutation proceeds, hot spots are likely to become “colder” (19). Somewhat surprisingly, we find also that some of the nucleotides become “hotter,” because loss of hot spots seems to be compensated by opposite changes elsewhere. For example, the suppression of the Ser-31 hot spot(s) was accompanied by a significant increased mutability of Met-33(I), Trp-35(III), and Ile-75(I). This compensatory device seems to take place at positions of otherwise moderate mutability. Hot spots are paramount for models of affinity maturation. Accumulation of favorable mutations require multiple cycles of hypermutation followed by selection (44, 45). When the distribution of mutants is considered to be random, degeneracy becomes unacceptable when, before antigen selection, there is more than one point mutation in the light (or heavy) chain every two rounds of DNA replication (17, 18). However, as mutations are not randomly distributed, the ratio between favorable and deleterious mutations is greatly altered. The higher the ratio, the larger the number of point mutations that can be accumulated before selection takes place. Future models of affinity maturation must take into consideration the effect of the skewed distribution of hot spots. To what extent it will be possible to predict the mutability of individual residues in any V-gene remains to be seen.
We have previously argued that the number of intrinsic mutations accumulated in individual transgene copies could not be the result of random accumulation of individual mutations (25). This raised the possibility that the mutations are not introduced at an average of one per gene, but that at least in some cells, multiple mutations are introduced in a single hypermutating round. The unequal rate of mutations in individual bases, allowing the accumulation of multiple mutations without unacceptable increase in the rate of degeneration, gives further support to such proposal. The proposal that each round of antigen selection takes place on cells, which on average have incorporated more than one point mutation in their antibody genes, still stands.
Acknowledgments
We thank T. Larson, J. M. Jarvis, G. King, and R. Pannell for the derivation of transgenic mice and hybridomas. We thank W. Gilks for the statistical analysis and M. Neuberger for helpful discussions and the Association for International Cancer Research and the Medical Research Council for financial support.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: WT, wild-type.
References
- 1.Griffiths G M, Berek C, Kaartinen M, Milstein C. Nature (London) 1984;312:271–275. doi: 10.1038/312271a0. [DOI] [PubMed] [Google Scholar]
- 2.Berek C, Berger A, Apel M. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 3.MacLennan I C, Liu Y J, Johnson G D. Immunol Rev. 1992;126:143–161. doi: 10.1111/j.1600-065x.1992.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 4.Jacob J, Przylepa J, Miller C, Kelsoe G. J Exp Med. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Küppers R, Zhao M, Hasmann M L, Rajewsky K. EMBO J. 1993;12:4955–4967. doi: 10.1002/j.1460-2075.1993.tb06189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pascual V, Liu Y-J, Magalski A, de Bouteiller O, Banchereau J, Capra J D. J Exp Med. 1994;180:329–339. doi: 10.1084/jem.180.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gearhart P J, Bogenhagen D F. Proc Natl Acad Sci USA. 1983;80:3439–3443. doi: 10.1073/pnas.80.11.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Both G W, Taylor L, Pollard J W, Steele E J. Mol Cell Biol. 1990;10:5187–5196. doi: 10.1128/mcb.10.10.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebecque S G, Gearhart P J. J Exp Med. 1990;172:1717–1727. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber J S, Berry J, Manser T, Claflin J L. J Immunol. 1991;146:3652–3655. [PubMed] [Google Scholar]
- 11.Rogerson B J. Mol Immunol. 1994;31:83–98. doi: 10.1016/0161-5890(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 12.Rada C, González-Fernández A, Jarvis J M, Milstein C. Eur J Immunol. 1994;24:1453–1457. doi: 10.1002/eji.1830240632. [DOI] [PubMed] [Google Scholar]
- 13.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Proc Natl Acad Sci USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berek C, Milstein C. Immunol Rev. 1988;105:5–26. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 15.Adetugbo K, Milstein C, Secher S D. Nature (London) 1977;265:299–304. doi: 10.1038/265299a0. [DOI] [PubMed] [Google Scholar]
- 16.Chui Y L, Lozano F, Jarvis J M, Pannell R, Milstein C. J Mol Biol. 1995;249:555–563. doi: 10.1006/jmbi.1995.0318. [DOI] [PubMed] [Google Scholar]
- 17.Weigert M. Prog Immunol. 1986;VI:138–144. [Google Scholar]
- 18.Allen D, Cumano A, Dildrop R, Kocks C, Rajewsky K, Rajewsky N, Roes J, Sablitzky F, Siekevitz M. Immunol Rev. 1987;96:5–22. doi: 10.1111/j.1600-065x.1987.tb00506.x. [DOI] [PubMed] [Google Scholar]
- 19.Secher D S, Milstein C, Adetugbo K. Immunol Rev. 1977;36:56–72. doi: 10.1111/j.1600-065x.1977.tb00382.x. [DOI] [PubMed] [Google Scholar]
- 20.Berek C, Milstein C. Immunol Rev. 1987;96:23–41. doi: 10.1111/j.1600-065x.1987.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 21.Sharpe M J, Milstein C, Jarvis J M, Neuberger M S. EMBO J. 1991;10:2139–2145. doi: 10.1002/j.1460-2075.1991.tb07748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Betz A G, Neuberger M S, Milstein C. Immunol Today. 1993;14:405–411. doi: 10.1016/0167-5699(93)90144-a. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez F A, Milstein C. Proc Natl Acad Sci USA. 1993;90:9862–9866. doi: 10.1073/pnas.90.21.9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Betz A G, Milstein C, Gonzalez F A, Pannell R, Larson T, Neuberger M S. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 25.Yelamos J, Klix N, Goyenechea B, Lozano F, Chui Y L, Gonzalez F A, Pannell R, Neuberger M S, Milstein C. Nature (London) 1995;376:225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]
- 26.Wagner S D, Milstein C, Neuberger M S. Nature (London) 1995;376:732. doi: 10.1038/376732a0. [DOI] [PubMed] [Google Scholar]
- 27.Jolly C J, Wagner S D, Rada C, Klix N, Milstein C, Neuberger M S. Semin Immunol. 1996;8:159–168. doi: 10.1006/smim.1996.0020. [DOI] [PubMed] [Google Scholar]
- 28.Milstein, C. & Neuberger, M. S. (1996) Adv. Protein Chem., in press. [DOI] [PubMed]
- 29.Reynaud C A, Garcia C, Hein W R, Weill J C. Cell. 1995;80:115–125. doi: 10.1016/0092-8674(95)90456-5. [DOI] [PubMed] [Google Scholar]
- 30.Rogozin I B, Kolchanov N A. Biochim Biophys Acta. 1992;1171:11–18. doi: 10.1016/0167-4781(92)90134-l. [DOI] [PubMed] [Google Scholar]
- 31.Milstein C, Even J, Berek C. Biochem Soc Symp. 1986;51:173–182. [PubMed] [Google Scholar]
- 32.Golding G B, Gearhart P J, Glickman B W. Genetics. 1987;115:169–176. doi: 10.1093/genetics/115.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolchanov N A, Solovyov V V, Rogozin I B. FEBS Lett. 1987;214:87–91. doi: 10.1016/0014-5793(87)80018-2. [DOI] [PubMed] [Google Scholar]
- 34.González-Fernández A, Gupta S K, Pannell R, Neuberger M S, Milstein C. Proc Natl Acad Sci USA. 1994;91:12614–12618. doi: 10.1073/pnas.91.26.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pettersson S, Sharpe M J, Gilmore D R, Surani M A, Neuberger M S. Int Immunol. 1989;1:509–516. doi: 10.1093/intimm/1.5.509. [DOI] [PubMed] [Google Scholar]
- 36.Galfré G, Milstein C. Methods Enzymol. 1981;73:3–46. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez F A, Gilmore D, Milstein C. Eur J Immunol. 1994;24:2918–2921. doi: 10.1002/eji.1830241151. [DOI] [PubMed] [Google Scholar]
- 38.Sigal F. Non-Parametric Statistic for the Behavioral Sciences. London: McGraw–Hill; 1956. [Google Scholar]
- 39.Bootsma D, Hoeijmakers J H J. Nature (London) 1993;363:114–155. doi: 10.1038/363114a0. [DOI] [PubMed] [Google Scholar]
- 40.Hanawalt P C. Science. 1994;266:1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 41.Hanawalt P C. Mutat Res. 1995;336:101–113. doi: 10.1016/0921-8777(94)00061-a. [DOI] [PubMed] [Google Scholar]
- 42.Neuberger M S, Milstein C. Curr Opin Immunol. 1995;7:248–254. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 43.Peters A, Storb U. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 44.Kepler T B, Perelson A S. J Theor Biol. 1993;164:37–64. doi: 10.1006/jtbi.1993.1139. [DOI] [PubMed] [Google Scholar]
- 45.Celada F, Seiden P E. Eur J Immunol. 1996;26:1350–1358. doi: 10.1002/eji.1830260626. [DOI] [PubMed] [Google Scholar]