

Abstract
Functional human immunodeficiency virus type -1 env clones have been widely used for vaccine design, neutralization assays, and pathogenesis studies. However, obtaining bona fide functional env clones is a time consuming and labor intensive process. A new high throughput method has been developed to characterize HIV-1 env genes. Multiple rev/env gene cassettes were obtained from each of seven HIV-1 strains using single genome amplification (SGA) PCR. The CMV promoter was amplified separately by PCR. A promoter PCR (pPCR) method was developed to link both PCR products using an overlapping PCR method. Pseudovirions were generated by cotransfection of pPCR products and pSG3Δenv backbone into 293T cells. After infecting TZM-bl cells, 75 out of 87 (86%) of the rev/env gene cassettes were functional. Pseudoviruses generated with pPCR products or corresponding plasmid DNA showed similar sensitivity to six HIV-1 positive sera and three monoclonal antibodies, suggesting neutralization properties are not altered in pPCR pseudovirions. Furthermore, sufficient amounts of pseudovirions can be obtained for a large number of neutralization assays. The new pPCR method eliminates cloning, transformation, and plasmid DNA preparation steps in the generation of HIV-1 pseudovirions, this allows for quick analysis of multiple env genes from HIV-1 infected individuals.
Keywords: HIV-1, neutralization, envelope, pseudovirion, PCR
1. Introduction
Envelope genes of human immunodeficiency virus type 1 (HIV-1) have been extensively studied using pseudovirions by cotransfection of a backbone of an env-defective clone and an env expression plasmid (Helseth et al., 1990). This allows for the study of env genes from a large number of HIV-1 isolates (Derdeyn et al., 2004; Gao et al., 1996; Wei et al., 2003; Li et al., 2005; Li et al., 2006). Traditionally, env genes are amplified from either proviral DNA or viral RNA through RT-PCR and the products are then cloned into expression vectors for generation of pseudovirions. However, the env gene products amplified through bulk PCR may be affected by recombination or resampling during bulk PCR amplification (Fang et al., 1998; Liu et al., 1996). Since minor sequence changes can affect the biological functions of envelope glycoproteins (Cordonnier et al., 1989; Kalia et al., 2005; LaBranche et al., 1995; Morris et al., 1994; Shimizu et al., 1999; Shioda et al., 1994), it is important to avoid studying env genes containing artificial elements generated during PCR in vitro.
Non recombinant env genes can be obtained using limiting dilution or single genome amplification (SGA) methods (Liu et al., 1996; Palmer et al., 2005; Simmonds et al., 1990). Since only one amplifiable viral genome is amplified in each PCR reaction in either method, the products are not affected by recombination or resampling. However, because the PCR products are obtained through multiple reactions, numerous purifications, ligations, transformations, and plasmid DNA preparations are required to test the functionality of the env PCR products. As a result, this cloning step is time consuming, labor intensive, and costly.
The use of Env pseudotype viruses in a single round infection system has greatly improved the accuracy and simplicity of the evaluation of neutralization activity in vaccinated humans and experimental animals (Derdeyn et al., 2004; Gao et al., 1996; Helseth et al., 1990; Li et al., 2005; Wei et al., 2003). Given the extent of viral diversity that is seen among patients and even within a single individual, a large number of env clones are needed to understand neutralization profiles. For this reason, two panels of subtype B and C viruses have been proposed for use as standards for the evaluation of anti-HIV-1 neutralization activity in anti-HIV-1 sera (Li et al., 2005; Li et al., 2006). Analysis of a large number of Env pseudovirions from many different individuals using autologous and heterologous sera may lead to identification of signature sequences of critical neutralization epitopes and assist vaccine design. Therefore, a more efficient system is needed to generate a large number of Env pseudovirions. This study describes a promoter PCR (pPCR) method that can significantly decrease the labor, time, and cost needed to obtain a large numbers of Env pseudovirions by eliminating the cloning step.
2. Methods and Materials
2.1 Amplification of HIV-1 env genes
Seven plasma samples were collected from HIV-1 positive individuals enrolled in a study of current HIV-1 strains in Ndola, Zambia. The study was approved by the ethics committee of the Tropical Disease Research Centre, the Duke University Institutional Review Board, and the National Institutes of Health. Viral RNA was extracted from the plasma and eluted into 55 μl of elution buffer using QiaAmp Viral RNA Mini kit (Qiagen; Valencia, CA). Reverse transcription was performed with 25 μl of vRNA and 25 pmol primer OFM19 5′-GCACTCAAGGCAAGCTTTATTGAGGCTTA-3′ in 100 μl using Superscript III (Invitrogen; Carlsbad, CA). Single genome amplification (SGA) of the cDNA was performed to obtain the rev/env cassette and to avoid artificial recombination and resampling of the viral genomes (Palmer et al., 2005). The cDNA was diluted 1:3, 1:9, and 1:27 (15 reactions per dilution) to determine the dilution with a positive rate of 30% or less. One microliter of each diluted cDNA was used for the first round amplification with primers OFM19 and VIF1 5′-GGGTTTATTACAGGGACAGCAGAG–3′ (Derdeyn et al., 2004). First round PCR was carried out with 1 unit of Platinum Taq Polymerase High Fidelity (Invitrogen; Carlsbad, CA), and 10 pmol of each primer in 20 μl. The PCR thermocycling conditions were as follows: one cycle at 94°C for 2 min; 35 cycles of a denaturing step at 94°C for 15 sec, an annealing step at 55°C for 30 sec, an extension step at 68°C for 4 min; and one cycle of an additional extension at 68°C for 10 min. First round PCR products (2 μl) were used for the second round PCR with primers env1Atopo 5′-CACCGGCTTAGGCATCTCCTATGGCAGGAAGAA-3′ and envN 5′-CTGCCAATCAGGGAAGTAGCCTTGTGT-3′. The second round PCR was carried out in 50 μl total volume with 2.5 units of Platinum Taq Polymerase High Fidelity and 15 pmol of each primer. The PCR thermocycling conditions were as follows: one cycle at 94°C for 2 min; 45 cycles of a denaturing step at 94°C for 15 sec, an annealing step at 55°C for 30 sec, an extension step at 68°C for 4 min; and one cycle of an additional extension at 68°C for 10 min.
Similarly, HIV-1 rev/env cassettes were amplified from five functional env clones, CAP210.2.00.E8, DU422.1, ZM53.MPB12, ZM109.FPB4, and TRO.11, (Li et al., 2005; Li et al., 2006). Amplification was performed with 2 units of Platinum Taq Polymerase High Fidelity (Invitrogen; Carlsbad, CA), 5 pmol of each primer (env1Atopo and envN), and 100 pmol of each plasmid template in a 50 μl volume. The PCR thermocycling conditions were as follows: one cycle at 94°C for 2 min; 20 cycles of a denaturing step at 94°C for 30 sec, an annealing step at 55°C for 30 sec, an extension step at 68°C for 4 min; and one cycle of an additional extension at 68°C for 10 min. All PCR products were visualized on a 0.7% agarose gel and purified with the QiaQuick PCR Purification kit (Qiagen; Valencia, CA).
2.2. Amplification of CMV promoter
The CMV promoter was amplified from the pcDNA 3.1D/V5-His-TOPO cloning vector (Invitrogen; Carlsbad, CA). The PCR was carried out in a total volume of 50 μl with 2 units of Platinum Taq Polymerase High Fidelity, 100 pmol plasmid, and 10 pmol of each primer (CMVenv 5′-AGTAATCAATTACGGGGTCATTAGTTCAT-3′ and CMVenv1A 5′-CATAGGAGATGCCTAAGCCGGTGGAGCTCTGCTTATATAGACCTC-3′). Each sample was subjected to one cycle at 94°C for 2 min; 30 cycles of a denaturing step at 94°C for 30 seconds, an annealing step at 55°C for 30 seconds, an extension step at 68°C for 4 minutes; and one cycle of an additional extension at 68°C for 10 min.
2.3 Promoter PCR (pPCR)
Overlapping PCR was performed using 100 ng of amplified HIV-1 env, 50 ng of CMV promoter, primers CMVenv and env1M 5′-TAGCCCTTCCAGTCCCCCCTTTTCTTTTA-3′ (20 pmol each) in a 50μl reaction with Platinum Taq Polymerase High Fidelity. The samples were subjected to one cycle at 94°C for 2 min; 20 cycles of 94°C for 30 seconds, 55°C for 30 seconds, 68°C for 4 minutes; and one cycle of an additional extension at 68°C for 10 min.
2.4 Single round infectivity assay
pPCR products were cotransfected with env-deficient HIV-1 backbone pSG3Δenv into 293T cells in a 24 well tissue culture plate using FuGENE6 transfection reagent (Roche Diagnostics; Indianapolis, IN) according to manufacturer instructions. Briefly, pPCR DNA (200 ng) or plasmid DNA (200 ng) and pSG3Δenv DNA (200 ng) were mixed with 1.2 μl of FuGENE6 (FuGENE:DNA ratio at 3 μl:1 μg) in a total volume of 20 μl with serum free DMEM, incubated for 30 minutes and added to 293T cells (70% confluence) seeded one day earlier at 5 ×104 per well. Transfected cells were maintained in DMEM with 10% FBS and Gentamicin. Forty-eight hours after transfection, supernatants were harvested. Equal volumes of pseudovirions were added to TZM-bl cells with DEAE (5 μg/ml) in a 96 well tissue culture plate (200 μl). Cultures were incubated for 48 hrs at 37°C with 5% CO2.
2.5 Luciferase Assay
Supernatants (100 μl) from infected TZM-bl cells were removed and 100 μl of Bright-Glo Luciferase Assay substrate with buffer (Promega; Madison, WI) was added to the cells. Following a 2-minute incubation, 100 μl of cell lysates were added to a solid black 96 well plate. Luminescence was measured with a Wallac 1420 Multilabel Counter (PerkinElmer: Waltham, MA). The TCID50 was determined as described previously (Li et al., 2005).
2.6 Neutralization Assay
Neutralization activity was measured as a reduction in luciferase activity after a single round infection of TZM-bl cells as previously described. (Li et al., 2005; Li et al., 2006; Montefiori, 2004). Equal amounts of pseudovirions (200 TCID50) were used in each reaction. Neutralization titers of pPCR pseudovirions were determined against six HIV-1 positive sera, one normal serum control, and three broadly neutralizing mAbs (4E10, 2F5, and 2G12).
2.7 Western Blot assay
Forty eight hours after cotransfection, the 293T cells were lysed with 250 μl of lysis buffer (50mM Tris-HCl, 150mM NaCl, 20mM EDTA, 1% Triton-X100, 0.1% SDS, pH 7.4). Cell lysates were mixed with denaturing sample buffer (24% Tris-HCl pH6.8, 20% SDS, 30% glycerol, 1.6% β-mercaptoethanol, 0.06% Bromophenol blue). Samples were boiled for 10 minutes and then loaded on a 4-15% gradient NuPAGE© SDS-PAGE gel (Invitrogen; Carlesbad, CA). Samples were normalized by the same amount of p24 protein. After the samples were transferred to a nitrocellulose membrane, the membrane was blocked (PBS containing 1% casein and 0.01% NaN3) for 1 hr. The blot was then reacted with an HIV-1 infected patient serum (1:500) and mouse mAb 13D5 to the HIV-1 Env protein (1:3 000). Finally, the membrane was reacted with an IRDye800 conjugated affinity purified goat anti-human antibody (Rockland Immunochemicals; Gilbertsville, PA) and an Alexa-Fluor 680 conjugated goat anti-mouse antibody (Invitrogen; Carlesbad, CA). Fluorescence was detected with an Odyssey Infrared Imaging system (LiCor Biosciences; Lincoln, NE).
2.8 Sequence analysis
Sequence analysis of PCR products was performed by cycle-sequencing and dye terminator methods with an ABI 3100 genetic analyzer (Applied Biosystems; Foster City, CA). Individual sequence fragments for each env SGA were assembled and edited using the Sequencher program 4.7 (Gene Codes; Ann Arbor, MI). Deduced Env amino acid sequences were aligned using CLUSTAL W (Thompson et al., 1994).
2.9 Statistical analysis
Linear regression analysis was performed using SigmaPlot 8.0. (Systat Software, Inc; Point Richmond, CA). Luciferase data was analyzed using the two-tail Student T test with Excel (Microsoft Office 2004, Redmond, WA).
3. Results
To establish the promoter PCR (pPCR) method, the HIV-1 env gene (3 121 bp) and CMV promoter (655 bp) were amplified separately from known functional env clones TRO.11, CAP210.2.00.E8, DU422.1, ZM53.MPB12, and ZM109.FPB4 (Li et al., 2005; Li et al., 2006) The CMV promoter was added to the 5′ end of the env PCR product by overlapping PCR (Fig. 1). The pPCR product was purified and cotransfected with an env-deficient proviral clone (pSG3Δenv) into 293T cells. The pseudovirions were harvested from the supernatants of the transfected cells 48 hrs after infection. An equal volume of supernatants containing pseudovirions generated with pPCR products or corresponding plasmid DNA were used to infect the TZM-bl cells. The luciferase activity of pPCR pseudovirions was on average 1.6 fold lower than that of their corresponding plasmid but significantly higher than that of the backbone control (Fig. 2). This suggests the pPCR method can be used to evaluate the biological functions of HIV-1 env genes by adding the CMV promoter at its 5′ end without a cloning step.
Figure 1.
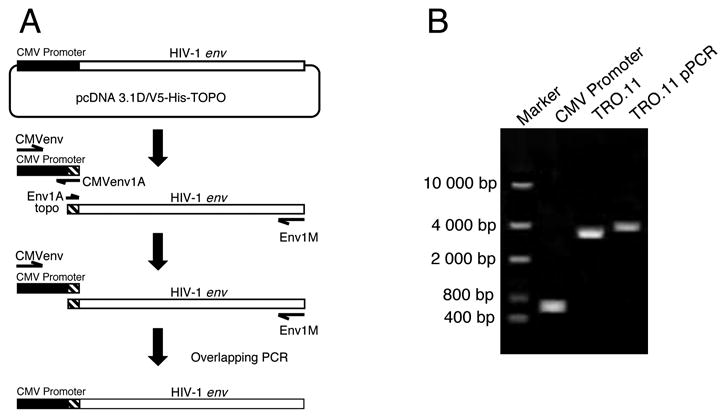
Generation of promoter PCR (pPCR) amplicons. (A) Schematic presentation of the pPCR procedure. The CMV promoter and the HIV-1 rev/env cassette were amplified separately. The CMV promoter was then added to the HIV-1 rev/env gene at the 5′ end by overlapping PCR. (B) PCR and pPCR amplification of the CMV promoter and the env gene from a functional env control clone TRO.11 visualized on a 0.7% agarose gel.
Figure 2.
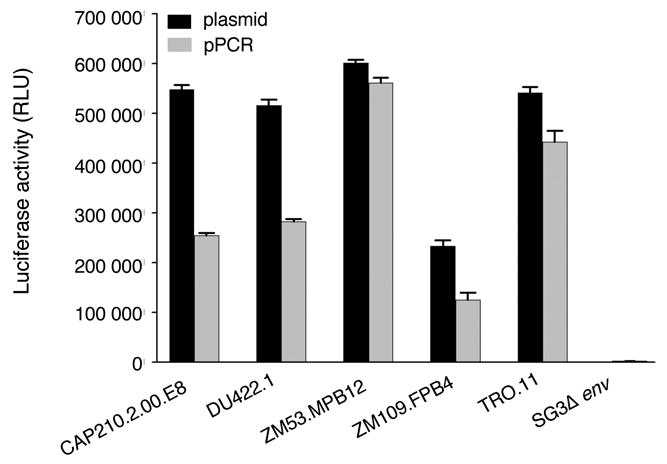
Infectivity of pseudovirions derived from pPCR DNA or corresponding functional env clones. The pPCR product (1000 ng) or corresponding env plasmid (1000 ng) was cotransfected with pSG3Δenv into 293T cells in a T25 flask. Supernatants were harvested 48 hrs after transfection and were used to infect TZM-bl cells. The luciferase activity was measured two days after infection. The data is shown as the mean ± standard error (n = 24 independent assays).
The new pPCR method was then tested with a patient plasma sample (05ZM373). Viral RNA was extracted and reverse transcribed into cDNA. A total of 48 PCR reactions were performed and five SGAs were obtained. The CMV promoter was added to all env PCR products at the 5′ end by pPCR (Fig. 3A). After purification, the pPCR products were cotransfected with pSG3Δenv backbone clone into 293T cells. Supernatants (20 μl) from transfected cells were used to infect TZM-bl cells. Luciferase activity was determined 48 hrs post infection. Four pPCR pseudovirions yielded positive responses. Three had slightly higher infectivity than the pPCR TRO.11 control, while 05ZM373.2 was less infectious than the TRO.11 control (Fig. 3B). 05ZM373.1 did not yield infectious pseudovirions and the luciferase activity was similar to the background level of pSG3Δenv and CMV promoter controls.
Figure 3.
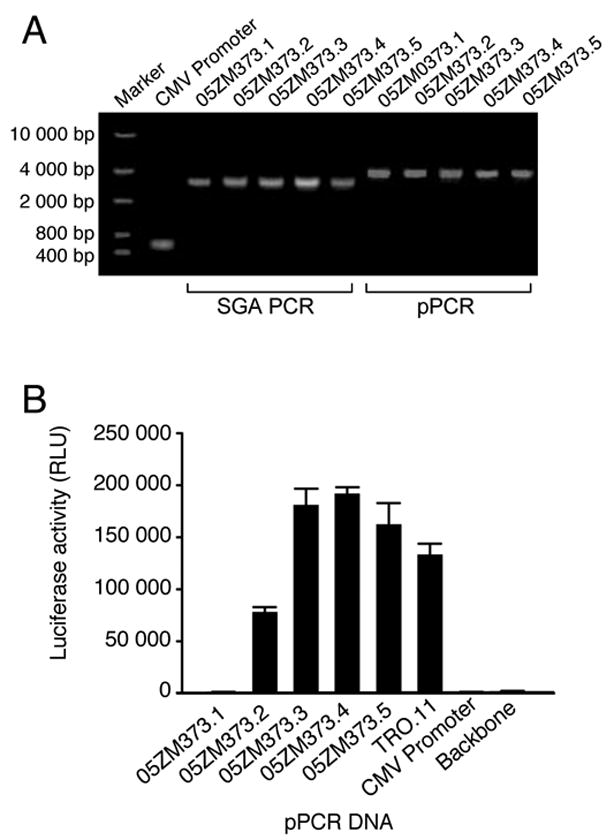
Infectivity of pseudovirions derived from pPCR from patient 05ZM373. (A) PCR and pPCR amplification of env genes from patient 05ZM373 plasma. Five HIV-1 rev/env cassettes were obtained using the single genome amplification (SGA) method. The CMV promoter was amplified and then added to all five env genes at the 5′ end by pPCR. (B) The pPCR DNA was cotransfected with pSG3Δenv into 293T cells. Pseudovirions were harvested 48 hrs after transfection and were used to infect TZM-bl cells. The luciferase activity was measured two days after infection. The data is shown as the mean ± standard error (n = 4 independent assays).
Western blot analysis was then performed to determine if the low or no infectivity of 05ZM373 pPCR pseudovirions was due to the low level of Env protein expression. The transfected 293T cells were lysed and equal amounts of cell lysates (normalized by p24 content) were separated by SDS-PAGE. Env proteins were detected for four pPCR pseudotype viruses. However, a lower level of Env protein was detected for 05ZM373.2 (Fig. 4). No Env protein was detected in 05ZM373.1. The infectivity of pPCR pseudovirions was in good agreement with the level of Env protein expression (Figs. 3B and 4). The precursor and cleaved Gag proteins were detected at comparable levels among all samples. Although the Env expression level of pPCR transfection was much lower than that of plasmid transfection (Fig. 4), the infectivity of the pseudovirions derived from pPCR products was not significantly effected (less than two fold) compared to plasmid-derived pseudovirions (Fig. 2). This result suggests over-expression of Env protein by plasmids is not required for the generation of infectious viruses with pPCR products.
Figure 4.
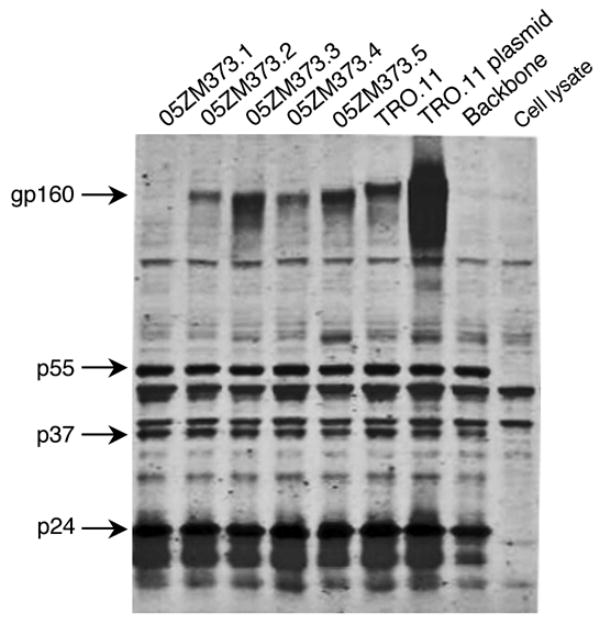
Western blot analysis of HIV-1 protein expression. The 293T cells transfected with pPCR products and pSG3Δenv were lysed 48 hrs after transfection. The viral proteins were separated on a 4–12% gradient reducing gel, transferred to nitrocellulose, and were reacted with an HIV-1 infected patient serum and a mouse mAb 13D5 to the Env protein. Viral proteins were visualized with secondary antibodies IRDye800 conjugated goat anti-human and Alexa-Fluor680 goat anti-mouse using a LiCor Odyssey Infrared Imaging system.
To investigate if variable levels of infectivity and Env protein expression were determined by Env amino acid sequences, all five 05ZM373 SGAs were sequenced. Open reading frames (ORFs) were intact in the four env genes that were positive in the luciferase assay. A premature stop codon (position 149) was found in the nonfunctional env gene (05ZM373.1), thus explaining the lack of Env protein expression and infectivity of 05ZM373.1 pseudovirions. It was not clear why protein expression and infectivity were lower for 05ZM373.2. To determine the reproducibility and fidelity of pPCR products, the same env gene was amplified by the pPCR method in five independent reactions. The pPCR products were sequenced and all five were identical, suggesting the pPCR method does not introduce detectable differences in sequence (data not shown).
In addition to evaluating the functionality of Env proteins from HIV-1 infected individuals, pseudovirions are also widely used for neutralization assays. To determine the sensitivity of 05ZM373 pPCR pseudovirions to neutralizing antibodies, their neutralization profiles were determined for six HIV-1 positive sera, a normal control serum, and three monoclonal antibodies (2G12, 2F5, and 4E10). Although four pPCR pseudovirions were from the same patient, three showed similar neutralization sensitivity to all sera and mAbs but 05ZM373.2 was slightly more resistant to all sera and 4E10 (Table 1).
Table 1.
Comparison of neutralization sensitivity of pseudovirions to HIV-1 positive sera and mAbs
| Pseudovirus | ID50 in TZM-bl cells
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reciprocal serum dilution
|
μg/ml
|
|||||||||
| BB8 | BB12 | BB28 | BB55 | BB70 | BB106 | Normal Sera | 2G12 | 2F5 | 4E10 | |
| 05ZM373.2 | 34 | 375 | 215 | 276 | 56 | 74 | <20 | >50 | >50 | >50 |
| 05ZM373.3 | 43 | >540 | 320 | 406 | 68 | 124 | <20 | >50 | >50 | 40 |
| 05ZM373.4 | 160 | >540 | 374 | >540 | 93 | 184 | <20 | >50 | >50 | 26 |
| 05ZM373.5 | 48 | >540 | 430 | 490 | 204 | 150 | <20 | >50 | >50 | 29 |
To confirm these results, viral RNA was obtained from six additional patient plasma samples (05ZM375, 05ZM379, 05ZM381, 05ZM394, 05ZM401, 05ZM413). Between five and twenty-one SGAs were obtained from each subject (Table 2). The CMV promoter was successfully added to most of env PCR products (33%–100%) at the 5′ end by pPCR. The functional env rate was 86% (73% – 100%) among 87 pPCR products from different HIV-1 strains. Phylogenetic analysis of all SGA sequences showed that they were subtype C viruses (data not shown). These results indicate the pPCR method is more efficient than the traditional cloning method to test functionality of multiple HIV-1 env genes from the same infected individual.
Table 2.
Summary of functional analysis of pPCR pseudovirions
| Virus | No. of SGA | No. of pPCR | No. of functional env genes (%) |
|---|---|---|---|
| 05ZM373 | 5 | 5 | 4 (80) |
| 05ZM375 | 21 | 21 | 18 (86) |
| 05ZM379 | 16 | 14 | 12 (86) |
| 05ZM381 | 16 | 12 | 11 (91) |
| 05ZM394 | 15 | 5 | 5 (100) |
| 05ZM401 | 19 | 15 | 11 (73) |
| 05ZM413 | 18 | 15 | 14 (93) |
|
| |||
| Total | 110 | 87 | 75 (86) |
Dose dependent luciferase activity was investigated by cotransfection of variable amounts of env pPCR products with the same amount of pSG3Δenv (400 ng) into 293T cells in a 24 well plate. The pPCR DNA was serially diluted 1:2 (800 ng - 12.5 ng). Luciferase activity decreased proportionally with decreasing pPCR DNA amounts. Linear regression analysis showed a good correlation (R2 = 0.9958) between DNA concentrations and luciferase activities among the tested concentrations (Fig. 5). Since 200 ng of pPCR DNA yielded only slightly less luciferase activity than 400 ng (p = 0.192) and significantly higher luciferase activity than 50 ng of pPCR DNA (p < 0.002), 200 ng of DNA was routinely used for initial screening of functional env genes.
Figure 5.
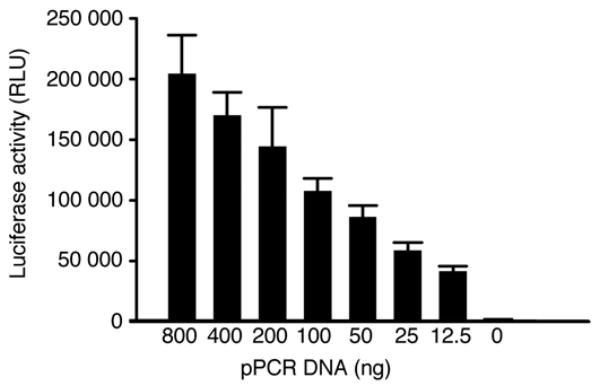
Dose dependent infectivity of pPCR pseudovirions. The pPCR products were 1:2 serially diluted (800 ng − 12.5 ng) and cotransfected with pSG3Δenv into 293T cells. The infectivity was determined by measuring luciferase activity. The data is shown as the mean ± standard error (n = 6 independent assays).
Large amounts of infectious virus stocks are required to determine neutralization characteristics for each pseudovirion with multiple HIV-1 positive sera and neutralizing mAbs. To determine if sufficient amounts of pseudovirus stocks can be generated using the pPCR method, four different transfection conditions were tested with different numbers of cells (Table 3). Transfection in T25 or T75 flasks yielded slightly higher luciferase activity than in 24 well or 6 well plates. However, the TCID50 titers were much higher in T25 or T75 flasks than in 24 well or 6 well plates. Since the medium volume in T75 was more than other cell culture containers used in this study, the total TCID50 was as high as 2 million. Therefore, many neutralization assays can be performed from one transfection with the pPCR product in a T75 flask.
Table 3.
Comparison of TCID50 titers of pseudovirions harvested from different cell cultures.
| pPCR DNA | Luciferase Activity (RLU) | TCID50/ml | Volume | Total TCID50 | |
|---|---|---|---|---|---|
| 24 well | 200 ng | 296 610 | 13 975 | 0.5 ml | 6 988 |
| 6 well | 200 ng | 262 924 | 10 687 | 2 ml | 21 374 |
| T25 | 1 000 ng | 460 090 | 69 877 | 5 ml | 349 385 |
| T75 | 4 000 ng | 423 603 | 91 376 | 24 ml | 2 193 024 |
To determine if pPCR pseudovirions have altered neutralization characteristics, pPCR products were obtained from five functional molecular clones and neutralization profiles were compared between pseudovirions derived from pPCR DNA transfections and corresponding HIV-1 env plasmid counterparts. Pseudovirions were generated with five pPCR-plasmid pairs and their neutralization profiles were determined with six HIV-1 positive sera and a normal control serum in addition to three monoclonal antibodies (2G12, 2F5, and 4E10). The neutralization titers in each pPCR-plasmid pair were comparable for both sera and monoclonal antibodies (Table 4). These results indicate pseudovirions derived from the pPCR products or traditional plasmids have similar neutralization characteristics.
Table 4.
Comparison of neutralization titers of pseudovirions generated from HIV-1 env plasmids and pPCR products
| Pseudovirus | ID50 in TZM-bl cells
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reciprocal serum dilution
|
μg/ml
|
||||||||||
| BB8 | BB12 | BB28 | BB55 | BB70 | BB106 | Normal Sera | 2G12 | 2F5 | 4E10 | ||
| CAP210.2.00.E8 | plasmid | <20 | 24 | 52 | 46 | 292 | <20 | <20 | >50 | >50 | <2 |
| pPCR | <20 | <20 | 25 | <20 | 147 | <20 | <20 | >50 | >50 | <2 | |
| DU422.1 | plasmid | 35 | 320 | 93 | 67 | 279 | 74 | <20 | >50 | >50 | <2 |
| pPCR | 30 | 249 | 79 | 37 | 197 | 44 | <20 | >50 | >50 | <2 | |
| ZM53.MPB12 | plasmid | <20 | <20 | 390 | <20 | <20 | 22 | <20 | >50 | >50 | 6 |
| pPCR | <20 | <20 | 279 | <20 | <20 | <20 | <20 | >50 | >50 | 11 | |
| ZM109.FPB4 | plasmid | 103 | 49 | 44 | 46 | 282 | <20 | <20 | >50 | >50 | 3 |
| pPCR | 69 | 38 | 31 | 40 | 168 | <20 | <20 | >50 | >50 | 2 | |
| TRO.11 | plasmid | >540 | >540 | 40 | 208 | 104 | 90 | <20 | <2 | >50 | <2 |
| pPCR | 293 | 402 | 21 | 152 | 41 | 77 | <20 | <2 | >50 | <2 | |
4. Discussion
The newly developed promoter PCR (pPCR) method can be used for high throughput functional analysis of HIV-1 env genes by directly adding a cytomegalovirus (CMV) immediate enhancer/promoter to the 5′ end of the HIV-1 rev/env gene PCR products. Since the steps of ligation, transformation, and plasmid DNA preparation are eliminated in the pPCR method, a large number of quasispecies HIV-1 env genes from a single individual can be quickly analyzed (Table 5). Coupled with the SGA method to obtain HIV-1 env genes directly from patient samples, pPCR also avoids analysis of artificial env genes generated either through recombination between different viral genomes or resampling of viral genomes during bulk PCR amplification (Fang et al., 1998; Liu et al., 1996). The high success rate of incorporated CMV promoter from a single PCR reaction greatly reduces time required to prepare DNA for transfection compared to traditional cloning methods (Table 5). Similar infectivity and neutralization profiles of pseudovirions generated with pPCR products and matched cloned plasmids indicate this method is reliable for determining neutralization characteristics of interested HIV-1 env genes. This new method can substantially reduce time and resources needed for characterization of HIV-1 env gene functions.
Table 5.
Comparison of different methods used for functional analysis of HIV-1 env quasispecies populations from one infected individual
| Bulk PCR | SGAa | pPCR | |
|---|---|---|---|
| PCR reaction | one | multiple | multiple |
| Ligation | single | multiple | no |
| Transformation | single | multiple | no |
| Plasmid DNA preparation | multiple | multiple | no |
| Recombination | yes | no | no |
| Resampling | yes | no | no |
| Time needed for transfection | days | weeks | hours |
single genome amplification
A virus stock containing as high as two million TCID50 can be obtained by transfecting 4 μg of pPCR DNA together with the pSG3Δenv backbone clone into 293T cells in a T75 flask. Since 200 TCID50 are normally used for one reaction in a standard neutralization assay in 96 well plates (Li et al., 2005), pseudovirions from one transfection can be used for many neutralization assays. In addition to function studies of multiple env genes from the same individual, the Env pseudovirions can be used for detailed analysis of neutralization profiles using a relatively large panel of mAbs and anti-HIV-1 sera. The new method may also be used to determine neutralization escape mutants among wild-type viruses by analyzing a large number of quasispecies viral populations from longitudinally collected samples.
Recently, it has been reported that the biological phenotype of polioviruses is determined not only by the sequences present in the viral genome, but also by quasispecies diversity of the virus population (Vignuzzi et al., 2006). Studies of multiple functional HIV-1 env genes individually and as a quasispecies viral population may shed new light on the understanding of biological functions, viral fitness, neutralization, and neutralization escape. Furthermore, the pPCR method can also be used to quickly characterize other proteins without cloning.
Acknowledgments
We thank Yi Yang for technical assistance. This work was supported by the NIH, NIAID grants CHAVI U01 AI067854, AI 055386, Duke University CFAR Sequencing Core Facility P30 AI64518, CIPRA RO3 AI54155; Bill & Melinda Gates foundation GHAVE 38619; J.L.K was supported by NIH training grant 5T32 AI007392
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cordonnier A, Montagnier L, Emerman M. Single amino-acid changes in HIV envelope affect viral tropism and receptor binding. Nature. 1989;340:571–4. doi: 10.1038/340571a0. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–22. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- Fang G, Zhu G, Burger H, Keithly JS, Weiser B. Minimizing DNA recombination during long RT-PCR. J Virol Methods. 1998;76:139–48. doi: 10.1016/s0166-0934(98)00133-5. [DOI] [PubMed] [Google Scholar]
- Gao F, Morrison SG, Robertson DL, Thornton CL, Craig S, Karlsson G, Sodroski J, Morgado M, Galvao-Castro B, von Briesen H, et al. Molecular cloning and analysis of functional envelope genes from human immunodeficiency virus type 1 sequence subtypes A through G. The WHO and NIAID Networks for HIV Isolation and Characterization. J Virol. 1996;70:1651–67. doi: 10.1128/jvi.70.3.1651-1667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helseth E, Kowalski M, Gabuzda D, Olshevsky U, Haseltine W, Sodroski J. Rapid complementation assays measuring replicative potential of human immunodeficiency virus type 1 envelope glycoprotein mutants. J Virol. 1990;64:2416–20. doi: 10.1128/jvi.64.5.2416-2420.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Gupta P, Montelaro RC. Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J Virol. 2005;79:2097–107. doi: 10.1128/JVI.79.4.2097-2107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBranche CC, Sauter MM, Haggarty BS, Vance PJ, Romano J, Hart TK, Bugelski PJ, Marsh M, Hoxie JA. A single amino acid change in the cytoplasmic domain of the simian immunodeficiency virus transmembrane molecule increases envelope glycoprotein expression on infected cells. J Virol. 1995;69:5217–27. doi: 10.1128/jvi.69.9.5217-5227.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human Immunodeficiency Virus Type 1 env Clones from Acute and Early Subtype B Infections for Standardized Assessments of Vaccine-Elicited Neutralizing Antibodies. J Virol. 2005;79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Salazar-Gonzalez JF, Derdeyn CA, Morris L, Williamson C, Robinson JE, Decker JM, Li Y, Salazar MG, Polonis VR, Mlisana K, Karim SA, Hong K, Greene KM, Bilska M, Zhou J, Allen S, Chomba E, Mulenga J, Vwalika C, Gao F, Zhang M, Korber BT, Hunter E, Hahn BH, Montefiori DC. Genetic and Neutralization Properties of Acute and Early Subtype C Human Immunodeficiency Virus Type 1 Molecular env Clones from Heterosexually Acquired Infections in Southern Africa. J Virol. 2006;80:11776–11790. doi: 10.1128/JVI.01730-06. Published online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SL, Rodrigo AG, Shankarappa R, Learn GH, Hsu L, Davidov O, Zhao LP, Mullins JI. HIV quasispecies and resampling. Science. 1996;273:415–6. doi: 10.1126/science.273.5274.415. [DOI] [PubMed] [Google Scholar]
- Montefiori DC. Evaluating neutralizing antibodies against HIV,SIV, and SHIV in luciferase reporter gene assays. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, Coico R, editors. Current protocols in immunology. John Whiley & Sons; New York, NY: 2004. pp. 12.11.1–12.11.15. [DOI] [PubMed] [Google Scholar]
- Morris JF, Sternberg EJ, Gutshall L, Petteway SR, Jr, Ivanoff LA. Effect of a single amino acid substitution in the V3 domain of the human immunodeficiency virus type 1: generation of revertant viruses to overcome defects in infectivity in specific cell types. J Virol. 1994;68:8380–5. doi: 10.1128/jvi.68.12.8380-8385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Jr, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol. 2005;43:406–13. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Haraguchi Y, Takeuchi Y, Soda Y, Kanbe K, Hoshino H. Changes in and discrepancies between cell tropisms and coreceptor uses of human immunodeficiency virus type 1 induced by single point mutations at the V3 tip of the env protein. Virology. 1999;259:324–33. doi: 10.1006/viro.1999.9764. [DOI] [PubMed] [Google Scholar]
- Shioda T, Oka S, Ida S, Nokihara K, Toriyoshi H, Mori S, Takebe Y, Kimura S, Shimada K, Nagai Y, et al. A naturally occurring single basic amino acid substitution in the V3 region of the human immunodeficiency virus type 1 env protein alters the cellular host range and antigenic structure of the virus. J Virol. 1994;68:7689–96. doi: 10.1128/jvi.68.12.7689-7696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Balfe P, Ludlam CA, Bishop JO, Brown AJ. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990;64:5840–50. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–8. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–12. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]