

Abstract
Purpose
To address the functional role of radiation-induced TGF-β signaling in normal epithelial background, we selected spontaneously immortalized lung epithelial cell line derived from the normal lung tissue of dominant-negative mutant of TGF-β RII (ΔRII) transgenic mouse that expressed conditionally ΔRII under the control of metallothionein promoter (MT-1) and assessed it's impact on radio-sensitivity.
Method and Materials
Spontaneously immortalized lung epithelial cell culture (SILECC) was established and all analyses were performed within 50 passages. Colony-forming and TUNEL assays were used to assess the clonogenic inhibition and apoptosis respectively. Western blot analysis was performed to assess the kinetics of p21, bax and RII proteins. TGF-β responsive promoter activity was measured using dual-luciferase reporter assay.
Results
Exposure to ZnSO4 inhibited TGF-β signaling induced either by recombinant TGF-β1 or ionizing radiation. SILECC treated either with ZnSO4 or neutralizing antibody against TGF-β showed a significant increase in radio-resistance when compared to untreated cells. Furthermore, the expression of the ΔRII inhibited the radiation-induced up-regulation of the TGF-β effector gene p21waf1/cip1..
Conclusions
Our findings imply that inhibition of radiation-induced TGF-β signaling via abrogation of RII function enhances radio-resistance of the normal lung epithelial cells, and this can be directly attributed to the loss of TGF-β signaling function.
Keywords: TGF-β1 signaling, normal lung cells, ionizing radiation, radiation resistance, p21waf1/cip1
Introduction
Transforming growth factor β (TGF-β) family of multifunctional cytokines has a wide range of physiological as well as pathological effects on cellular processes ranging from proliferation and differentiation to apoptosis (1, 2). Originally named for their transforming activities in in-vitro assays, TGF-β isoforms now unequivocally demonstrate both tumor suppressor and oncogenic activities. Dysregulation of these processes can result in various fibrotic as well as malignant diseases. While in normal tissues suppressor activities of TGF-β dominate, during tumorigenesis the changes in TGF-β expression-associated cellular responses tilt the balance in favor to its oncogenic activities (3).
Cellular proliferation is complex involving both stimulatory and inhibitory signals. Abnormal proliferation observed in cancer cells is caused by mutations that either increase positive signals or decrease negative growth control signals, or both. TGF-β isoforms (TGF-β1, TGF-β2, and TGF-β3) are 25 kDa homodimer polypeptides that regulate cell proliferation and differentiation (4). TGF-β molecules are potent inhibitors of growth in a variety of cells including epithelial, endothelial and lymphoid cells (5). TGF-β is expressed in growth-arrested cells and in the G1 phase of the cell cycle and contributes to an orderly progression through the cell cycle (6). TGF-β signals by binding to transmembrane serine-threonine kinases termed as receptor I (RI) and receptor II (7). Genetic evidence shows that both receptors are required for signaling of TGF-β (7). A third receptor (RIII) is not believed to be directly involved in TGF-β signaling but acts to present TGF-β to RII (7).
Upon TGF-β treatment, RI is recruited to the RII to form receptor-ligand complex on the cell surface and is activated subsequently by type II receptor kinase upon phosphorylation of its juxtamembrane GS domain. This activates the RI, which then serves as a docking site for Smad-2 or Smad-3 proteins associated with the chaperone protein SARA (Smad anchor for receptor activation) (8). Following phosphorylation by RI, Smad 2/3-protein complex dissociates from the RI, associates with Smad 4 and translocates into nucleus. The Smad complex then activates transcription of target genes (such as p21waf1/cip1 and other CDK inhibitors) through an intermediate transcription factor or by binding to DNA directly. Thus, the translocated Smads are speculated to be key effectors for TGF-β mediated growth inhibition [reviewed in ref (7)].
Although Smad signaling has a central role in the TGF-β pathway, Smad-independent TGF-β signaling has also been documented (9, 10). For instance, PAK2 (p21-activated kinases) is activated by TGF-β in a Smad 2- and Smad 3-independent manner and is specific to fibroblast cells, but not to epithelial cells indicating that the response to the TGF-β is differentially regulated in different cell types (8).
It is well known that radiation induces TGF-β1 isoform in various cell types (11–13). Interestingly, radiation down-regulates TGF-β3 isoform and does not alter the expression of TGF-β2 isoform (12, 13). TGF-β1 is implicated primarily for negative growth regulation (14) as well as in cell death (15, 16). Based on exclusive induction of TGF-β1 by radiation and its implication on negative growth regulation and apoptosis, we hypothesized that radiation induces endogenous TGF-β protein that will exert clonogenic inhibition and cause apoptosis in cells with intact TGF-β signaling components. This hypothesis was demonstrated and proved by our group earlier (17, 18).
Lung tumors are known to harbor aberrant expression of RII and lack or contain mutations in Smad-4 expression (19, 20). The finding that RII/Smad4 expression is low in many lung cancer cell lines raises the possibility that absence of RII/Smad4 expression may cause lack of response to TGF-β and this can play a critical role in resistance to chemotherapy and radiation. In various cell types lacking RII expression, ectopic re-expression of RII led to restoration of TGF-β signaling (21). Zhao et al reported that the RII is indispensable for mediating both the mitogenic and antiproliferative effects of TGF-β in lung fibroblasts (22).
We have previously reported that restoration of TGF-β signaling enhances sensitivity to ionizing radiation in pancreatic cancer background (17). In the same study, we reported that in pancreatic cancer cell line, MIA PaCa-2, radiation induces TGF-β signaling, up-regulates p21waf1/cip1, Bax and activates caspases (17). By loss-of-function approach using dominant-negative mutant for RII in normal mouse embryonic fibroblasts, we demonstrated that these fibroblast cells were resistant to radiation-induced apoptosis (17). To address the functional role of radiation-induced TGF-β signaling in normal epithelial background, we selected spontaneously immortalized lung epithelial cell line and disrupted the RII receptor gene and assessed it's impact on radio-sensitivity. To achieve this, we isolated and established a mouse lung cell line derived from the normal lung tissue of dominant-negative mutant of TGF-β RII (ΔRII) transgenic mouse that conditionally expressed ΔRII, controlled by zinc-regulated promoter. Here, we report that the RII-defective spontaneously immortalized epithelial normal lung cells are significantly more radio-resistant compared to the primary lung cells that express functional endogenous RII with intact TGF-β signaling.
Materials and Methods
Generation of primary mouse cell culture from ΔRII transgenic mouse
The AM3 transgenic DNR mice breeders in the strain FVB/N were kindly provided by Dr. Lalage Wakefield (NCI, Bethesda, Maryland). The mice were housed in a pathogen free environment and cared for according to the guidelines stipulated in the “Guide for the care and use of laboratory animals”, DHHS publication NIH-85-23. They were crossbred to obtain sizeable ΔRII mice population. Primary lung cells were isolated from DNR transgenic mice and cultured as per the protocols described previously (23, 24). Briefly, cells were re-suspended in Waymouth medium containing 2.5% fetal bovine serum, antibiotics, 50mg/ml amphotericin B and soybean trypsin inhibitor and seeded into chamber slides coated with air-dried rat-tail collagen. After two hours of recovery and attachment, hormones and growth factors were added to the media. After the epithelial cells reached an exponential growth phase with no fibroblast cells present (around 5 passages), cultures were grown in regular DMEM (GIBCO BRL) containing 10% fetal bovine serum (GIBCO) and 1% Penicillin/Streptomycin at 37°C and 5% CO2. Since the lung primary cultures exceeded 10 passages, we named these cells as Spontaneously Immortalized Lung Epithelial Cell Culture (SILECC). All experiments were begun after 10 passages and completed within 50 passages.
Immunocytochemistry
Cytokeratin expression was determined in SILECC by immunocytochemical analysis, as described by us previously (17, 25) using pan-Cytokeratin antibody (Santa Cruz, CA), and the Elite ABC kit (Vector Laboratories, Burlingame, CA). A549 lung adenocarcinoma cells were used as a positive control for cytokeratin.
Irradiation
A 100 kV industrial X-ray machine (Phillips, Netherlands) was used to irradiate the cultures at room temperature. The dose rate with a 2-mm aluminum plus 1-mm beryllium filter was 3.85 Gy per minute at a focus-surface distance of 20 cm.
Semi-quantitative Reverse Transcription-PCR
Total RNA was isolated from exponentially growing untreated and ZnSO4 treated SILECC using TRIzol reagent (GIBCO-BRL). One μg of total RNA was reverse transcribed into cDNA using random hexamer primers and reverse transcriptase in a 20 μl reaction mix according to the manufacturers' recommendations (Perkin-Elmer). Five μl of this cDNA was used as the starting template for the primary PCR (35 cycles). The 25 μl of PCR mix (Perkin-Elmer) contained 0.5 U Taq polymerase, reaction buffer, 1.5mM MgCl2, 200 μM deoxynucleotide triphosphates and 1.0 μM of forward and reverse primers flanking the cDNA sequence of RII gene as reported by us previously (26). The primers were designed to amplify 123 bp fragment of the human RII sequence between nucleotides −7 and +573, encoding extra-cellular and trans-membrane domains (Gene Bank accession #M85079). The primer sequences were as follows: 5′- GTCTGCCATGGGTCGGGGGC-3′ (sense) and 5′- GTGACTATCATGTCGTTATT-3′ (anti-sense) (Figure 1). The control β-actin gene was amplified from the same pool of cDNA. The PCR products were resolved on ethidium bromide stained agarose gel. Densitometry was performed to obtain semi-quantitative expression of RII as the ratio between RII and β-actin.
Figure 1. Schematic representation of the ΔRII transgene.
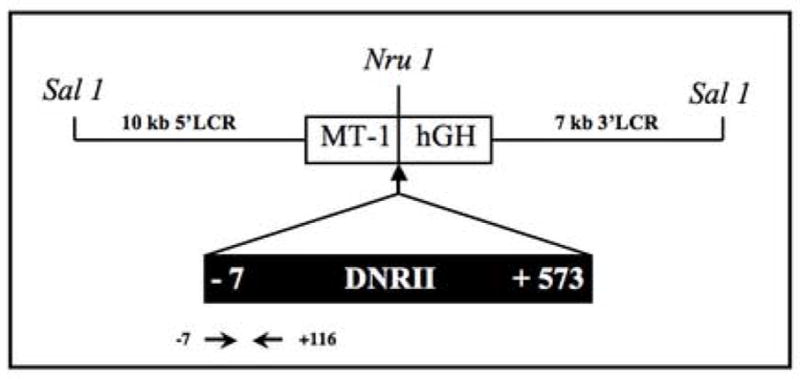
A 19.1 kb SalI DNA fragment containing ΔRII (DNR) was released from pMT-LCR-ΔRII and used for microinjections. MT-1, mouse metallothionein I promoter; hGH, poly(A) region of human growth hormone gene; 10 kb 5 LCR and 7 kb 3 LCR, 10 kb EcoRI fragment of the 5 LCR and 7 kb EcoRI fragment of the 3 LCR of the mouse MT I and II gene locus (28). Location of primers for PCR is shown.
Western Blot Analysis
Three types of treatments were performed. The SILECC were subjected to 5 Gy irradiation alone, ZnSO4 alone, and ZnSO4 treatment immediately followed by 5 Gy radiation. Control cells were left untreated. Protein extracts were prepared at 1, 3, 6, 12, and 24 hours after treatment. Total protein extracts from SILECC were subjected to Western blot analysis as described previously (25), using anti-TGF-β RII antibody (sc-400: Santa Cruz, CA) and polyclonal anti-human TGF-β RII (residues 1–28) antibody (Upstate Biotechnology); p21waf1/cip1 (sc-6246) (Santa Cruz, CA); or Bax (6A7) (BD-Pharmingen, San Jose, CA) antibodies. Anti-β-actin antibody (Sigma Chemical Co) was used as an internal loading control. These proteins were detected using Enhanced Chemiluminescence method (Amersham).
Dual - Luciferase Reporter Assay
TGF-β responsive promoter activity was measured using the 6XSBE-Luc, a Smad2/3- and Smad4-dependent luciferase reporter kindly provided by Dr. Scott Kern (Johns Hopkins University). A 6XSBE-Luc (1.6 μg/ml media) and a pRL-TK expression plasmid (0.4 μg/ml, Promega) were co-transfected into lung cells using Lipofectamine Plus transfection reagent (Gibco BRL) according to manufacturer's protocol. Twenty-four hours after transfection, the cells were treated with combination of radiation (5 Gy) or human recombinant TGF-β (10 ng/ml), with or without ZnSO4 (0.12 μM). To block radiation-induced TGF-β, neutralizing antibody against TGF-β1, (AF-101-NA; R&D systems) was added (500 ng/ml media) to the cells 1 hour prior to irradiation. Firefly luciferase activity was assayed 24 hours after irradiation using a TD-20/20 Luminometer (Turner Designs) and compared to the activity obtained for the empty vector. All results were normalized against the activity of the co-transfected Renilla luciferase vector pRL-TK.
Colony forming assay
For clonogenic cell survival studies, two different cell concentrations in quadruplet sets were used for each radiation dose. Cell lines were left untreated or exposed to 1 to 6 Gy dose of radiation. After incubation for 10 or more days, colonies were stained with crystal violet and the colonies containing more than 50 cells were counted. The surviving fraction (SF) was calculated as a ratio between the number of colonies formed and the product of the number of cells plated with the plating efficiency. The curve was plotted using X-Y log scatter (Delta Graph® 4.0) and was fitted by using the formula for the single hit multi-target (SHMT) model to obtain the D0 and SF2 (17).
Quantitation of Apoptosis
Apoptosis was quantified by TUNEL staining (Roche Diagnostics) which detects DNA strand breaks by terminal transferase-mediated dUTP-digoxigenin end labeling. Briefly, SILECC were seeded in chamber slides and after 24 hrs the cells were treated with 5 Gy dose of radiation or human recombinant TGF-β (10ng/ml) with or without ZnSO4 (0.12 μM). To block radiation-induced TGF-β, a neutralizing antibody to TGF-β1 (AF-101-NA) (R&D systems) was added (500ng/ml media) to the cultures prior to irradiation. Twenty-four hours after treatment, the DNA was then tailed with digoxigenin-dUTP and conjugated with an anti-digoxigenin fluorescein. The cells were counterstained with propidium iodide and antifade. The stained specimen was observed in triple band-pass filter using Nikon-microphot epifluorescence microscope. To determine the percentage of apoptotic cells, four independent experiments in total were performed and a minimum of 1000 cells were scored in each experiment.
Statistical Analysis
Descriptive statistics, such as the calculation of means and standard deviations were used to examine the data from the separate experiments. To examine the difference in survival curves by dose between the “radiation alone” group and the “radiation plus ZnSO4” treatment group, a multiple linear regression model was used. The logarithm of the surviving fraction was the outcome variable and dependent variables included group, dose, and the interaction between group and dose. The non-parametric Kruskall-Wallis test was used to determine if there were differences in apoptotic cell frequency. All tests were 2-sided and p-values less than 0.05 were considered significant.
Results
Expression of dominant-negative mutant for TGF-β RII transgene (ΔRII) is regulated by ZnSO4
To determine whether ZnSO4 regulates the expression of transgene in SILECC, RNA and protein expression of dominant-negative mutant of TGF-β RII was determined using RT-PCR and Western blotting respectively. RT-PCR results demonstrated the induction of expression of ΔRII at either 0.5 or 2.5 μM of ZnSO4 treatment (Figure 2A) with no presence of the product in the untreated lane, suggesting that ZnSO4 tightly regulates the transgene ΔRII expression in SILECC.
Figure 2. Regulation of the expression of ΔRII by ZnSO4 treatment as assessed by RT-PCR and Western blot analysis.
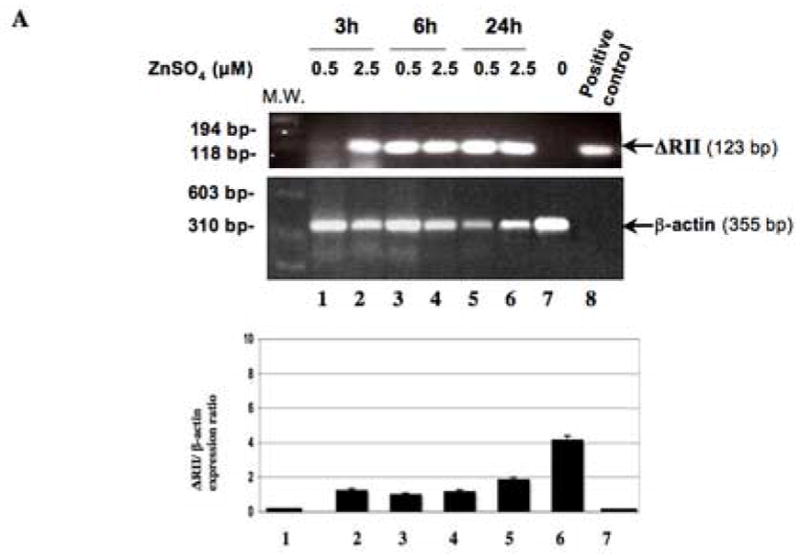
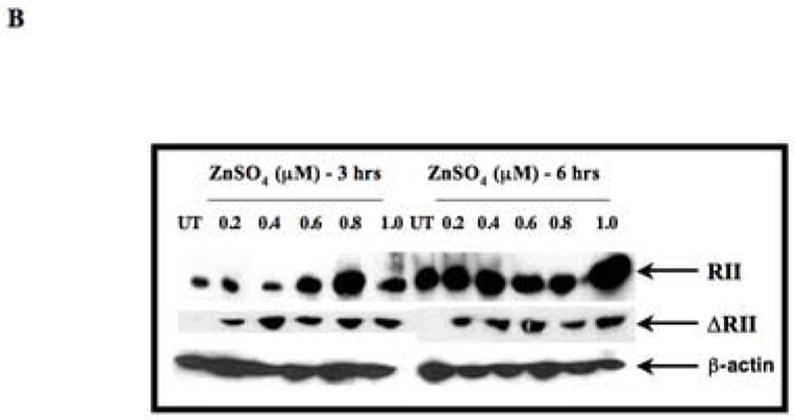
(A) RT-PCR was used to detect the expression of the ΔRII mRNA from SILECC cells in the presence (lanes 1–6) or absence (lane 7) of ZnSO4. Lane 8 represents positive control. Total mRNA was isolated from SILECC that was treated with ZnSO4 for the time intervals indicated. PCR (35 cycles) was performed using the reverse transcription reaction and appropriate primers. Gel was stained with ethidium bromide and the intensities of bands were quantified by densitometry. Black bar graph represents ΔRII expression normalized to the β-actin. (B) Western blot analysis was performed to detect ΔRII and endogenous ΔRII protein expression. Cells were left untreated (UT) or treated with ZnSO4 for the time intervals indicated. The blots were probed using sc-400 anti-RII antibody (SantaCruz), anti-RII antibody targeting amino terminus of RII protein (Upstate Biotechnology) and β-actin as a loading control.
To further confirm the expression of ΔRII at the protein level and to establish a dose-response relationship, SILECC were treated with different concentrations of ZnSO4 ranging from 0.2 to 1 μM and total proteins were extracted and separated by SDS-PAGE. Using both antibodies against RII (sc-400 and polyclonal anti- RII antibody), a 32 kDa ΔRII protein was detected at all the concentrations of ZnSO4 tested at 3 hours, 6 hours and 24 hours after treatment, but no protein was detected in the untreated cells. Mouse β–actin was used on the same blot as a loading control (Figure 2B). Interestingly, the levels of endogenous RII (at 70 kDa) were found to be elevated in ZnSO4-treated SILECC. This is in concordance with the previous study in which it was demonstrated that the presence of ΔRII protein led to an increase in endogenous receptors RI, RII and RIII proteins and this increase does not interfere with the dominant-negative function of ΔRII protein (27, 28). These findings demonstrate that both RNA and protein levels of ΔRII in SILECC can be directly regulated by ZnSO4 exposure.
SILECC expresses cytokeratin
Having demonstrated the regulation of transgene ΔRII by ZnSO4, we next determined the expression of cytokeratin in order to ascertain that the SILECC are of the epithelial origin. Even after 50 passages, the SILECC derived from mouse normal lung epithelial tissue of ΔRII transgenic mice maintained an epithelial morphology (Figure 3A) since these cells expressed cytokeratin (Figure 3B). Thus, the presence of cytokeratin expression together with morphological data showing polygonal shapes typical of epithelial cells suggest that SILECC behave as a normal epithelial lung cells.
Figure 3. Cytokeratin expression in SILECC.
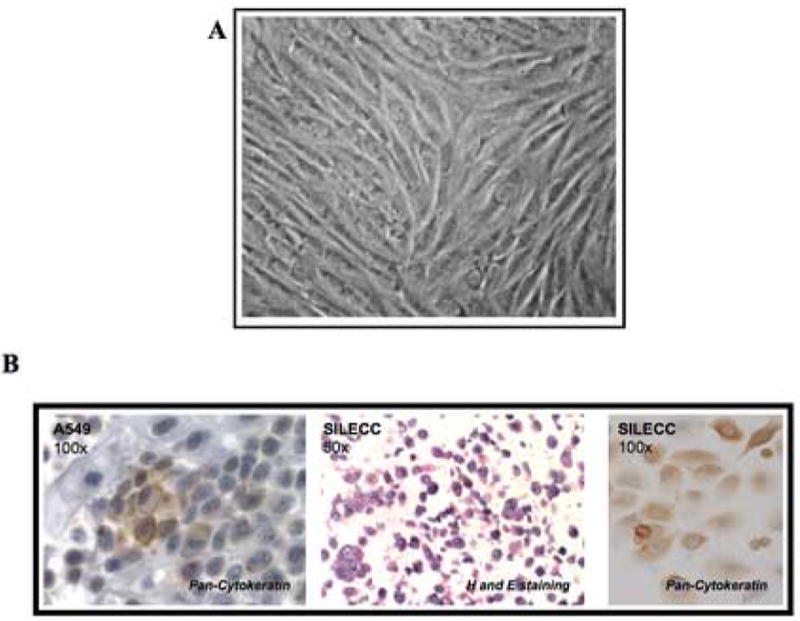
(A) Microphotograph of SILECC cells in standard growing conditions. Magnification: x150. (B) SILECC were stained with Hematoxylin and eosin staining and pan-Cytokeratin and A549 (lung adenocarcinoma cells) were stained with pan-Cytokeratin as positive control.
Recombinant TGF-β or radiation failed to induce TGF-β signaling in SILECC treated with ZnSO4 but not in untreated SILECC
To assess the effect of ΔRII protein expression on TGF-β signaling in SILECC cells, TGF-β responsive SBE-Luc reporter activity was analyzed by transient transfection approach (Figure 4A). TGF-β responsive promoter activity (as assessed by Smad binding element activity) was expressed as the ratio of Firefly luciferase activity over the Renilla luciferase activity. The human recombinant TGF-β1 up-regulated the TGF-β responsive SBE-promoter activity while treatment with ZnSO4 in combination with recombinant TGF-β abrogated the TGF-β responsive SBE-promoter activity, suggesting that ΔRII protein induced by ZnSO4 effectively blocked the TGF-β signaling function.
Figure 4. Inhibition of TGF-β signaling by ZnSO4 in SILECC.
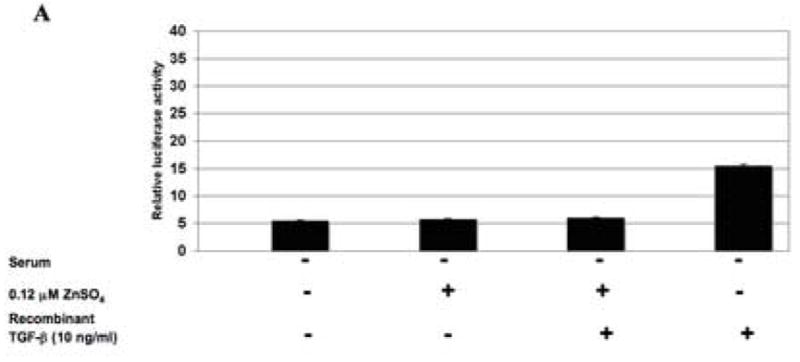
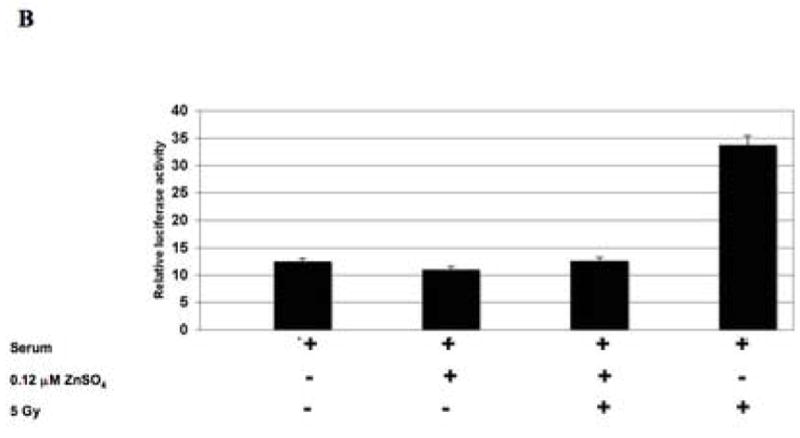
(A) The SILECC cells were co-transfected with pRL-TK and 6XSBE luciferase constructs and treated for 24 hours with ZnSO4 and/or recombinant human TGF-β1 in the absence of serum. (B) SILECC cells were transfected with the same constructs, treated for 24 hours with ZnSO4 and/or irradiated with 5 Gy in the presence of serum. TGF-β responsive promoter activity (6XSBE-Luc) was expressed as the ratio of firefly luciferase activity to Renilla luciferase activity. The data presented here is the mean of three independent experiments. Error bars represent standard error.
Based on abrogation of TGF-β function by ΔRII protein, we hypothesized that radiation-induced TGF-β signaling function will be abrogated by ΔRII protein. To test this hypothesis, SILECC were treated with 5 Gy dose of ionizing radiation in combination with or without ZnSO4. Irradiation caused induction of TGF-β responsive SBE-promoter activity and this activity was abrogated by the expression of ΔRII induced by ZnSO4 (Figure 4B). These results strongly suggest that both the TGF-β and radiation activate the TGF-β signaling pathway and this can be potentially abrogated by ZnSO4-induced expression of ΔRII protein.
The transgene ΔRII protein protects normal lung epithelial cells from radiation-induced clonogenic inhibition and apoptosis
We have previously reported that restoration of TGF-β signaling led to enhanced sensitivity to ionizing radiation in TGF-β defective pancreatic cancer cells (17). In this present study, we addressed the functional role of TGF-β signaling in radiation-inducible clonogenic inhibition and apoptosis in the normal cell background. SILECC were irradiated with or without ZnSO4 to assess the response to radiation by colony-forming assay for clonogenic inhibition and TUNEL assay for apoptosis. In colony-forming assays, the SF2 value of exponentially growing irradiated SILECC was 0.355 with a D0 value of 1.71 Gy (Figure 5). On the other hand, the SF2 for SILECC with ZnSO4 treatment was 0.515 with a D0 value of 2.78 Gy, suggesting that compared to SILECC with no ΔRII protein expression, the SILECC expressing ΔRII protein were significantly more resistant to ionizing radiation. Statistical analysis using a multiple regression model indicated that the differences between two slopes were statistically significant (p<0.0001). Interestingly, both curves failed to exhibit a shoulder since both had n=1.1 suggesting that there was no accumulation of sub lethal damage. Consistent with the observation on clonogenic inhibition, TUNEL analysis indicated that the SILECC with ΔRII protein expression were significantly more resistant (p<0.0001) to ionizing radiation-inducible apoptosis than SILECC with no ZnSO4 treatment (29), both at 24 and 48 hours. Interestingly, treatment with neutralizing antibodies against TGF-β caused reversal of radiation-induced apoptosis in SILECC (p<0.0001), both at 24 and 48 hours. Together, these findings demonstrated that radiation resistance in normal cells can be directly attributed to the loss of TGF-β signaling function.
Figure 5. Expression of ΔRII increases radio-resistance of SILECC.
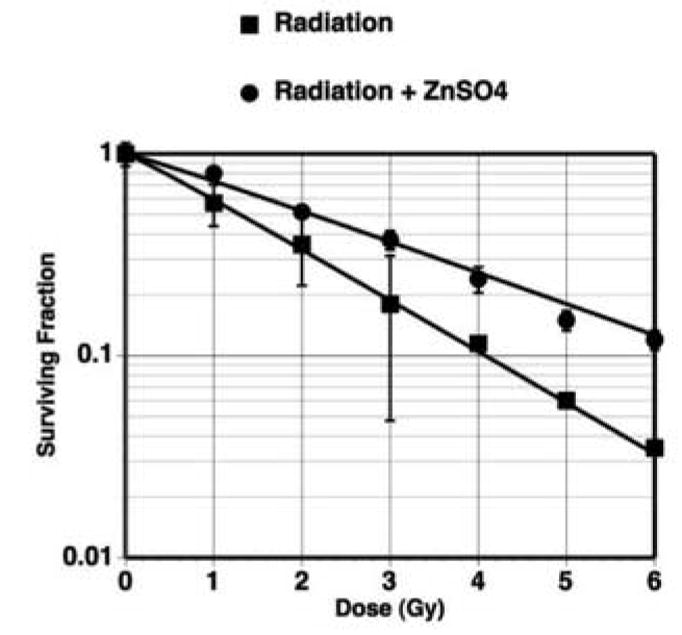
Clonogenic survival of SILECC exposed to radiation in the presence or absence of ZnSO4 was assessed by colony-forming assay and analyzed by single-hit multi-target model curve fit.
Radiation-induced p21 up-regulation is inhibited by ZnSO4 treatment
p21waf/cip1 is a p53 response gene, whose product inhibits the function of cyclin-dependent kinases (CDKs) during G1 phase of the cell cycle (30, 31). SILECC harbor wild-type p53 gene function. It is also known that TGF-β signaling through Smad complex activates the transcription of target genes such as p21waf1/cip1 and other CDK inhibitors, which are speculated to be key effectors for TGF-β mediated growth inhibition (32). Further, we reported that radiation-induced TGF-β induces Bax protein (17). We speculated that the increased radiation resistance in ZnSO4 treated-SILECC might be mediated by abrogation of TGF-β induced expression of p21waf1/cip1 and Bax by ΔRII. Western blot analysis showed a basal level of p21waf1/cip1 in both untreated and ZnSO4 treated SILECC. As shown in Figure 7, the levels of p21waf1/cip1 were up-regulated after 1 and 3 hours in response to radiation plus ZnSO4 treatment, then down-regulated at 6 and 12 hours and were completely absent at 24 hours. However, with radiation alone, significant and sustained induction of p21waf1/cip1 was observed at all time points. Further, no change in Bax levels were observed between treated and untreated groups. In this normal lung epithelial cell line, induction of Bax may not be transcriptionally regulated by radiation. This is concordance with the previous report that in no induction of Bax was detected in normal human diploid fibroblast (33) or in normal prostate epithelial and stromal cells (34). Rather, radiation may cause activation of Bax via conformational changes by exposing the NH2-terminus and the COOH-terminus to mitochondria. Under such conformational activation of Bax no changes may be observed at either RNA or protein expression level. These data suggest that the down-regulation of p21waf1/cip1 protein in irradiated plus ZnSO4 treated SILECC was due to the abrogation of the TGF-β signaling by the expression of ΔRII, since endogenous wild-type p53 transactivation function will be disrupted due to reduced phosphorylation of Serine 18 caused by disruption of TGF-β signaling (35).
Figure 7. Radiation-induced p21waf1/cip1 up-regulation is inhibited by treatment with ZnSO4.
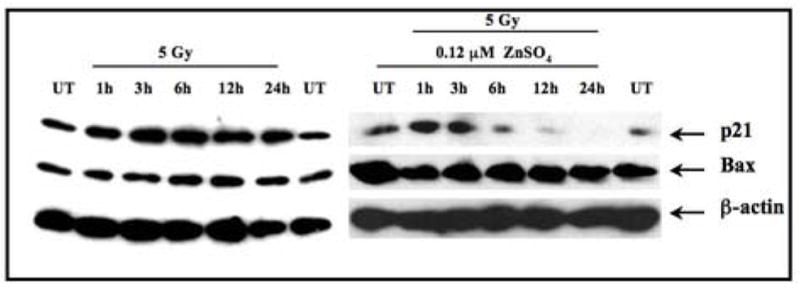
Antibodies to p21waf1/cip1 were used in Western blotting of total lysates of SILECC. Cells were left untreated, treated with ZnSO4 alone or 5 Gy alone or ZnSO4 exposure followed by 5 Gy of radiation. The blots were probed with antibody against p21waf1/cip1 and Bax. β-actin was used as a loading control.
Discussion
In the present study, we adopted a novel ex-vivo approach to investigate the functional role of TGF-β signaling in radiation-induced apoptosis and clonogenic inhibition in established spontaneously immortalized primary lung epithelial cell line derived from the normal lung epithelial tissue of the transgenic mouse expressing dominant-negative mutant of RII under the control of metallothionein promoter (28). The advantage of this approach is that unlike transformed tumor cells, these cells are normal except that they contain the silent ΔRII that could be induced when necessary with zinc. By this approach, no other factors associated with the transformed tumor cell phenotype or paracrine stromal-epithelial interactions would interfere with TGF-β signaling. Particularly, paracrine stromal-epithelial interaction is a well-established event in tumorigenesis (36, 37). Recent report indicates that specific inactivation of RII in fibroblasts, but not in epithelial cells in transgenic mice brought about an increase in proliferation of adjacent epithelia (36). Hence, this novel approach establishes an important model to understand the role of radiation-induced TGF-β in normal lung epithelium.
We have confirmed that the SILECC maintain normal epithelial morphology. These cells showed the polygonal shapes typical of epithelial cells. No altered cell morphology and cytoskeletal organization was observed. It is well documented that cytokeratin polypeptides constitute the intermediate filament cytoskeleton of epithelial cells (38). Further, the patterns of cytokeratin expression represent markers of epithelial differentiation status. We found that SILECC had an abundant level of cytokeratin suggesting that this normal lung cell culture is of epithelial origin. Abundant staining of cytokeratin found in SILECC using pan-Cytokeratin antibody can also be attributed to the fact that normal lung tissue has been known to harbor expression of various forms of cytokeratin polypeptides (38).
We have previously shown that RII expression is decreased in highly tumorigenic pancreatic cell line MIA PaCa-2 and the expression of functional RII restores TGF-β mediated growth inhibition caused by both recombinant TGF-β and radiation (17). In this present study, using a novel system to regulate TGF-β signaling in normal epithelial background through controlled expression of ΔRII, we demonstrated that abrogation of endogenous TGF-β function causes the following responses: (a) increased proliferative potential as assessed by growth curves (data not shown); (b) inhibition of radiation-induced or recombinant TGF-β-induced Smad signaling as assessed by TGF-β responsive reporter assay; (c) resistance to radiation-induced clonogenic inhibition; and (d) resistance to recombinant TGF-β or radiation-induced apoptosis. Our findings are consistent with the study by Bottinger et al (28) where primary pancreatic acinar cell cultures from control mice showed a marked inhibition as judged by [3H] thymidine incorporation when treated with all isoforms of TGF-β. At the same time the acinar cells from transgenic mice line AM3 were insensitive to all the isoforms of TGF-β but not activin indicating that DNR expression only inactivates TGF-β signaling, but not the signaling by other members of the TGF-β superfamily (28). Further, these findings agree with our previous report on abrogation of radiation-induced TGF-β responsive promoter activity with enhanced resistance to radiation-induced apoptosis in primary mouse embryonic fibroblasts (MEF) transfected with RII dominant negative mutant (17). It is of note that the treatment with the neutralizing antibodies against TGF-β also reversed radiation-induced apoptosis indicating that these changes were indeed dependent on TGF-β signaling in SILECC. Altogether, these observations correspond well with our previous reports where the expression of the ΔRII significantly diminished radiation-induced apoptosis in MEF cells, and the treatment of the pancreatic MIA PaCa-2/RII cells with neutralizing anti-TGF-β antibodies caused these cells to increase their radiation resistance (17).
In our previous reports (17, 18), radiation failed to induce p21waf/cip1 protein expression in MIA PaCa cells probably because the p53 and TGF-β pathways are not functional in these cells. However, restoration of TGF-β signaling in MIA PaCa cells led to p21waf/cip1 protein elevation after radiation. This induction was abrogated when the cells were exposed to anti-TGF-β neutralizing antibody. Similarly, in our present study we found that the expression of the ΔRII inhibited the radiation-induced p21waf/cip1 up-regulation suggesting that enhanced radio-resistance of the SILECC may be conferred by abrogation of the TGF-β-induced expression of p21waf/cip1. Since p21waf/cip1 is regulated by p53, it stands to reason to suggest that there is crosstalk between p53 and TGF-β signaling pathways. It has been documented that p53 function is required for TGF-β response by cooperating with Smads (39). Also, it was reported that transcriptional activation of p21waf/cip1 by TGF-β requires p53. Ewan et al demonstrated that abrogation of TGF-β function could lead to decreased p53 Ser-18 phosphorylation in the irradiated mammary epithelial tissue (35). In our previous reports, we documented the induction of p21waf/cip1 when TGF-β signaling was restored in mutant p53 MIA PaCa-2 pancreatic cancer cells (17). In addition, we demonstrated that abrogation of endogenous TGF-β function by ΔRII in p53 functional MEFs caused enhanced resistance to radiation-induced apoptosis (17). These previous observations taken together with this present study demonstrate that abrogation of TGF-β function may impair p53 mediated p21waf/cip1 up-regulation either due to impaired p53 transactivation function (owing to reduced levels of phospho-p53) or due to impaired Smad: p53 binding ratio to p21waf/cip1 promoter for its activation.
In summary, the findings of this study underscore the significance of TGF-β function since targeting the dysregulation of TGF-β function with simultaneous retention of function of all other pivotal pathways in the normal epithelial cell background can impact significant cellular response leading to enhanced cell proliferation and resistance to killing effects of radiation.
Figure 6. The transgene ΔRII protects SILECC from radiation-induced apoptosis.
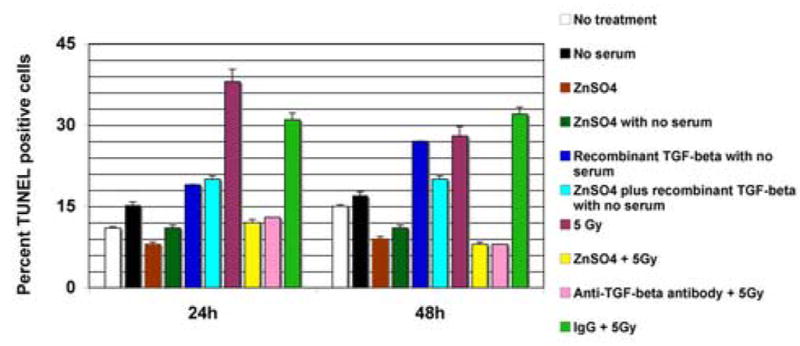
SILECC were exposed to the conditions indicated and TUNEL assay was performed after 24 h and 48 h of treatment. The bar graph shows the percentage of TUNEL-positive cells. Data represent a mean of four experiments. Error bars represent standard error.
Acknowledgments
The authors are grateful to Dr. Wakefield L.M. of the Laboratory of Cell Regulation and Carcinogenesis, National Cancer Institute, Bethesda, MD for providing AM3 transgenic DNR mice breeders. The authors gratefully acknowledge Dr Craig C. Wood of the Center for Health Research of the Geisinger Clinic for his technical assistance in the statistical analysis of the experimental data. This work was supported by grants from NCI (R01CA86937) and Kentucky Lung Cancer Program award to MMA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Whitman M. Smads and early developmental signaling by the TGFbeta superfamily. Genes Dev. 1998;12:2445–2462. doi: 10.1101/gad.12.16.2445. [DOI] [PubMed] [Google Scholar]
- 3.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst. 2000;92:1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 4.Roberts AB, Sporn MB. The transforming growth-betas. In: Sporn MB, Roberts AB, editors. Peptide growth factors and their receptors. Heidelberg: Springer-Verlag; 1991. [Google Scholar]
- 5.Lyons RM, Moses HL. Transforming growth factors and the regulation of cell proliferation. Eur J Biochem. 1990;187:467–473. doi: 10.1111/j.1432-1033.1990.tb15327.x. [DOI] [PubMed] [Google Scholar]
- 6.Howell GM, Sun L, Ziober BL, et al. The role of growth regulatory aberrations in progression of human colon carcinoma. Cancer Metastasis Rev. 1993;12:275–286. doi: 10.1007/BF00665958. [DOI] [PubMed] [Google Scholar]
- 7.Wrana JL, Attisano L, Wieser R, et al. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 8.Wilkes MC, Murphy SJ, Garamszegi N, et al. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol Cell Biol. 2003;23:8878–8889. doi: 10.1128/MCB.23.23.8878-8889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engel ME, McDonnell MA, Law BK, et al. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 10.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. Embo J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suardet L, Li C, Little JB. Radio-induced modulation of transforming growth factor beta1 sensitivity in a p53 wild-type human colorectal-cancer cell line. Int J Cancer. 1996;68:126–131. doi: 10.1002/(SICI)1097-0215(19960927)68:1<126::AID-IJC22>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 12.O'Malley Y, Zhao W, Barcellos-Hoff MH, et al. Radiation-induced alterations in rat mesangial cell Tgfb1 and Tgfb3 gene expression are not associated with altered secretion of active Tgfb isoforms. Radiat Res. 1999;152:622–628. [PubMed] [Google Scholar]
- 13.Wang J, Robbins ME. Radiation-induced alteration of rat mesangial cell transforming growth factor-beta and expression of the genes associated with the extracellular matrix. Radiat Res. 1996;146:561–568. [PubMed] [Google Scholar]
- 14.Zimmerman CM, Padgett RW. Transforming growth factor beta signaling mediators and modulators. Gene. 2000;249:17–30. doi: 10.1016/s0378-1119(00)00162-1. [DOI] [PubMed] [Google Scholar]
- 15.Shima Y, Nakao K, Nakashima T, et al. Activation of caspase-8 in transforming growth factor-beta-induced apoptosis of human hepatoma cells. Hepatology. 1999;30:1215–1222. doi: 10.1002/hep.510300503. [DOI] [PubMed] [Google Scholar]
- 16.Motyl T, Gajkowska B, Ploszaj T, et al. Expression and subcellular redistribution of Bax during TGF-beta1-induced programmed cell death of HC11 mouse mammary epithelial cells. Cell Mol Biol (Noisy-le-grand) 2000;46:175–185. [PubMed] [Google Scholar]
- 17.Ahmed MM, Alcock RA, Chendil D, et al. Restoration of transforming growth factor-beta signaling enhances radiosensitivity by altering the Bcl-2/Bax ratio in the p53 mutant pancreatic cancer cell line MIA PaCa-2. J Biol Chem. 2002;277:2234–2246. doi: 10.1074/jbc.M110168200. [DOI] [PubMed] [Google Scholar]
- 18.Alcock RA, Dey S, Chendil D, et al. Farnesyltransferase inhibitor (L-744,832) restores TGF-beta type II receptor expression and enhances radiation sensitivity in K-ras mutant pancreatic cancer cell line MIA PaCa-2. Oncogene. 2002;21:7883–7890. doi: 10.1038/sj.onc.1205948. [DOI] [PubMed] [Google Scholar]
- 19.Hougaard S, Norgaard P, Abrahamsen N, et al. Inactivation of the transforming growth factor beta type II receptor in human small cell lung cancer cell lines. Br J Cancer. 1999;79:1005–1011. doi: 10.1038/sj.bjc.6690161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakowlew SB, Moody TW, You L, et al. Reduction in transforming growth factor-beta type II receptor in mouse lung carcinogenesis. Mol Carcinog. 1998;22:46–56. doi: 10.1002/(sici)1098-2744(199805)22:1<46::aid-mc6>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Wu G, Willson JK, et al. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J Biol Chem. 1994;269:26449–26455. [PubMed] [Google Scholar]
- 22.Zhao Y, Young SL. Requirement of transforming growth factor-beta (TGF-beta) type II receptor for TGF-beta-induced proliferation and growth inhibition. J Biol Chem. 1996;271:2369–2372. doi: 10.1074/jbc.271.5.2369. [DOI] [PubMed] [Google Scholar]
- 23.Logsdon CD. Pancreatic duct cell cultures: there is more to ducts than salty water. Gastroenterology. 1995;109:1005–1009. doi: 10.1016/0016-5085(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 24.Logsdon CD, Williams JA. Pancreatic acinar cells in monolayer culture: direct trophic effects of caerulein in vitro. Am J Physiol. 1986;250:G440–447. doi: 10.1152/ajpgi.1986.250.4.G440. [DOI] [PubMed] [Google Scholar]
- 25.Fralix KD, Ahmed MM, Mattingly C, et al. Characterization of a newly established human pancreatic carcinoma cell line, UK Pan-1. Cancer. 2000;88:2010–2021. doi: 10.1002/(sici)1097-0142(20000501)88:9<2010::aid-cncr5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Venkatasubbarao K, Ahmed MM, Mohiuddin M, et al. Differential expression of transforming growth factor beta receptors in human pancreatic adenocarcinoma. Anticancer Res. 2000;20:43–51. [PubMed] [Google Scholar]
- 27.Tang B, de Castro K, Barnes HE, et al. Loss of responsiveness to transforming growth factor beta induces malignant transformation of nontumorigenic rat prostate epithelial cells. Cancer Res. 1999;59:4834–4842. [PubMed] [Google Scholar]
- 28.Bottinger EP, Jakubczak JL, Roberts IS, et al. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. Embo J. 1997;16:2621–2633. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abe T, Ouyang H, Migita T, et al. The somatic mutation frequency of the transforming growth factor beta receptor type II gene varies widely among different cancers with microsatellite instability. Eur J Surg Oncol. 1996;22:474–477. doi: 10.1016/s0748-7983(96)92824-3. [DOI] [PubMed] [Google Scholar]
- 30.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 31.Hu YX, Watanabe H, Ohtsubo K, et al. Infrequent expression of p21 is related to altered p53 protein in pancreatic carcinoma. Clin Cancer Res. 1998;4:1147–1152. [PubMed] [Google Scholar]
- 32.Hu PP, Datto MB, Wang XF. Molecular mechanisms of transforming growth factor-beta signaling. Endocr Rev. 1998;19:349–363. doi: 10.1210/edrv.19.3.0333. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, Tran BN, Worley LA, et al. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46:1561–1564. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- 34.Bromfield GP, Meng A, Warde P, et al. Cell death in irradiated prostate epithelial cells: role of apoptotic and clonogenic cell kill. Prostate Cancer Prostatic Dis. 2003;6:73–85. doi: 10.1038/sj.pcan.4500628. [DOI] [PubMed] [Google Scholar]
- 35.Ewan KB, Henshall-Powell RL, Ravani SA, et al. Transforming growth factor-beta1 mediates cellular response to DNA damage in situ. Cancer Res. 2002;62:5627–5631. [PubMed] [Google Scholar]
- 36.Bhowmick NA, Chytil A, Plieth D, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 37.Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr Opin Genet Dev. 2005;15:97–101. doi: 10.1016/j.gde.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlage WK, Bulles H, Friedrichs D, et al. Cytokeratin expression patterns in the rat respiratory tract as markers of epithelial differentiation in inhalation toxicology. I. Determination of normal cytokeratin expression patterns in nose, larynx, trachea, and lung. Toxicol Pathol. 1998;26:324–343. doi: 10.1177/019262339802600307. [DOI] [PubMed] [Google Scholar]
- 39.Cordenonsi M, Dupont S, Maretto S, et al. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]