

Abstract
Mice prematurely expressing human CR2 (hCR2) in the B cell lineage have a defective B cell ontogeny and immune response. Our recent analysis of this phenotype suggested that signalling through hCR2 and presumably mouse CD19 on the B cell surface, during bone marrow development, could result in the observed changes in B cell function in these mice. To test this hypothesis, we back crossed hCR2high transgenic mice onto the CD19−/− background. CD19−/−hCR2high mice were found to possess even fewer mature B cells than their CD19+/+hCR2high littermates, demonstrating that loss of CD19 exascerbated the effects elicited through hCR2. This data suggests that CD19 provides a survival signal during B cell development in this model. Next, we examined if the removal of the main ligand for CR2, namely C3d, through back-crossing onto the C3−/− background could restore normal B cell development. However, we found only minor recovery in peripheral B cell numbers and no obvious change in function. This was despite a 3-fold increase in the level of hCR2 expression on B cells isolated from the spleen or bone marrow of C3−/−hCR2high mice when compared with C3 sufficient littermates. These data demonstrate that hCR2 is integrated in mouse B cell signalling and that the downstream effects of hCR2 expression during early B cell development are partially but not completely due to interaction with C3 fragments and signaling through CD19 in the bone marrow environment.
Keywords: Transgenic/Knockout, Complement, B Lymphocytes, B cell development
1. Introduction
The expression of a functional B cell receptor (BCR), including the pre-BCR during the late pro-B cell stage, is essential to provide the signals that allow a B cell to mature and survive (Kraus et al., 2004; Melchers et al., 1999; Reichlin et al., 2001; Rolink and Melchers, 1996). Signals from the BCR are highly regulated through interaction with other membrane expressed receptors and proteins. Complement receptor 2 (CR2; CD21) is one such regulator. Through its association with CD19, as well as CD81 and CD225 (Leu 13) in a signaling complex, CR2 has been shown to be a key modifier of B cell development and function (Fearon and Carroll, 2000; Fearon and Carter, 1995; Matsumoto et al., 1991).
In the normal situation, expression of CR2 on human and mouse B lymphocytes is not detectable on pre-B or immature B cells, becoming detectable on transitional B cells and well expressed on mature B cells (Takahashi et al., 1997; Tedder et al., 1984). A key role for CR2 in the development and maintenance of the humoral response to T-dependant (TD) antigens (Ags) was first hinted at by several in vivo studies using CR1/2 blocking Abs and a study using CR2-IgG fusion protein. (Gustavsson et al., 1995; Hebell et al., 1991; Heyman et al., 1990; Thyphronitis et al., 1991). The independent generation by gene targeting of 3 lines of CR1/2 deficient mice confirmed these earlier findings as well as illustrating the necessity for CR2 expression on both B cells and the highly specialized follicular dendritic cell (FDC) population (Ahearn et al., 1996; Croix et al., 1996; Del Nagro et al., 2005; Fang et al., 1998; Haas et al., 2002; Molina et al., 1996). CR2 has been shown to facilitate the activation of B cells to TD antigens through a variety of mechanisms. These include enhanced presentation of BCR-bound Ag by class II MHC (Cherukuri et al., 2001a) and prolonging BCR signaling via lipid rafts (Cherukuri et al., 2001b) as well as the provision of co-stimulatory signals (Fearon and Carroll, 2000; Fearon and Carter, 1995). Additionally, CR1/2 mice have been shown to manifest a defect in response to T – independent (TI) Ag, underlining the importance of this receptors function in the breadth of B cell responses (Haas et al., 2002).
The potency of the co-stimulatory role of CR2 in lowering the threshold for activation of B cells after BCR/antigen co-ligation was first illustrated by Fearon and colleagues, who showed that B cells responded significantly better to a C3d linked antigen than to native antigen alone (Dempsey et al., 1996). CR2, in both mouse and man, binds with high affinity to the C3 breakdown fragment C3d (as well as iC3b and C3dg, (Cole et al., 1985; Farries et al., 1990; Iida et al., 1983; Kalli et al., 1991; Molina et al., 1994; Weis et al., 1984) which remains covalently bound to complement activating surfaces or antigen(Law and Dodds, 1997). Consistent with a key role for C3 fragments in amplifying the immune response via the CR2/CD19, C3−/− mice have weak humoral immune responses and defects in germinal center formation, (Fischer et al., 1996; Wessels et al., 1995). These data clearly demonstrated a direct link between the complement system and the humoral immune response as well as underline the importance of BCR/CR2/C3d interaction in regulating the level of signal finally transmitted to the B cell.
After ligation of CR2 with C3d, the majority of the B cell signaling activity generated is thought to be derived through association with CD19 (Fearon and Carter, 1995; Tedder et al., 1994). CD19, a member of the Ig superfamily, is expressed from the early pre-B cells developmental stage until plasma cell differentiation (Bradbury et al., 1993; Tedder and Isaacs, 1989; Tedder et al., 1994). From the moment it is expressed, CD19 has been shown to have regulatory function in the B cell (in pre-BCR signaling (Krop et al., 1996) and recombinase gene expression in pro-B cells (Billips et al., 1995)). However, absence of CD19 does not appear to influence B cell numbers until after B cells leave the bone marrow, where CD19−/− mice show marked decrease in B cell numbers and significant defects in B cell development (Engel et al., 1995; Rickert et al., 1995; Sato et al., 1995). On the other hand, transgenic mice that over express human CD19 are hyperresponsive to transmembrane signals. They display a marked alteration of B cell development from the point of IgM expression forward that results in reduced B cell numbers in periphery (Engel et al., 1995; Rickert et al., 1995; Sato et al., 1996; Zhou et al., 1994). These studies demonstrate that both the lack of, as well as excess CD19, have a marked impact on B cell survival, and that regulation of the signals received through CD19 and/or CR2/CD19 are likely to be critical to B cell function.
Previously, we have created two types of hCR2 transgenic (tg) mice in order to directly investigate the role of CR2 in B cell fate (Marchbank et al., 2002; Marchbank et al., 2000). The first tg line generated, which utilized a human P1 phage clone containing the entire human CR2 gene and all of the known transcriptional regulatory regions, showed normal developmental stage expression of hCR2 on mouse B cells. Despite a very low level of B cell surface expression, hCR2 reconstituted mCR1/2−/− mice demonstrated a partial restoration of the humoral immune response to sheep red blood cells (SRBC) as compared with non-tg mCR1/2−/− littermate controls (Marchbank et al., 2000). The second tg line expresses hCR2 cDNA under the control of a B cell-specific λ-promoter/enhancer minigene (lambda hCR2; (Marchbank et al., 2002)). In contrast to mice expressing hCR2 under its own promoter, expression of hCR2 was detected during the early stages of B cell development and resulted in a partial block in B cell development with a subsequent 40–60% reduction in mature B cell numbers. Furthermore, despite association with mCD19 and ability to flux Ca2+, mature B cells in the lambda hCR2 tg mice failed to respond adequately in vivo when challenged with TD and TI antigens (Birrell et al., 2005; Kulik et al., 2007; Marchbank et al., 2002). Our recent analysis suggests that this is very likely due to a result of intrinsic changes in signaling pathways downstream of BCR and CR2/CD19 cross-linking. Furthermore, these changes appear to protect hCR2 tg mice from the onset of organ specific auto-immune disease (Kulik et al., 2007). Interestingly, the absence of CR1/2 or C4 has been shown to result in increased autoantibody production in lupus prone mice (Fischer et al., 1996). This led to the conclusion that a central role for complement and CR1/2 was to signal the elimination of self-reactive B cells and thereby maintain B cell tolerance in the periphery (Carroll, 2000; Prodeus et al., 1998). Our data are consistent with this suggestion, in that many of the defects noted in the hCR2 tg mice could be the result of increased selective pressure or more stringent tolerance induction as a result of increased signaling through CD19/CR2 in conjunction with C3d binding in the bone marrow environment and/or the periphery. In order to investigate this hypothesis and to gain greater understanding of the B cell defects noted in the hCR2 tg mice, we bred the hCR2 tg mice onto the CD19−/− and C3−/− backgrounds.
2. Materials and Methods
2.1 Cells
Peripheral blood lymphocytes (PBL) from mice were collected into 20 μl of heparin via a tail bleed and washing once in cold PBS. Bone marrow B cells were collected by flushing mouse femurs with cold PBS. Isolated spleens were ground into single cell suspensions using frosted glass slides and transferred to 15 ml conical tubes on ice. Large debris settled after a 10 min incubation and the supernatant was transferred to a new tube. Cells were pelleted and washed once with staining buffer (PBS, 1% FBS, 0.02% sodium azide). All mouse cell samples were incubated with 0.5–1ml of red blood cell (RBC) lysis buffer and incubated at room temperature for 1–2 mins. The cells were then washed with 1 ml staining buffer 1–2 times. Cells were then counted and 1–3 × 106 cells/ml used per analysis. Cells were then stained as described below.
2.2 Antibodies
Purified and biotin conjugated 171 (anti-hCR2; (Guthridge et al., 2001)), as well as purified and biotin-IgG1 (isotype control) were produced in the laboratory following standard methods. Anti-mCD16/mCD32 (2.4G2 Fc block), anti-mCR2 (7G6), anti-hCR2/PE-(B-Ly-4), Biotin or fluorescein (FITC) conjugated 145-2C11 (anti-CD3ε), anti-CD19-PE, anti-CD24-FITC, biotin anti-CD23; biotin anti-CD138 (syndecan-1); anti-CD1d-PE and allophycocyanin (APC) or PerCP or FITC-conjugated anti-mCD45R, B220 (RA3-6B2) were all obtained from BD Pharmingen. Cy5.5-conjugated goat anti mouse IgM, goat anti-mouse IgG-PE, donkey anti-rabbit-HRPO, SA-APC and SA-PE were obtained from Jackson ImmunoResearch Laboratories (Stratech).
2.3 Mice
The human CR2 transgenic mice used herein were generated and screened by PCR as previously described (Marchbank et al., 2002). The mice used in this study are mCR1/2 sufficient C57BL/6 or Wild type (WT) background. The C3 deficient line, C3−/−C57BL/6 was a kind gift of Prof Michael Carroll. The CD19−/−C57BL/6 mice were a kind gift of Prof Robert Rickert. C3−/−hCR2high and CD19−/−hCR2high mice were generated by backcrossing the 2 parent strains and mice used in this study are to F4 on that backcross. All animal experiments were carried out in accordance with local ethic committee and Home Office guidelines.
2.4 SDS-PAGE and Western blot analysis for C3
Mice were tail bled into tubes containing 20 μl of heparin, cells were pelleted at 300g and heparinised sera collected. Sera was diluted 1/100 in sample buffer containing 0.05% v/v β2-mercaptoethanol and run out on 10% SDS-PAGE gels, blotted onto nitrocellulose, and blocked with 5% dried milk/PBS. The blots were incubated overnight at 4°C with polyclonal rabbit anti-mC3 (1/3000 in 5% dried milk/PBS), a generous gift of Dr. Carmen van den Berg (Cardiff University, Cardiff, UK), washed three times in PBS/0.1% Tween 20. Blots were then incubated with donkey anti-rabbit-HRP (1/3000 in 5% dried milk/PBS), washed twice with PBS/0.1% Tween 20 and with PBS only. Blots were developed using an enhanced chemiluminescence (ECL) substrate (Pierce, Cramlington, UK) according to the manufacturer’s specifications.
2.5 Flow Cytometry
After RBC lysis, cells were washed and resuspended in staining buffer supplemented with 10 μg/ml of 2.4G2 antibody in order to block Fc receptors. After a fifteen min incubation on ice, 100 μl of cells were added 1 to 1 staining buffer containing primary Ab (0.1–3 μg/ml). Directly conjugated B220 or anti-CD19 antibodies were used to identify B cells, as appropriate to the analysis. Cells were incubated for 30 min on ice in the dark. After incubation, cells were washed in staining buffer 3 times and then incubated with the appropriate streptavidin conjugated fluorochrome to detect biotin labeled primary Abs as appropriate. Following incubation, cells were washed as above and then resuspended in staining buffer containing 1% formaldehyde. Flow cytometry was performed using a Becton-Dickinson FACScalibur (Oxford, UK).
2.6 Ex vivo serum add back
Splenocytes were collected from C3−/−hCR2high mice using standard techniques. Blood was collected from the in house mouse colony and allowed to clot for 1hr at RT. Sera was then stored at −80°C until required. Cells were incubated at 37°C for 2hr with a ¼ dilution of indicated sera in 1 ml of RPMI. Cells were then washed, stained with B220-FITC (to identify B cells). Cells were also stained with 1 μg/ml biotin-171 (anti-hCR2) or 0.5 μg/ml 1D3-PE (anti-CD19) or 0.5 μg/ml biotin-7E9 (anti-mCR1/2) or 0.3 μg/ml goat anti-mouse IgM-Cy5.5. Cells were then washed before incubation with SA-APC at 1/1000 dilution in staining buffer were appropriate. Cells were washed and resuspended in staining buffer containing 1% formaldehyde prior to analysis via flow cytometry.
2.7 In vivo C3 add back
3 mg of purified human C3 (a kind gift from Dr C. Harris, Cardiff, UK) in PBS or PBS alone was injected intraperitoneally into two groups of 6 age, sex and weight matched C3−/−hCR2high mice. BM and Spleens were collected 4 hrs post injection as described above and B cells were then identified using B220-FITC. Cells were also stained with 10 μl/ml B-Ly-4-PE (anti-hCR2) as described above. Cells were washed and resuspended in staining buffer containing 1% formaldehyde prior to analysis via flow cytometry. 10,000 B220-FITC positive events were counted; the staining analysis was carried out in duplicate and the average used.
2. 8 Sheep red blood cell (SRBC) immunizations and serum immunoglobulin assays
SRBCs (TCS Bioscience, UK) were washed 3 times and resuspend to 1 × 108 SRBC/ml in PBS. Mice were injected intraperitoneally at days 0 and 28 with 500 μl of SRBC suspension. Serum was collected at days 0,14,21,28 and 35. Detection of Ab to SRBC was carried out using flow cytometry. SRBC were washed 3 times in PBS and re-suspended at a 0.05% v/v solution in flow buffer. 100 μl of SRBC solution was plated on ELISA plates and incubated with heat inactivated (56°C for 30 min) mouse anti-sera. Standard curves were generated with doubly diluted control anti-sera to confirm that test samples fell within the linear range of the assay for anti-SRBC IgG. Test samples were incubated in triplicate at room temperature for 30 min and then wash 3x in flow buffer. Plates were then incubated with 1/250 dilution of anti-mouse IgG-PE for 30 min at room temperature. SRBC were washed 3 x and then immediately analysed by flow cytometry. Between 5–7 mice were used in each group as stated.
3. Results
3. 1 CD19 deficient hCR2high mice exhibit severely reduced numbers of mature B cells
We have established that mice prematurely expressing human CR2 (hCR2) in the B cell lineage have a defective B cell ontogeny and immune response (Birrell et al., 2005; Marchbank et al., 2002). We have also shown that signaling through hCR2, and presumably mouse CD19, can occur in immature B cells from hCR2high mice (Kulik et al., 2007). This finding could explain many of the observed changes in B cell function in these mice. To investigate whether removal of signals through CD19 could restore B cell numbers and function back to wild type levels, we backcrossed the hCR2high mice onto the CD19−/− background (Figure 1A). However, we found that the absence of CD19 did not rescue B cells numbers in hCR2high B6 mice. In fact, we found that mCD19−/−hCR2high mice had significantly fewer B cells than wt B6 mice or mCD19+/+hCR2high mice (Figure 1B; 7.1% ± 2.2, n = 7 vs. 49.6% ± 4.2, n = 10 or 20.9 ± 7.1, n = 14; in both cases p < 0.0005 using the Mann-Whitney test, respectively). Examination of the remaining B cells in the CD19−/−hCR2high mice revealed that the surface expression of hCR2 was significantly less than that found on B cells from CD19+/+hCR2high mice, on average 40% lower (Figure 1C; MFU 102.6 ± 4.5, n = 5 vs 157 ± 21.5, n = 8.; p< 0.0005 using the Students T test, respectively). This data suggests that CD19 provides a survival or tonic signal to allow B cells, which express hCR2 to transit through and exit the bone marrow environment.
Fig 1. Analysis of CD19−/−hCR2+ tg mouse B cells.
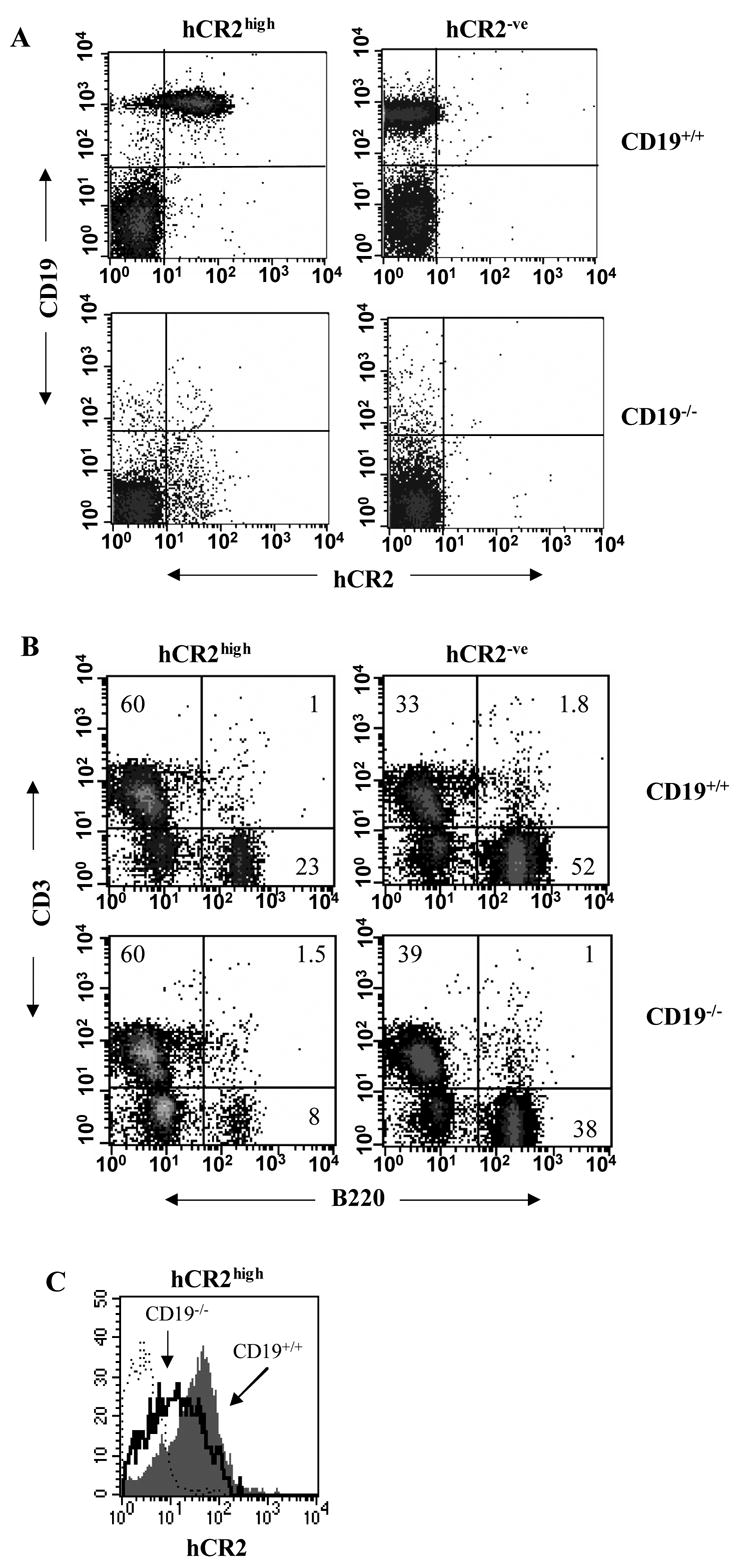
Splenocytes were isolated from littermates of each genotype as indicated in materials and methods. A) Cells were stained for CD19 and hCR2 to confirm the genotype of the mice B) Cells were stained with B220 and anti-CD3ε to identify B and T cells, respectively. The percentage of cells falling into each quadrant is indicated. Results are representative of 10 WT (CD19+/+hCR2−/−), 14 CD19+/+hCR2high, 5 CD19−/−hCR2−/− and 7 CD19−/−hCR2high littermates. C) Expression level of hCR2 was determined on B220 positive splenocytes in the presence (gray filled histogram) or absence (solid black line) of CD19. Background binding of anti-hCR2 to splenocytes from CD19+/+hCR2−/− (dashed line) is also included. Results are representative of 5 CD19−/−hCR2high and 8 CD19+/+hCR2high littermates.
3. 2 Peripheral B cell numbers in hCR2high mice are improved in the absence of C3
The data from the CD19−/− mice support a role for signaling through CR2 during B cell development being partly responsible for the observed phenotype in the hCR2 tg mice but not in the way originally conceived. To investigate this further, we turned our attention to C3, and the main ligands for CR2. We have recently shown that strong signaling can occur through hCR2 on immature B cells after cross linking with C3dg in the presence of a sub-optimal signal through sIgM (Kulik et al., 2007). Thus, one explanation for the phenotype observed in the hCR2 tg mice is that they receive a signal through cross linking of hCR2 by C3dg in the bone marrow environment. To examine this hypothesis, we backcrossed the hCR2high mice onto the C3−/− background (Figure 2). Analysis of C3−/−hCR2high mice revealed a small, but significant, increase in the number of B cells found in peripheral blood compartment compared with C3+/+hCR2high littermates. However, B cell numbers still remain significantly lower in C3−/−hCR2high mice than found in hCR2−ve or WT mice, regardless of C3 status (Table 1). B cell numbers in the spleen and bone marrow of C3−/−hCR2high and C3+/+hCR2high were essentially the same. We have previously found that there is an increase CD43+CD25+cells in the bone marrow (BM) of hCR2high mice (Marchbank et al., 2002). The number of B cells positive for CD43+CD25+ in the BM of C3−/−hCR2high and C3+/+hCR2high was found to be essentially the same (data not shown). This result suggested that the absence of C3 does have a subtle effect on B cell development in the hCR2high mice, but it does not eliminate the phenotypes seen as a result hCR2 expression on BM B cells.
Fig 2. Analysis of C3−/−hCR2+ sera by Western blot.
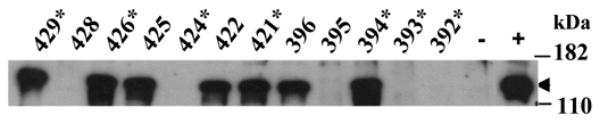
Heparinised sera was collected from backcrossed hCR2high and C3−/− mice. Diluted sera were run-out on a 10% reducing SDS-PAGE gel and blotted onto nitrocellulose as described in materials and methods. Mouse C3 was detected using a rabbit anti-mouse C3 polyclonal antibody follow by a HRPO conjugated secondary. ECL western blotting substrate was used to visualize bound antibody. Wild type C57BL/6 sera was used as positive control (+) and sera from the original C3−/− knockout colony was used as negative control (−). Individual mouse codes are noted and an asterisk denotes hCR2 positive mice. The arrow points to the C3 α chain and molecular weight markers are shown.
Table 1.
Peripheral B cell numbers are partially recovered in C3−/−hCR2high mice.
| hCR2high | hCR2−ve | |||
|---|---|---|---|---|
| C3−/− | C3+/+ | C3−/− | C3+/+ | |
| PBL | 37.4 ± 7.6* | 29.9 ± 7.0 | 66.3 ± 7.1 | 69.9 ± 7.0 |
| Spleen | 60.0 ± 3.7 | 59.8 ± 2.6 | 64.1 ± 1.3 | 68.5 ± 1.1 |
| BM | 17.3 ± 1.4 | 18.8 ± 7.2 | 25.4 ± 2.4 | 26.9 ± 4.0 |
Mice were bred onto the C3−/− background. Cells from BM, spleen and blood were collected as described in the methods section. Shown are the relative percentage of B220+ cells in each tissue falling in the lymphocyte gate (FSC v SSC; anti-CD3e negative in similar manner to Figure 1B) on standard flow cytometry. ** p < 0.01, N>6.
3.3 Splenic B cell subpopulation differences are partially recovered in C3−/−hCR2high mice
One of the hallmarks of the B cell defect in the hCR2high mice is a marked redistribution of B cells in the spleen (Marchbank et al., 2002). Particularly, marginal zone (MZ) B cells were found to be greatly expanded at the expense of the transitional and follicular B cell subpopulations. Analysis of the B cell subpopulations in C3−/−hCR2high mice revealed a partial or full recovery in the follicular (CD23hiCD24loCD1d−) and transitional B cell (CD23hiCD24hiCD1dint) populations, with the MZ B cell (CD23−CD24loCD1dhi) pool shrinking towards the normal wild type percentage (Figure 3). However, the MZ population was still significantly expanded at the expense of the follicular compartment in the C3−/−hCR2high mice compared to wild type mice. These data suggest that interaction with of hCR2 with mouse C3 or its breakdown components is having a partial but not obligatory effect on B cell development in these mice.
Fig 3. B cell subpopulation distribution is restored towards wild type levels in C3−/− hCR2high mice.
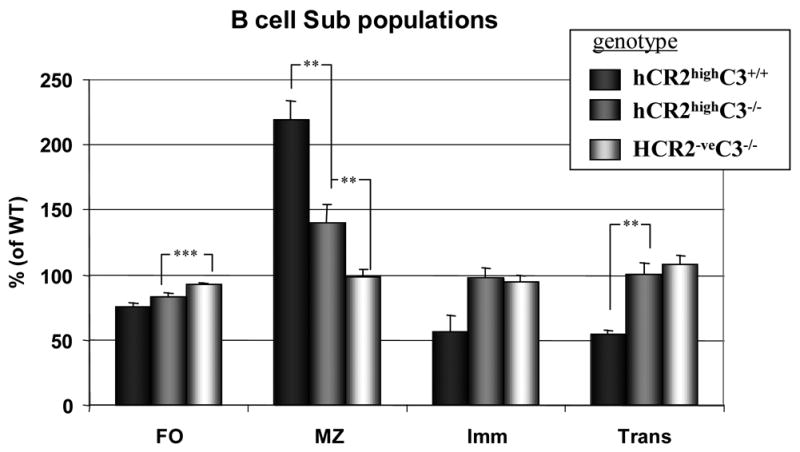
Splenocytes were isolated from littermates of mice with the genotype indicated. Cells were stained with B220-PerCP, Biotin anti-CD23 + SA-APC, anti-CD1d-PE and anti-CD24-FITC to allow B cells to be sub divided into Follicular (FO; B220+CD23hiCD24dullCD1ddull), Marginal Zone (MZ; B220+CD23dullCD24intCD1dhi), Immature (Imm; B220+CD23dullCD24hiCD1dint) and Transitional (Trans; B220+CD23hiCD24hiCD1dint) B cell sub populations. The number of cells falling into each of these groups is expressed as a percentage of mean values for 12 age and sex matched wild type mice. Each bars represent the mean and S.E.M. from 5 C3+/+hCR2high (black filled), 12 C3−/−hCR2high (dark gray filled) and 12 C3−/−hCR2 negative (light gray filled) mice for each sub population. The Mann-Whitney test was used to determine the p values; ** p<0.005; ***p<0.0001.
3. 4 Removal of C3 does not recover the immune response in hCR2high mice
We have shown above that removal of the main ligand for hCR2 had a restorative effect on B cell numbers in the periphery and were interested to establish whether removal of C3 could return B cell function in response to TD antigen back towards WT levels. Although the absence of C3 is detrimental to the humoral immune response, the use of high dose of antigen or adjuvant has been shown to allow a substantial immune response to be detected in C3−/− mice (Fischer et al., 1996; Wessels et al., 1995). Thus, WT, hCR2high, C3−/− and C3−/−hCR2high mice were injected with SRBC in complete Freunds adjuvant (CFA) and the immune response monitored. Immune responses in the C3−/−, hCR2high and C3−/−hCR2high mice were weak and largely similar during the primary response (data not shown). After an antigen boost, it became evident that the immune response in the C3−/−hCR2high mice was not improved in comparison to hCR2high mice on the wild type background (Figure 4A). Indeed, the immune response in C3−/− mice was significantly greater than that seen in the C3−/−hCR2high mice or C3+/+hCR2high littermates. Analysis of the percentage of GC (data not shown) and plasma cells at day 36 after primary injection in each group of animals also confirmed that C3−/−hCR2high mice failed to mount any significant response to SRBC when compared to both wild type and C3−/− mice (Figure 4B). These data demonstrate that absence of C3 mediated signals through hCR2 during B cell development does not translate into an increased ability to respond to antigen.
Fig 4. hCR2 tg mice have an unresponsive phenotype regardless of C3 status.
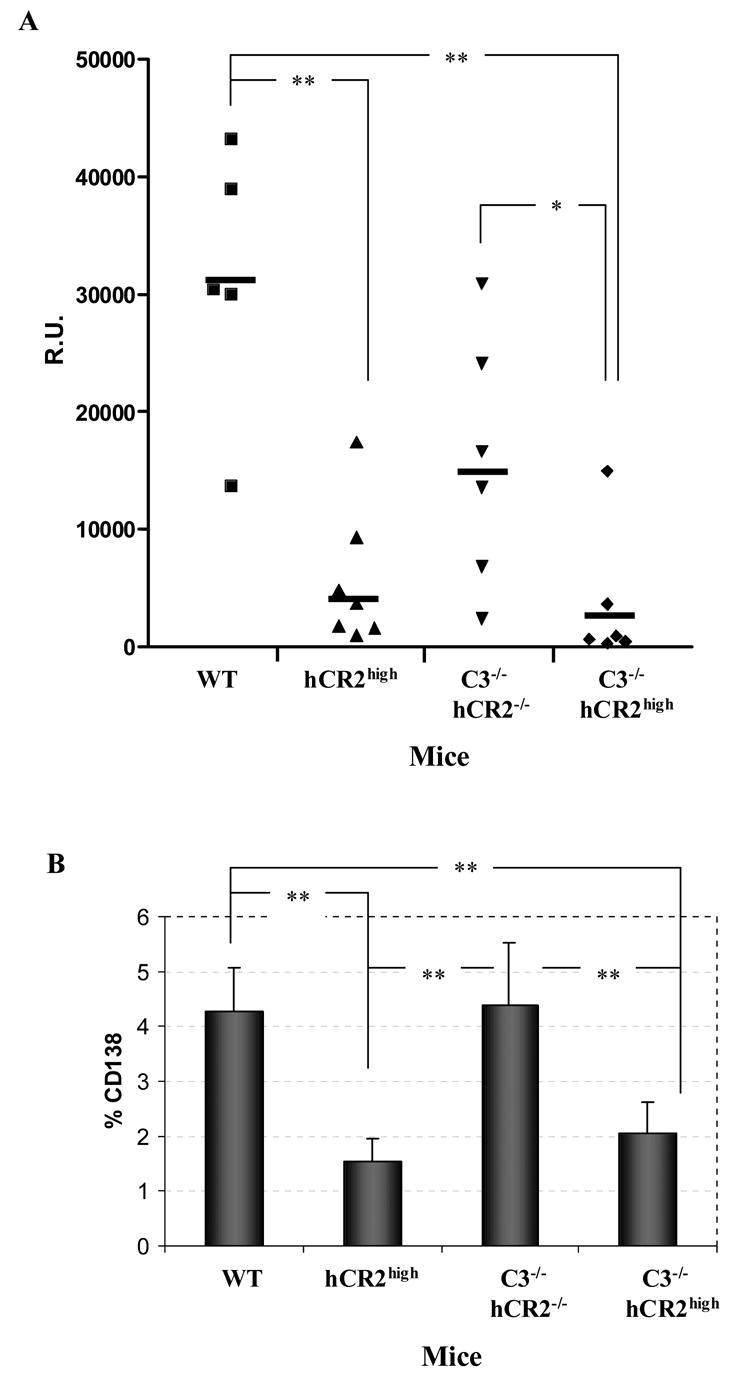
Mice were injected with sheep red blood cells (SRBC) as described in material and methods. One week after an antigen boost, serum and splenocytes were collected from mice. (A) Anti-SRBC titre was established by flow cytometric analysis as described in materials and methods. Reference serum was used to generate a standard curve from which R.U. could be calculated. (B) The percentage plasma cells (B220+CD138+) in the spleen were established after immunization of each genotype. Age and sex matched mouse groups include: 5 WT (Wild type; C3+/+hCR2−/−); 7 hCR2high (C3+/+hCR2high); 6 C3−/− hCR2−/− and 6 C3−/−hCR2high. The Mann-Whitney test was used to determine the p values; *p<0.05, ** p<0.005.
3.5 Absence of C3 results in a 3 fold increase in B cell expression of hCR2
We have previously shown that hCR2 expression levels drop significantly as the B cells mature in the bone marrow from B220lo to B220hi and are reduced further still as B cells migrate into the periphery (Birrell et al., 2005). However, on analysis of B cells isolated from C3−/−hCR2high mice, we found that this reduction in hCR2 expression levels was no longer fully apparent (Figure 5A). Furthermore, despite peripheral B cells in the C3−/−hCR2high mice having equivalent surface IgM levels they displayed a 3 fold increase in hCR2, a 1.5 fold increase in mCR1/2 and a significant increase in CD19 expression levels when compared to B cells isolated from mice which expressed normal levels of C3 (Figure 5B–F). In the case of mCR1/2 and CD19, these data are in complete agreement with those previously reported on the C3−/− background (Hasegawa et al., 2001). Thus, the increase in hCR2 expression levels in C3−/−background suggests that it has been integrated into the mechanisms that modulate the expression levels of endogenous co-receptor proteins on the B cell surface.
Fig 5. B cell expression levels of hCR2 increase in the absence of C3.
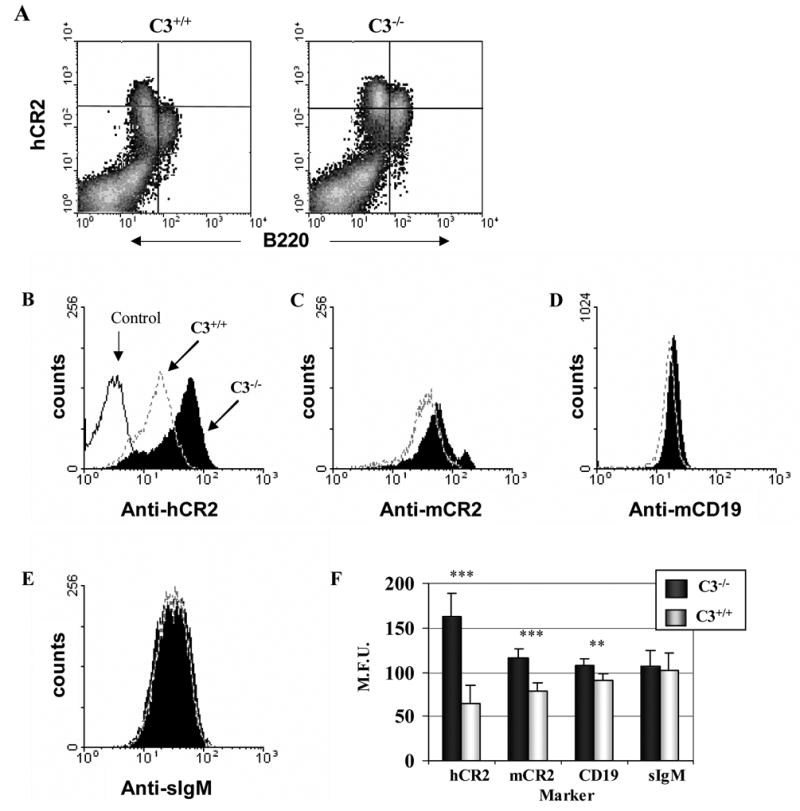
(A) Bone marrow cells were collected from C3+/+hCR2high and C3−/−hCR2high mice as described in materials and methods. Cells were stained for B220 and hCR2 (using biotin-b171+ SA-PE). The results shown are of the closest to mean values, for each genotype, of the 6 mice analysed. (B–F) Splenocytes were isolated as described in materials and methods. Cells were stained with B220 and various B cell markers as depicted. Shown are B220+ cells from C3+/+hCR2high (dashed line) and C3−/−hCR2high (solid filled histogram) mice stained with (B) anti-hCR2 (b171 +SA-PE). Background staining of the anti-hCR2 antibody on C3+/+hCR2−ve mice is represented by the solid line. (C) anti-mCR1/2 (b7E9 + SA-PE) (D) anti-CD19-PE (E) goat anti-IgM-Cy5.5. (F) Shows a bar chart of the average staining for these markers with S.E.M. of 6 age and sex matched C3+/+hCR2high (light grey) and C3−/−hCR2high (solid black) littermates. The Mann-Whitney test was used to determine the p values; *p<0.05, ** p<0.005; ***p<0.0005.
3.6 Administration of C3 to C3−/−hCR2high B cells reduces cell surface expression of co-receptor proteins
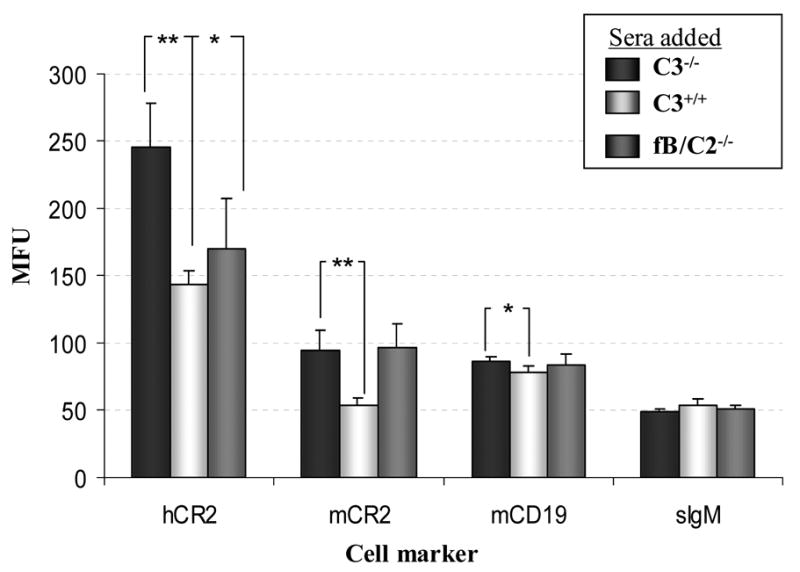
In order to confirm the requirement for C3 in the modulation of hCR2 expression levels on the B cell surface. C3−/−hCR2high splenocytes were incubated ex vivo with either wild type, C3−/− or fB/C2−/− sera for 2 hours. We found a marked reduction in hCR2, mCR1/2 and mCD19 expression levels in the presence of active murine C3 (Figure 6). In the absence of alternative pathway activation but the presence of native C3 (fB/C2−/− sera) there was still a small but significant reduction in hCR2 expression levels, whilst mCR1/2 and CD19 levels remained unchanged. Next, we administered purified hC3 to C3−/−hCR2high mice to establish whether presence of human C3 in the murine system would replicate the results ex vivo. We found a small but significant decrease in bone marrow (Figure 7A; Bly4, 153 ± 8.1 vs 121 ± 3.0; PBS vs hC3, respectively) and splenic B cell (Figure 7B; Bly4, 149 ± 9.4 vs 117 ± 3.5; PBS vs hC3, respectively) expression of hCR2 four hour after administration of hC3. mCD19 levels were decreased significantly (p<0.04) in the spleen (Figure 7B; 1D3, 87.4 ± 1.3 vs 78.5 ± 2.9; PBS vs hC3, respectively) and trended lower in the bone marrow environment (Figure 7A; 1D3, 86.1 ± 1.8 vs 82.1 ± 2.4; PBS vs hC3, respectively). As expected, expression levels of mIgM on mature peripheral B cells were found to be equivalent in this experiment (Figure 7B; GaM-IgM, 44.1 ± 1.7 vs 41.6 ± 2.2; PBS vs hC3, respectively). Analysis of mCR1/2 in peripheral B cells was also found to be unaltered and may reflect differences in affinity between mouse and human CR2 for hC3 and its breakdown fragments. Overall, these data demonstrate that hCR2 expression levels on the B cell surface are inversely linked to C3 levels and that there is probably a feedback signal generated through ligation of CR2 and CD19 by naturally occurring C3 breakdown fragments.
Fig 6. Incubation of C3−/−hCR2high B cells ex vivo with mouse C3 containing sera rapidly reduces B cell expression of hCR2.
Isolated splenocytes from four C3−/−hCR2high mice were incubated for 120 minutes with C3−/− sera (black bar); normal mouse sera (C3+/+, light gray bar) or C2/fB−/− (dark gray bar) and then flow cytometry was carried out to determine the effects on cell surface markers. The mean and S.E.M. is shown for each marker analysed. Results shown are representative of 2 independent experiments.
Fig 7. In vivo reconstitution of C3−/−hCR2high mice with hC3 results in reduction in hCR2 expression levels.
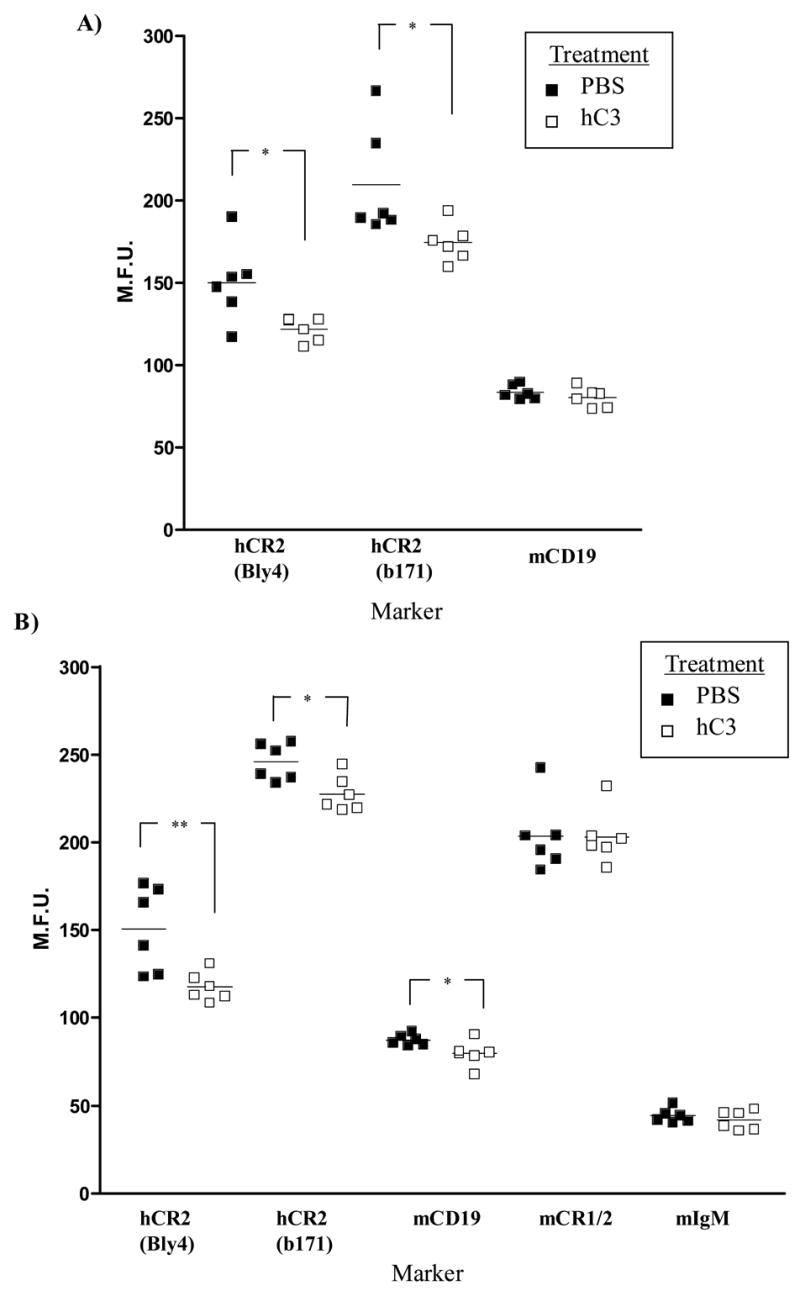
Age and sex matched C3−/−hCR2high mice were injected I.P. with 3mg of purified hC3 in PBS (open square, 6 mice) or PBS alone (filled square, 6 mice). 4 hours post injection, bone marrow and spleen were harvested as described in materials and methods. (A) shows bone marrow staining and (B) shows staining of splenocytes. B cells were identified using B220-FITC and levels of cell markers established using the appropriate antibodies and standard flow cytometry. The Mann-Whitney test was used to determine the p values; *p<0.05, ** p<0.01.
4. Discussion
The generation of mice expressing human CR2 under the control of a lambda light chain promoter-enhancer mini gene has resulted in mice that have an altered B cell ontogeny and a severely muted response to foreign or self antigen (Birrell et al., 2005; Marchbank et al., 2002). Through the reduction or removal of signaling capability of the CD19/CR2 complex by cross breeding the CD19−/− mice with the hCR2 tg mice, we hoped to remove a negative signal we envisaged was being delivered as a consequence of hCR2 interacting with CD19, after its ligation on the B cell surface, during early B cell development in the hCR2high mice. Unexpectedly, we found that CD19−/−hCR2high mice had a more pronounced reduction in B cell numbers than their CD19−/− littermates and CD19+/+hCR2high mice. This suggested that signals delivered via CD19 are protective in hCR2high mice during this stage of B cell development. The B cell populations in the CD19−/− mice are largely unaltered in the bone marrow environment but a marked loss of B cell numbers is noted in the periphery, approximately at the point of sIgD and mCR1/2 expression (Engel et al., 1995; Rickert et al., 1995; Sato et al., 1995). Thus, it appeared possible that the point of endogenous mCR1/CR2 expression was linked to a negative effect on B cell numbers in the periphery. In the case of hCR2 tg mice, the point of hCR2 expression also appears to be associated with the ‘negative effects’ on B cells which is greater after removal of CD19. However, the analysis of CR1/2−/−CD19−/− did not find any recovery in B cell numbers at this point when compared to CD19−/− mice and suggests that mCR1/2 is not directly involved in the loss of B cells(Hasegawa et al., 2001). It is worth noting that the CR1/2 mice used in that study expressed a hypomorphic mCR1/2 that still associates with CD19 and after binding with C3d could allow this molecule to be involved in signaling. The data herein continues to suggest that hCR2 expression results in a negative signal with regard to B cell development, at the point of its expression, that CD19 counteracts. The down regulation of hCR2 expressions levels is consistent with this idea and suggests the B cell attempts to reduce the comparatively increased negative signal generated through hCR2 in the absence of CD19 (Figure 1C). The potential for direct signaling through CR2 has not been totally ruled out (Delcayre et al., 1987; Frade et al., 1992) and despite overwhelming data confirming the positive signaling potential of CR2 (Fearon and Carroll, 2000; Fearon and Carter, 1995), negative signaling in response to CR2 ligation has also been demonstrated (Chakravarty et al., 2002; Lee et al., 2005). CR2 dependent BCR signal amplification has been shown to depend on CD19 (Lyubchenko et al., 2005), but CD19 independent effects could result from CR2 being free to associate and/or interfere with other signaling proteins or processes that it would not normally have access to or depend on CD19 during this phase of B cell development. Furthermore, the role of the other two identified members of the CR2 signaling complex, namely CD81 and CD225, are obvious candidates for modulation of hCR2 signals. Indeed, CD81−/− mice do demonstrate a down modulation of both B cell function and expression of CD19 (Maecker and Levy, 1997; Miyazaki et al., 1997; Tsitsikov et al., 1997). Although these mice display apparently normal B cell development, these studies highlight a potent signaling partner for hCR2 in the CD19−/−hCR2high mice that could be involved in the modulation of signaling in the hCR2 tg mice.
As CD19 apparently exerts a positive influence on B cell survival during B cell development in hCR2 tg mice, our attention shifted to the interaction of CR2 binding to its main ligand, C3d. The removal of C3 resulted in an almost 3 fold increase in B cell surface expression level of hCR2 compared to hCR2 expression levels in the presence of C3 (Figure 4A). A significant increase in B cell surface expression of mouse CR1/2 and CD19 on the C3−/− background was also noted (Figure 4B and C) and has been previously reported by Hasegawa et al(Hasegawa et al., 2001). This data demonstrates that despite hCR2 being expressed by an exogenous promoter (a mouse Vλ2 promoter-Vλ2–4 enhancer minigene (Marchbank et al., 2002)), hCR2 is being treated or responded to in a manner very similar to that of the endogenous murine members of the CR2/CD19 signaling complex. The fact that addition of C3 containing sera can result in rapid down regulation of members of the CR2/CD19 signaling complex suggests that tight regulation or feedback on expression levels of these molecules exist. This data fits with the idea that a common regulatory loop exists between C3, CR1/2 and CD19 that influences B cell signaling thresholds as suggested by Tedder and colleagues (Hasegawa et al., 2001). Our bone marrow data demonstrates the regulation of hCR2 expression levels in the presence of C3 is at the transition between B220lo and B220hi status and thus, is likely to be concurrent with the expression of sIgM or pre-BCR. Recently, it was shown that after antigen binding to BCR, the BCR itself could form a platform for activation of the classical complement pathway which resulted in the deposition of C3 fragments on the antigen which would presumably allow direct binding with CR1/2 and provide co-stimulatory signals into the B cell in a cell bound amplification loop (Manderson et al., 2006; Rossbacher and Shlomchik, 2003). It is also worth remembering that CR2 was shown to bind to sepharose bound inactive C3 (Cole et al., 1985). Thus, the ability of fB/C2 deficient sera to cause down regulation of hCR2 expression (Figure 6) demonstrates that the natural generation of inactive C3 could provide ligand to interact with CR2 as well as suggesting that the generation of C3d is not necessarily required for this outcome. Therefore, it is possible that continual activation or natural turn over and the subsequent interaction of C3 breakdown fragments with CR2 could exist as a mechanism to provide the required low level survival signals (Kraus et al., 2004) and/or tolerising signals essential to normal mature B cell function (Carroll, 2000; Prodeus et al., 1998). An extension of this hypothesis to the hCR2 tg mice would suggest that C3, activated in the presence of the pre-BCR, could be interacting with hCR2 in the BM and presumably results in signaling into the B cell. We have previously demonstrated CR2 and sub-optimal BCR cross linking will signal Ca2+ flux in immature B cells (Kulik et al., 2007). Furthermore, complement activation mediated by antigen engaged mIgM has been clearly demonstrated (Manderson et al., 2006; Rossbacher and Shlomchik, 2003) and it is thus conceivable that the pre-BCR, as a function of its role in providing the positive selection signal, may be in a conformation to facilitate such complement activation. Deficiencies of early classical complement pathway proteins, such as C1q and C4, are strongly associated with the development of the autoimmune disorder systemic lupus erythematosus (SLE; (Pickering et al., 2000)), which supports a role for complement activation in the induction, and maintenance of B cell tolerance. To date this has been ascribed to the role of complement in clearing apoptotic cells, but it is possible that C1q binding and complement activation on the newly formed BCR is essential for its selection and acts as a secondary test of its functionality. Thus, in the case of the hCR2 tg mice, the B cells are possibly receiving additional or conflicting signals which are altering those normally present after a limited C activation in the bone marrow at the point of pre-BCR expression. Corroborating evidence for aberrant BCR signaling being the likely mechanism behind the altered phenotype in the hCR2 tg mice can be found from the fact that we have previously found altered patterns of tyrosine phosphorylated proteins in B cells from hCR2 tg mice before and after BCR cross linking (Kulik et al., 2007). Additionally, mobilization of Ca2+ in immature B cells has previously been shown to increase to levels beyond that seen in mature B cells as a means of amplifying weak signals in the BM environment, presumably to delete autoreactive B cells from the repertoire (Benschop et al., 2001) and thus, additional signal amplification via hCR2 in the bone marrow is likely to disrupt these highly sensitive signaling networks. Due to the complexity of these pathways an array-based approaches is being undertaken in an attempt to identify changes in signaling and associated proteins.
Despite B cell subpopulations in the splenic environment being returned towards normal or WT values, removal of C3 and its breakdown products only resulted in a small recovery in numbers of peripheral blood B cells in these mice compared with hCR2high mice on the C3 sufficient background. Additionally, the lack of response to antigen was maintained in the C3−/−hCR2high mice, being almost identical to age matched C3+/+hCR2 tg mice and significantly reduced compared to C3−/− littermates. Clearly, lack of C3 does not completely reverse the phenotype or the reduction in B cell numbers noted in hCR2high mice and thus, hCR2 binding to mouse C3 or its breakdown fragments cannot be the only factor in the observed defect. There is still evidence of a previously noted down turn in hCR2 expression levels as B cells mature into the peripheral tissue despite the absence of the C3 ((Birrell et al., 2005);Figure 5A), suggesting the possibility that hCR2 interacts with other molecules as the B cell matures in the mouse. Evidence from analysis of myeloma cell adhesion to BM stroma cells has suggested that CR2 could interact with CD23 (Huang et al., 1995), and highlights one potential complement independent means of CR2 engagement. IFN-α, recently shown to bind to CR2 within a similar range of affinity as C3d, gp350 of EBV and CD23 (Asokan et al., 2006), represents another key molecule that could interact with hCR2 on the mature B cell. Interestingly, a link between IFN-α expression and sensitivity to auto-immune disease has been discovered (Baechler et al., 2003; Santiago-Raber et al., 2003). This data, in combination with the association of CR2 structure/function and expression levels with SLE, suggests that interactions between CR2 and IFN-α may be of importance in the onset of autoimmune disease. Thus, even though the main ligand for CR2 binding and signaling is removed from this environment, other molecules could interact with hCR2 and provide sufficient signals to perturb normal BCR function.
Overall, the work presented herein demonstrates that CD19 is likely to modulate/dampen or reduce the signal given by hCR2 in the bone marrow and removal of C3 does reduce some but not all the perceived signals that are induced through hCR2 at the point of BCR expression. It seems likely that the mechanisms by which the B cell interprets signals through the BCR/CR2/CD19 complex upon ligand binding within the bone marrow and beyond are the critical factors in unraveling the mystery behind the marked changes noted in B cells which express hCR2 during early B cell development. Furthermore, these data suggest that an understanding of the interaction of hCR2 with mouse B cell signaling components within immature B cells should provide a means to dissect and identify the proteins involved in setting the thresholds for activation of B cells.
Acknowledgments
These studies were supported a Wellcome Trust Research Career Development Fellowship (KJM) and by NIH R0-1 CA53615 (VMH), Medical Scientist Training Program grant T32 GM008497 (KAK). The authors would like to thank Dr Isabel Pappworth, Dr Brad Spiller, Dr Claire Harris and Prof. Paul Morgan for helpful discussion and the staff at the B.S.U. at Cardiff University who helped make these studies possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahearn JM, Fischer MB, Croix D, Goerg S, Ma M, Xia J, Zhou X, Howard RG, Rothstein TL, Carroll MC. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–62. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- Asokan R, Hua J, Young KA, Gould HJ, Hannan JP, Kraus DM, Szakonyi G, Grundy GJ, Chen XS, Crow MK, Holers VM. Characterization of human complement receptor type 2 (CR2/CD21) as a receptor for IFN-alpha: a potential role in systemic lupus erythematosus. J Immunol. 2006;177:383–94. doi: 10.4049/jimmunol.177.1.383. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop RJ, Brandl E, Chan AC, Cambier JC. Unique signaling properties of B cell antigen receptor in mature and immature B cells: implications for tolerance and activation. J Immunol. 2001;167:4172–9. doi: 10.4049/jimmunol.167.8.4172. [DOI] [PubMed] [Google Scholar]
- Billips LG, Nunez CA, Bertrand FE, 3rd, Stankovic AK, Gartland GL, Burrows PD, Cooper MD. Immunoglobulin recombinase gene activity is modulated reciprocally by interleukin 7 and CD19 in B cell progenitors. J Exp Med. 1995;182:973–82. doi: 10.1084/jem.182.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell L, Kulik L, Morgan BP, Holers VM, Marchbank KJ. B cells from mice prematurely expressing human complement receptor type 2 are unresponsive to T-dependent antigens. J Immunol. 2005;174:6974–82. doi: 10.4049/jimmunol.174.11.6974. [DOI] [PubMed] [Google Scholar]
- Bradbury LE, Goldmacher VS, Tedder TF. The CD19 signal transduction complex of B lymphocytes. Deletion of the CD19 cytoplasmic domain alters signal transduction but not complex formation with TAPA-1 and Leu 13. J Immunol. 1993;151:2915–27. [PubMed] [Google Scholar]
- Carroll MC. The role of complement in B cell activation and tolerance. Adv Immunol. 2000;74:61–88. doi: 10.1016/s0065-2776(08)60908-6. [DOI] [PubMed] [Google Scholar]
- Chakravarty L, Zabel MD, Weis JJ, Weis JH. Depletion of Lyn kinase from the BCR complex and inhibition of B cell activation by excess CD21 ligation. Int Immunol. 2002;14:139–46. doi: 10.1093/intimm/14.2.139. [DOI] [PubMed] [Google Scholar]
- Cherukuri A, Cheng PC, Pierce SK. The role of the CD19/CD21 complex in B cell processing and presentation of complement-tagged antigens. J Immunol. 2001a;167:163–72. doi: 10.4049/jimmunol.167.1.163. [DOI] [PubMed] [Google Scholar]
- Cherukuri A, Cheng PC, Sohn HW, Pierce SK. The CD19/CD21 complex functions to prolong B cell antigen receptor signaling from lipid rafts. Immunity. 2001b;14:169–79. doi: 10.1016/s1074-7613(01)00098-x. [DOI] [PubMed] [Google Scholar]
- Cole JL, Housley GA, Jr, Dykman TR, MacDermott RP, Atkinson JP. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc Natl Acad Sci U S A. 1985;82:859–63. doi: 10.1073/pnas.82.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croix DA, Ahearn JM, Rosengard AM, Han S, Kelsoe G, Ma M, Carroll MC. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J Exp Med. 1996;183:1857–64. doi: 10.1084/jem.183.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Nagro CJ, Kolla RV, Rickert RC. A critical role for complement C3d and the B cell coreceptor (CD19/CD21) complex in the initiation of inflammatory arthritis. J Immunol. 2005;175:5379–89. doi: 10.4049/jimmunol.175.8.5379. [DOI] [PubMed] [Google Scholar]
- Delcayre AX, Fiandino A, Barel M, Frade R. gp140, the EBV/C3d receptor (CR2) of human B lymphocytes, is involved in cell-free phosphorylation of p120, a nuclear ribonucleoprotein. Eur J Immunol. 1987;17:1827–33. doi: 10.1002/eji.1830171223. [DOI] [PubMed] [Google Scholar]
- Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–50. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 1998;160:5273–9. [PubMed] [Google Scholar]
- Farries TC, Seya T, Harrison RA, Atkinson JP. Competition for binding sites on C3b by CR1, CR2, MCP, factor B and Factor H. Complement Inflamm. 1990;7:30–41. doi: 10.1159/000463124. [DOI] [PubMed] [Google Scholar]
- Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000;18:393–422. doi: 10.1146/annurev.immunol.18.1.393. [DOI] [PubMed] [Google Scholar]
- Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu Rev Immunol. 1995;13:127–49. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- Fischer MB, Ma M, Goerg S, Zhou X, Xia J, Finco O, Han S, Kelsoe G, Howard RG, Rothstein TL, Kremmer E, Rosen FS, Carroll MC. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J Immunol. 1996;157:549–56. [PubMed] [Google Scholar]
- Frade R, Gauffre A, Hermann J, Barel M. EBV/C3d receptor (CR2) interacts by its intracytoplasmic carboxy-terminal domain and two distinct binding sites with the p53 anti-oncoprotein and the p68 calcium-binding protein. J Immunol. 1992;149:3232–8. [PubMed] [Google Scholar]
- Gustavsson S, Kinoshita T, Heyman B. Antibodies to murine complement receptor 1 and 2 can inhibit the antibody response in vivo without inhibiting T helper cell induction. J Immunol. 1995;154:6524–8. [PubMed] [Google Scholar]
- Guthridge JM, Young K, Gipson MG, Sarrias MR, Szakonyi G, Chen XS, Malaspina A, Donoghue E, James JA, Lambris JD, Moir SA, Perkins SJ, Holers VM. Epitope mapping using the X-ray crystallographic structure of complement receptor type 2 (CR2)/CD21: identification of a highly inhibitory monoclonal antibody that directly recognizes the CR2-C3d interface. J Immunol. 2001;167:5758–66. doi: 10.4049/jimmunol.167.10.5758. [DOI] [PubMed] [Google Scholar]
- Haas KM, Hasegawa M, Steeber DA, Poe JC, Zabel MD, Bock CB, Karp DR, Briles DE, Weis JH, Tedder TF. Complement receptors CD21/35 link innate and protective immunity during Streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity. 2002;17:713–23. doi: 10.1016/s1074-7613(02)00483-1. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujimoto M, Poe JC, Steeber DA, Tedder TF. CD19 can regulate B lymphocyte signal transduction independent of complement activation. J Immunol. 2001;167:3190–200. doi: 10.4049/jimmunol.167.6.3190. [DOI] [PubMed] [Google Scholar]
- Hebell T, Ahearn JM, Fearon DT. Suppression of the immune response by a soluble complement receptor of B lymphocytes. Science. 1991;254:102–5. doi: 10.1126/science.1718035. [DOI] [PubMed] [Google Scholar]
- Heyman B, Wiersma EJ, Kinoshita T. In vivo inhibition of the antibody response by a complement receptor-specific monoclonal antibody. J Exp Med. 1990;172:665–8. doi: 10.1084/jem.172.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Kawano MM, Mahmoud MS, Mihara K, Tsujimoto T, Niwa O, Kuramoto A. Expression of CD21 antigen on myeloma cells and its involvement in their adhesion to bone marrow stromal cells. Blood. 1995;85:3704–12. [PubMed] [Google Scholar]
- Iida K, Nadler L, Nussenzweig V. Identification of the membrane receptor for the complement fragment C3d by means of a monoclonal antibody. J Exp Med. 1983;158:1021–33. doi: 10.1084/jem.158.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli KR, Ahearn JM, Fearon DT. Interaction of iC3b with recombinant isotypic and chimeric forms of CR2. J Immunol. 1991;147:590–4. [PubMed] [Google Scholar]
- Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117:787–800. doi: 10.1016/j.cell.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Krop I, Shaffer AL, Fearon DT, Schlissel MS. The signaling activity of murine CD19 is regulated during cell development. J Immunol. 1996;157:48–56. [PubMed] [Google Scholar]
- Kulik L, Marchbank KJ, Lyubchenko T, Kuhn KA, Lyubchenko G, Haluszczak C, Gibson MG, Boackle SA, Holers VM. Intrinsic B Cell Hypo-Responsiveness in Mice Prematurely Expressing Human CR2/CD21 During B Cell Development. Eur J Immunol. 2007 doi: 10.1002/eji.200636248. In Press. [DOI] [PubMed] [Google Scholar]
- Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997;6:263–74. doi: 10.1002/pro.5560060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Haas KM, Gor DO, Ding X, Karp DR, Greenspan NS, Poe JC, Tedder TF. Complement component C3d-antigen complexes can either augment or inhibit B lymphocyte activation and humoral immunity in mice depending on the degree of CD21/CD19 complex engagement. J Immunol. 2005;175:8011–23. doi: 10.4049/jimmunol.175.12.8011. [DOI] [PubMed] [Google Scholar]
- Lyubchenko T, dal Porto J, Cambier JC, Holers VM. Coligation of the B cell receptor with complement receptor type 2 (CR2/CD21) using its natural ligand C3dg: activation without engagement of an inhibitory signaling pathway. J Immunol. 2005;174:3264–72. doi: 10.4049/jimmunol.174.6.3264. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Levy S. Normal lymphocyte development but delayed humoral immune response in CD81-null mice. J Exp Med. 1997;185:1505–10. doi: 10.1084/jem.185.8.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderson AP, Quah B, Botto M, Goodnow CC, Walport MJ, Parish CR. A novel mechanism for complement activation at the surface of B cells following antigen binding. J Immunol. 2006;177:5155–62. doi: 10.4049/jimmunol.177.8.5155. [DOI] [PubMed] [Google Scholar]
- Marchbank KJ, Kulik L, Gipson MG, Morgan BP, Holers VM. Expression of human complement receptor type 2 (CD21) in mice during early B cell development results in a reduction in mature B cells and hypogammaglobulinemia. J Immunol. 2002;169:3526–35. doi: 10.4049/jimmunol.169.7.3526. [DOI] [PubMed] [Google Scholar]
- Marchbank KJ, Watson CC, Ritsema DF, Holers VM. Expression of human complement receptor 2 (CR2, CD21) in Cr2−/− mice restores humoral immune function. J Immunol. 2000;165:2354–61. doi: 10.4049/jimmunol.165.5.2354. [DOI] [PubMed] [Google Scholar]
- Matsumoto AK, Kopicky-Burd J, Carter RH, Tuveson DA, Tedder TF, Fearon DT. Intersection of the complement and immune systems: a signal transduction complex of the B lymphocyte-containing complement receptor type 2 and CD19. J Exp Med. 1991;173:55–64. doi: 10.1084/jem.173.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchers F, ten Boekel E, Yamagami T, Andersson J, Rolink A. The roles of preB and B cell receptors in the stepwise allelic exclusion of mouse IgH and L chain gene loci. Semin Immunol. 1999;11:307–17. doi: 10.1006/smim.1999.0187. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Muller U, Campbell KS. Normal development but differentially altered proliferative responses of lymphocytes in mice lacking CD81. Embo J. 1997;16:4217–25. doi: 10.1093/emboj/16.14.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H, Holers VM, Li B, Fung Y, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Chaplin DD. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci U S A. 1996;93:3357–61. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H, Kinoshita T, Webster CB, Holers VM. Analysis of C3b/C3d binding sites and factor I cofactor regions within mouse complement receptors 1 and 2. J Immunol. 1994;153:789–95. [PubMed] [Google Scholar]
- Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, Goodnow CC, Carroll MC. A critical role for complement in maintenance of self-tolerance. Immunity. 1998;9:721–31. doi: 10.1016/s1074-7613(00)80669-x. [DOI] [PubMed] [Google Scholar]
- Reichlin A, Hu Y, Meffre E, Nagaoka H, Gong S, Kraus M, Rajewsky K, Nussenzweig MC. B cell development is arrested at the immature B cell stage in mice carrying a mutation in the cytoplasmic domain of immunoglobulin beta. J Exp Med. 2001;193:13–23. doi: 10.1084/jem.193.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–5. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- Rolink A, Melchers F. B-cell development in the mouse. Immunol Lett. 1996;54:157–61. doi: 10.1016/s0165-2478(96)02666-1. [DOI] [PubMed] [Google Scholar]
- Rossbacher J, Shlomchik MJ. The B cell receptor itself can activate complement to provide the complement receptor 1/2 ligand required to enhance B cell immune responses in vivo. J Exp Med. 2003;198:591–602. doi: 10.1084/jem.20022042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–88. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J Immunol. 1996;157:4371–8. [PubMed] [Google Scholar]
- Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc Natl Acad Sci U S A. 1995;92:11558–62. doi: 10.1073/pnas.92.25.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Kozono Y, Waldschmidt TJ, Berthiaume D, Quigg RJ, Baron A, Holers VM. Mouse complement receptors type 1 (CR1;CD35) and type 2 (CR2;CD21): expression on normal B cell subpopulations and decreased levels during the development of autoimmunity in MRL/lpr mice. J Immunol. 1997;159:1557–69. [PubMed] [Google Scholar]
- Tedder TF, Clement LT, Cooper MD. Expression of C3d receptors during human B cell differentiation: immunofluorescence analysis with the HB-5 monoclonal antibody. J Immunol. 1984;133:678–83. [PubMed] [Google Scholar]
- Tedder TF, Isaacs CM. Isolation of cDNAs encoding the CD19 antigen of human and mouse B lymphocytes. A new member of the immunoglobulin superfamily. J Immunol. 1989;143:712–7. [PubMed] [Google Scholar]
- Tedder TF, Zhou LJ, Engel P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol Today. 1994;15:437–42. doi: 10.1016/0167-5699(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Thyphronitis G, Kinoshita T, Inoue K, Schweinle JE, Tsokos GC, Metcalf ES, Finkelman FD, Balow JE. Modulation of mouse complement receptors 1 and 2 suppresses antibody responses in vivo. J Immunol. 1991;147:224–30. [PubMed] [Google Scholar]
- Tsitsikov EN, Gutierrez-Ramos JC, Geha RS. Impaired CD19 expression and signaling, enhanced antibody response to type II T independent antigen and reduction of B-1 cells in CD81-deficient mice. Proc Natl Acad Sci U S A. 1997;94:10844–9. doi: 10.1073/pnas.94.20.10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis JJ, Tedder TF, Fearon DT. Identification of a 145,000 Mr membrane protein as the C3d receptor (CR2) of human B lymphocytes. Proc Natl Acad Sci U S A. 1984;81:881–5. doi: 10.1073/pnas.81.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LJ, Smith HM, Waldschmidt TJ, Schwarting R, Daley J, Tedder TF. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent B-lymphocyte development. Mol Cell Biol. 1994;14:3884–94. doi: 10.1128/mcb.14.6.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]