

Abstract
We investigated structural determinants of fast inactivation and deactivation in sodium channels by comparing ionic flux and charge movement in skeletal muscle channels, using mutations of DIII-DIV linker charges. Charge altering and substituting mutations at K-1317, K-1318 depolarized the g(V) curve but hyperpolarized the h∞ curve. Charge reversal and substitution at this locus reduced the apparent voltage sensitivity of open- and closed-state fast inactivation. These effects were not observed with charge reversal at E-1314, E-1315. Mutations swapping or neutralizing the negative cluster at 1314, 1315 and the positive cluster at 1317, 1318 indicated that local interactions dictate the coupling of activation to fast inactivation. Gating charge was immobilized before channel entry into fast inactivation in hNaV1.4 but to a lesser extent in mutations at K-1317, K-1318. These results suggest that charge is preferentially immobilized in channels inactivating from the open state. Recovery of gating charge proceeded with a single, fast phase in the double mutation K-1317R, K-1318R. This mutation also partially uncoupled recovery from deactivation. Our findings indicate that charged residues near the fast inactivation “particle” allosterically interact with voltage sensors to control aspects of gating in sodium channels.
INTRODUCTION
Voltage-gated sodium channels regulate the excitatory phase of action potentials used in electrical signaling by excitable cells such as neurons and muscle fibers (1,2). Sodium channels in brain, cardiac, and skeletal muscle share sequence homology indicating a structure comprised of four domains each made up of six-transmembrane segments (3–5). This basic organization is common to other members of the voltage-gated ion channel superfamily, including potassium and calcium channels. Conserved sequences in these channels are thought to promote functions such as voltage sensitivity ascribed to the complement of positively charged amino acids in the fourth segment (3). The notion that S4 segments act as voltage sensors in channel gating has been supported by experiments with chemical modification (6–9), toxins (10), fluorescence labeling (11,12), and mutagenesis (13–15).
Certain functions of sodium channels have unique or domain-specific structural determinants. For example, fast inactivation is dependent on a conserved IFMT motif in the DIII-DIV cytoplasmic linker (16–19). Residues in the S4-S5 loops of DIII and DIV are thought to mediate hydrophobic interactions with that motif, or inactivation particle, to block the open pore (20–25). IFMT/receptor interaction appears to depend on the late movement of DIVS4 in response to membrane depolarization (26), explaining the observation that mutations in DIVS4 attenuate the voltage dependence of fast inactivation (27,28). With fast inactivation, gating charge is immobilized such that recovery of the gating charge is bimodal during repolarization of the membrane (29,30). Charge immobilized with fast inactivation has been localized to the voltage sensors in DIII and DIV (11).
The concentration of charged residues in the DIII-DIV linker approximates the charge density of the voltage sensors in domains I–IV (3). Charged residues are not part of the inactivation particle but may regulate other aspects of channel gating. DIII-DIV linker charge-altering mutations influence the kinetics of fast inactivation but do not prohibit its completion (31–33). In addition, charge substituting and reversing mutations of the negative cluster E-1314, E-1315 in hNaV1.4 slow deactivation from open and fast-inactivated states (33).
In this study we hypothesized that charge content in the linker regulates channel deactivation as a consequence of alteration of gating charge movement. To test this hypothesis, we compared the effects of mutations at E-1314, E-1315 and K-1317, K-1318 on ionic flux and charge immobilization. Each of these clusters is contained within the central, highly structured region of the linker adjacent to the IFMT motif (19; rNaV1.2). Our findings indicate that the positive cluster K-1317, K-1318 couples activation with fast inactivation and charge immobilization. Coupling is dependent on local interactions in this region of the DIII-DIV linker. In addition, the K-1317, K-1318 cluster regulates inactivated state deactivation through its impact on the immobilization of charge. Portions of this work have been reported in abstract form (34).
MATERIALS AND METHODS
Site-directed mutagenesis
Double mutations were prepared by site-directed mutagenesis at E-1314, E-1315 and at K-1317, K-1318 in human SCN4A using a polymerase chain reaction (PCR) overlap extension method (35). Appropriate single primers carrying both mutations were used with primers flanking a 930-basepair AvrII-SacII fragment. The PCR fragment was digested and cloned into pGH19/SCN4A. Charge swapping and neutralizing mutations at E-1314, E-1315/K-1317, K-1318 were constructed by using PCR overlap extension mutagenesis with the appropriate primers containing the mutation at K-1317, K-1318 and the E-1314K, E-1315K or E-1314Q, E-1315Q verified construct (33) as the template. The resulting clones were E-1314K, E-1315K/K-1317E, K-1318E, and E-1314Q, E-1315Q/K-1317Q, K-1318Q. All constructs were confirmed by sequencing. Plasmids were linearized with Not I and transcribed with T7 RNA polymerase for injection into oocytes.
Oocyte preparation
Xenopus laevis oocyte lobes were surgically removed after anesthetizing the animals with 0.17% tricaine (3-aminobenzoic acid ethyl ester; Sigma Chemical, St. Louis, MO) according to guidelines approved by Animal Use and Care Committees at ISU and at USU. Xenopus were isolated for several days after surgeries before return to the colony. Oocyte lobes were teased apart with forceps and treated for 30 min with 2 mg/ml collagenase (Sigma) in a solution containing (in mM): NaCl 96, KCl 2, MgCl2 20, HEPES 5, pH 7.4. Isolated oocytes were then rinsed with the same solution and incubated at 18°C in culture medium containing (in mM): NaCl 96, KCl 2, MgCl2 1, CaCl2 1.8, HEPES 5, sodium pyruvate 2.5, pH 7.4, with 100 mg/l gentamicin and 3% horse serum (Hyclone Laboratories, Logan, UT).
Oocytes were coinjected with mRNA for sodium channel α and β1 subunits (1:1 volume, α-subunit at 1 μg/μl, β1 subunit at 3 μg/μl) in a volume of 50 nl/oocyte, the day after isolation. For macropatch recordings, injections were made at the equatorial line separating animal and vegetal poles. At 3–7 days postinjection, vitelline membranes were removed with forceps in a hyperosmotic solution containing (in mM): NaCl 96, KCl 2, MgCl2 20, HEPES 5, mannitol 400, pH 7.4, before recordings. For gating current recordings, oocytes were injected in the vegetal pole and recordings were made from intact oocytes.
Macropatch recordings
On-cell macropatch recordings were used to obtain Na+ current data shown in Figs. 1–9. The bath solution contained (in mM): NaCl 9.6, KCl 88, EGTA 11, HEPES 5, pH 7.4 and recording pipette solution contained (in mM): NaCl 96, KCl 4, MgCl2 1, CaCl2 1.8, HEPES 5, pH 7.4. Voltage clamping and data acquisition were done as previously described (36) using EPC-9 patch-clamp amplifiers (HEKA, Lambrecht, Germany) controlled via Pulse software (HEKA) run by Macintosh G4 computers.
FIGURE 1.

Families of currents recorded from hNaV1.4 and mutations at the negative charge cluster E-1314, E-1315 and the positive charge cluster K-1317, K-1318 as shown in the diagram at lower left. Calibration: 3 ms, 3 nA hNaV1.4; 1.5 nA E-1314K, E-1315K; 1 nA K-1317E, K-1318E; E-1314Q, E-1315Q/K-1317Q, K-1318Q, and E-1314K, E-1315K/K-1317E/K-1318E; 750 pA K-1317Q, K-1318Q, and K-1317R, K-1318R.
FIGURE 2.

g(V) relations and Boltzmann fits for hNaV1.4 and mutations obtained from experiments as shown in Fig. 1. Values represent the mean ± SE from 12–19 experiments.
FIGURE 3.

h∞ relations and Boltzmann fits for hNaV1.4 and mutations in the DIII-DIV linker. Channels were subjected to 500-ms prepulses to voltages shown before assessment of channel availability with −20-mV test pulses. Values represent the mean ± SE from 12–19 experiments.
FIGURE 4.

Kinetics of activation (A) and open-state deactivation (B) in hNaV1.4 and mutations of the DIII-DIV linker. (A) Time for 10%–90% peak amplitude of sodium currents. (B) Open-state deactivation kinetics obtained using the protocol shown as the inset; channels were opened with 0-mV, 0.5-ms conditioning pulses before 20-ms commands to evoke tail currents. Values represent the mean ± SE from 14–19 experiments (A) or 18–26 experiments (B).
FIGURE 5.

Open-state fast inactivation kinetics obtained from experiments as shown in Fig. 1. Values represent the mean ± SE from 12–19 experiments.
FIGURE 6.

Double pulse protocol (A) used to elicit current traces as shown in (B) after a −130-mV interpulse command. Normalized recovery (C) is plotted along with single exponential fits for hNaV1.4. Calibration for traces shown in A: 10 ms; hNaV1.4, 1.5 nA, KKRR 400 pA, KKQQ 300 pA, EEKK, and KKEE 250 pA.
FIGURE 7.

Voltage dependence for recovery time constants in hNaV1.4 and mutations of the DIII-DIV linker. Values represent the mean ± SE of 14–20 experiments.
FIGURE 8.

Voltage dependence for recovery delay measurements in hNaV1.4 and mutations of the DIII-DIV linker. Values represent the mean ± SE of 14–20 experiments.
FIGURE 9.

Eyring model of inactivation comprising time constants of entry into fast inactivation (Fig. 5), recovery (Fig. 7), and closed-state fast inactivation. Values of closed-state fast inactivation time constants represent the mean ± SE in 8–14 experiments and are given elsewhere for entry and recovery transitions.
Data in experiments using tail current protocols were acquired at 10 μs per point, and data in experiments using other protocols were acquired at 20 μs per point. Bath temperature was maintained at 15°C ± 0.1°C for all experiments with a Peltier device and HCC-100A temperature controller (Dagan, Minneapolis, MN). Oocyte holding potential was −120 mV between trials and changed to −150 mV immediately before each protocol. Leak (p/4) and capacitance subtractions were done after patch formation and corrected before each voltage clamp experiment. Analyses and graphing were done using PulseFit (HEKA) and Igor Pro (WaveMetrics, Lake Oswego, OR).
Conductance/voltage (g(V)) relationships were derived using Eq. 1:
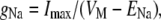 |
(1) |
where gNa is sodium conductance, Imax is calculated as peak current in response to the test pulse, VM is test pulse voltage, and ENa is the measured Na+ equilibrium potential. Activation (g(V)) and steady-state fast inactivation (h∞) curves were fit by a Boltzmann distribution according to Eq. 2:
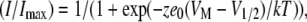 |
(2) |
where the normalized current amplitude I/Imax is measured from the response to the test pulse potential VM after a variable-voltage prepulse or from the holding potential, z is the apparent valence, e0 is elementary charge, V1/2 is midpoint voltage, k is the Boltzmann constant, and T is temperature in K.
Activation kinetics were determined by measuring the time for 10%–90% of peak inward current during a 20-ms depolarization from the holding potential to voltages ranging from −60 mV to 30 mV. Time constants of open-state fast inactivation were determined by fitting current decays to Eq. 3:
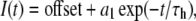 |
(3) |
where I(t) is current amplitude as a function of time, offset is plateau amplitude (asymptote), a1 is current amplitude at t = 0, and τh is the fast inactivation time constant.
Recovery from fast inactivation was measured using a double pulse protocol (37). To inactivate channels, 0-mV, 50-ms depolarizing pulses were used, followed by variable voltage and duration interpulses. For −190 mV to −130 mV, interpulse durations increased by 50-μs steps, and for −120 mV to −90 mV, 100-μs steps were used. These hyperpolarizing commands preceded a 10-ms recovery test pulse to 0 mV. Recovery currents were normalized to each initial depolarization, and recovery time constants were calculated from single exponential fits to recovery curves according to Eq. 3 with the parameters
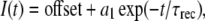 |
where τrec is the recovery time constant.
The kinetics of closed-state fast inactivation were determined from experiments in which channels were depolarized by prepulses of varying durations up to 300 ms and then subjected to 20-ms, −20-mV test pulses to assess channel availability. The decrement in peak current amplitude was normalized to the initial test pulse, and the decay in normalized current amplitude was fit with Eq. 3.
Descriptions of fast inactivation were further characterized by assuming a first-order, two-state Eyring model of the fast inactivation reaction (not inactivated ↔ inactivated) and fitting τ versus voltage curves to Eq. 4:
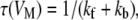 |
(4) |
where τ(VM) represents the time constant of progression to equilibrium of fast inactivation as a function of membrane potential, kf is the rate of the forward reaction, and kb is the rate of the reverse reaction. Reaction rates were determined according to Eqs. 5 and 6:
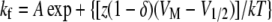 |
(5) |
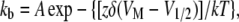 |
(6) |
where A is 1/2 rate at VO, z is the (apparent) total reaction valence (in electronic charge), δ is the fractional barrier distance, VM is membrane potential (in mV), V1/2 is the midpoint potential (in mV), k is the Boltzmann constant, and T is temperature in K.
Open-state deactivation kinetics, given as time constants (τD), were derived from the decay of tail currents using Eq. 3 with the following parameters:
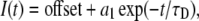 |
where τD is the deactivation time constant. We measured open-state deactivation at voltages from −190 mV to −70 mV. Over this voltage range, tail current decay in the DIII-DIV linker mutation IFM/QQQ was complete. Since fast inactivation is completely abolished in this mutation (36), complete decay of current at these voltages occurs as a consequence of deactivation.
To measure kinetics of inactivated-state deactivation, we used the double pulse protocol described above and extrapolated normalized recovery currents to the time at which current amplitude was zero, using a single exponential function. We calculated the x intercept of the recovery curve as the delay in the onset to recovery from fast inactivation (38).
Cut-open oocyte recordings
Oocytes were placed in a recording Perspex chamber, directly over a small opening surrounded by a Vaseline ring. The bottom thermal conductive chamber and the middle (guard) chamber were filled with external gating solution of the following composition (in mM): 120 NMG-MES (N-methyl D-glucamine, methanesulfonic acid), 10 HEPES, 2 Ca(OH)2, pH 7.4. Another chamber with a small Vaseline-lined opening was placed over the animal pole of the oocyte with gentle pressure. Glass electrodes containing platinum wires and filled with 3% agar in 500 mM NMG-MES were placed in the bottom, middle (guard), and top chambers. The other end of these electrodes was placed in a pool of 1 M NaCl containing chlorided silver wires connected to a Dagan CA-1B amplifier. Electrical access to the interior of the oocyte was obtained by perfusing the bottom of the oocyte with 0.5% saponin (Sigma) in internal solution composed of (in mM): 120 NMG-MES, 10 HEPES, 2 EGTA, pH 7.4. The animal pole was impaled with a borosilicate electrode filled with 3 M KCl, and membrane potential clamped to −100 mV.
Ionic currents were elicited by stepping membrane potential to voltages from −90 mV to 60 mV from a holding potential of −120 mV. From these experiments we calculated open-state fast inactivation kinetics from the decay of outward current flowing through sodium channels using Eq. 3.
Gating currents were isolated by adding 1 μM tetrodotoxin (TTX, Sigma) to the upper chamber, followed by a train of 20-ms step depolarizations to 0 mV. Charge/voltage (Q/V) relations were determined by integrating the outward gating current (IgON) to quantify charge movement. Charge immobilization was measured by integrating the fast component of the inward gating current (IgOFF) at the end of step depolarizations and comparing to IgON according to Eq. 7:
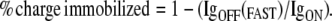 |
(7) |
Onset of gating charge immobilization was determined from experiments using variable duration pulses at voltages from −40 mV to 50 mV. The fast component of IgOFF was compared to IgON at pulse durations ranging from 0.25 ms to 15 ms using Eq. 7. Onset kinetics were determined by fitting the % charge immobilized curve to a single exponential function using Eq. 3.
Recovery of gating charge was determined in experiments using a double pulse protocol. The membrane was depolarized to 0 mV for 30 ms, followed by interpulses at voltages ranging from −120 mV to −70 mV for durations up to 50 ms, before a second depolarization to 0 mV. Recovery currents were determined by normalizing the IgON for first and second depolarizations of each sweep and remobilization parameters determined by fitting the recovery curve with Eq. 8:
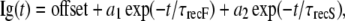 |
(8) |
where Ig(t) is normalized IgON as a function of time, offset is plateau amplitude (asymptote), a1 and a2 are normalized IgON at the beginning of the fast (F) and slow (S) phases of recovery, and τrecF and τrecS are the recovery time constants for gating charge remobilization.
Statistical significance was assessed with Instat 2.0 (GraphPad, San Diego, CA) using Student's unpaired t-tests or, in those cases where there was a statistically significant difference between standard deviations, Welch's alternative t-tests. Statistical significance of difference was accepted at p values ≤ 0.05. Significant differences between DIII-DIV linker mutants and hNaV1.4 for at least three consecutive voltages were taken as a significant difference in voltage dependence.
RESULTS
Macropatch recordings: conductance
To determine the role of charged residues proximal to the IFMT inactivation “particle”, mutations were constructed in human skeletal muscle sodium channel background (hNaV1.4) for two clusters of charge in the DIII-DIV linker (E-1314, E-1315 and K-1317, K-1318). These clusters are located on the C-terminal side of the IFMT inactivation particle (Fig. 1). Mutations at these clusters are denoted subsequently as the following: E-1314K, E-1315K = EEKK; K-1317Q, K-1318Q = KKQQ; K-1317R, K-1318R = KKRR; K-1317E, K-1318E = KKEE; E-1314Q, E-1315Q/K-1317Q, K-1318Q = EEQQ/KKQQ; E-1314K, E-1315K/K-1317E, K-1318E = EEKK/KKEE. In Fig. 1, families of currents elicited by step depolarization to voltages ranging from −90 mV to 60 mV from a holding potential of −150 mV are shown for wild-type and mutant channels.
Equilibrium properties of activation were calculated from Boltzmann fits to the normalized g(V) relations from these experiments. Charge reversal or substitution at K-1317, K-1318 significantly depolarized the midpoint of the g(V) curve (Table 1, Fig. 2 A). The quadruple mutations EEQQ, KKQQ and EEKK, KKEE also produced a depolarizing shift in V1/2 (Fig. 2 B). Apparent valence was decreased in KKQQ, KKEE, KKRR and EEKK, KKEE but was unaffected in EEKK and EEQQ, KKQQ. Boltzmann fits to the normalized steady-state fast inactivation curves (h∞) are shown in Fig. 3 and parameters given in Table 2. Mutations at K-1317, K-1318 significantly hyperpolarized the h∞ curve (effects largest in KKQQ) and decreased apparent valence (effects largest in KKRR). Similar effects were observed in EEQQ/KKQQ and EEKK/KKEE (Fig. 3 B), whereas EEKK was without effect on midpoint or valence of the h∞ curve. Taken together, our results suggest that positively charged residues near the IFMT inactivation particle influence equilibrium properties of sodium channel gating as a consequence of allosteric effects, not charge content.
TABLE 1.
Activation parameters for hNaV1.4 and DIII-DIV linker mutations
| Mutation | V1/2 (mV) | Activation valence | Rise time, 0 mV (μs) |
|---|---|---|---|
| hNaV1.4 | −37.5 ± 1.5 | 3.59 ± 0.10 | 198.9 ± 5.0 |
| EEKK | −34.0 ± 1.1 | 3.52 ± 0.09 | 216.0 ± 6.4* |
| KKQQ | −35.7 ± 0.8 | 3.27 ± 0.08* | 183.7 ± 4.0* |
| KKEE | −28.0 ± 1.0‡ | 2.99 ± 0.06‡ | 212.6 ± 6.0 |
| KKRR | −27.8 ± 1.4‡ | 2.95 ± 0.06‡ | 262.1 ± 11.0‡ |
| EEQQ/KKQQ | −33.6 ± 1.4* | 3.46 ± 0.1 | 210.6 ± 8.0 |
| EEKK/KKEE | −29.8 ± 1.1‡ | 3.20 ± 0.1† | 203.4 ± 6.2 |
p ≤ 0.05.
p ≤ 0.01.
p ≤ 0.001.
TABLE 2.
Inactivation parameters for hNaV1.4 and DIII-DIV linker mutations
| Mutation | V1/2 (mV) | Apparent valence | τH, 0 mV (ms) | τH, −80 mV (ms) | τH, −140 mV (ms) |
|---|---|---|---|---|---|
| hNaV1.4 | −88.4 ± 1.7 | 4.75 ± 0.17 | 0.44 ± 0.02 | 22.4 ± 1.5 | 1.73 ± 0.09 |
| EEKK | −93.4 ± 1.33* | 4.48 ± 0.14 | 0.51 ± 0.02* | 15.8 ± 1.3† | 2.45 ± 0.30* |
| KKQQ | −101.7 ± 1.38‡ | 3.46 ± 0.12† | 0.41 ± 0.03 | 9.1 ± 1.0‡ | 2.50 ± 0.15‡ |
| KKEE | −97.5 ± 1.50† | 3.64 ± 0.13‡ | 0.95 ± 0.05‡ | 4.2 ± 0.4‡ | 1.56 ± 0.11 |
| KKRR | −99.1 ± 2.22† | 2.41 ± 0.03‡ | 1.59 ± 0.09‡ | 4.9 ± 0.5‡ | 2.24 ± 0.16* |
| EEQQ/KKQQ | −97.0 ± 4.55† | 3.86 ± 0.39† | 0.49 ± 0.02 | 15.4 ± 1.2† | 2.89 ± 0.44* |
| EEKK/KKEE | −98.52 ± 6.50‡ | 3.76 ± 0.77† | 0.57 ± 0.03* | 10.5 ± 1.2† | 2.08 ± 0.12* |
p ≤ 0.05.
p ≤ 0.005.
p ≤ 0.0001.
Kinetics: activation, open-state deactivation, and fast inactivation
We sought to determine whether the charged residues surrounding the IFMT inactivation motif affected the rates of sodium channel gating transitions. Activation kinetics were determined as the 10%–90% rise time of Na+ currents from experiments as shown in Fig. 1. KKQQ significantly accelerated activation rise times at voltages from −50 mV to 20 mV (Fig. 4 A). In contrast, KKRR slowed activation at voltages more positive than −30 mV.
Open-state deactivation kinetics determined from time constants (τD) of tail current decays elicited by hyperpolarizing commands from −190 mV to −70 mV are shown in Fig. 4 B. KKEE and KKRR accelerated deactivation over a voltage range of −190 mV to −130 mV, and −150 mV to −90 mV, respectively. KKEE and the quadruple mutation EEKK/KKEE prolonged deactivation only at −80 mV and −70 mV compared to hNaV1.4. Other mutations were without effect on τD.
KKQQ accelerated open-state fast inactivation from −60 mV to −40 mV (Fig. 5 A). KKEE and KKRR elicited biphasic effects on open-state fast inactivation; τh was accelerated at −60 mV and −50 mV but was prolonged at voltages more positive than −30 mV. Thus, these two mutations reduced the apparent voltage dependence of open-state fast inactivation. EEKK slowed open-state fast inactivation at potentials more positive than −10 mV but did not dramatically alter voltage dependence (Fig. 5 B). Voltage dependence of open-state fast inactivation was not dramatically reduced by EEQQ/KKQQ or by EEKK/KKEE. These results suggest that charged residues in the linker modulate fast inactivation but are not essential to that process. Second, they suggest that a local interaction in the DIII-DIV linker is a determinant of the coupling of voltage-dependent activation to voltage-independent fast inactivation.
Recovery and recovery delay
Recovery from fast inactivation is preceded by a brief delay, indicating that channels must deactivate before they can recover from inactivation (37). Although we have previously shown that recovery delay is controlled, at least in part, by charged residues in the DIII and DIV voltage sensors, we wanted to determine whether those charges interact with the cluster of charges proximal to the IFMT motif. Recovery from fast inactivation and the delay preceding recovery were determined from a double pulse protocol (Fig. 6 A). Recovery currents (Fig. 6 B) were normalized to the peak amplitude in response to the first depolarization, as shown in Fig. 6 C.
Recovery and delay were unaffected by KKEE compared to hNaV1.4 (Figs. 7 A and 8 A, Table 2). KKQQ, KKEE, EEKK, EEQQ/KKQQ, and EEKK/KKEE slowed recovery (Fig. 7) and prolonged recovery delay (Fig. 8). In contrast, KKRR had biphasic effects on recovery. Recovery time constants were increased in KKRR compared to hNaV1.4 (slowed recovery) from −190 mV to −140 mV but were decreased (faster recovery) from −120 mV to −90 mV (Fig. 7 A). However, KKRR accelerated recovery delay at all voltages tested (Fig. 8 A) without altering the voltage-dependent nature of that transition. Thus, whereas most mutations produced effects on recovery predictable from recovery delay measurements, effects of KKRR on recovery were partially uncoupled from the deactivating transition.
Closed-state fast inactivation and Eyring model of fast inactivation
Residues in the DIV voltage sensor have been demonstrated to link activation to fast inactivation. We wanted to determine whether clusters of charged residues in the DIII-DIV linker near the inactivation “particle” participate in a dialog between the voltage sensor and the inactivation gate. Closed-state fast inactivation time constants were determined as stated in Materials and Methods and plotted along with time constants obtained for open-state fast inactivation (Fig. 5) and for recovery (Fig. 7). The resulting curve shown in Fig. 9 represents the voltage dependence of hNaV1.4 and charge cluster mutations for transit between fast inactivated (FI) and nonfast inactivated states. An Eyring model of FI/non-FI applied to these curves yielded values for reaction midpoint, valence, and fractional barrier distance, as given in Table 3. Parameters were most dramatically affected by KKRR and KKEE to hyperpolarize the reaction midpoint, decrease valence, and lower barrier distance. Thus, in KKRR and KKEE the kinetics of fast inactivation from closed and open states were relatively similar, unlike hNaV1.4. One interpretation of these results is that the cluster of positive charges around the IFMT motif interact with voltage-sensitive regions of the sodium channel to modulate fast inactivation but that such an interaction is not electrostatic in nature.
TABLE 3.
Parameters of two-state model of fast inactivation
| Mutation | Reaction midpoint (mV) | Maximum τ (ms) | Reaction valency (z) | Fractional barrier distance (δ) |
|---|---|---|---|---|
| hNaV1.4 | −85.4 | 23.3 | 1.99 | 0.49 |
| EEKK | −88.4 | 20.3 | 1.81 | 0.47 |
| KKQQ | −92.0 | 14.3 | 1.82 | 0.45 |
| KKEE | −107.0 | 6.2 | 1.28 | 0.65 |
| KKRR | −106.4 | 6.3 | 0.75 | 0.67 |
| EEQQ/KKQQ | −83.6 | 21.1 | 1.80 | 0.39 |
| EEKK/KKEE | −89.5 | 14.8 | 1.79 | 0.46 |
Charge movement and immobilization
To determine more closely the role of the cluster of charged residues near the inactivation “particle”, we measured the effects of linker mutations on charge movement and charge immobilization as more precise indicators of the movement of S4 voltage sensors during gating transitions. Gating currents were obtained with the cut-open oocyte technique after blocking ionic current with 1 μM TTX (Fig. 10 A). Q(V) relations were determined from experiments in which channels were depolarized from −90 mV to 60 mV from a holding potential of −120 mV. The midpoint of the Q(V) curve was left shifted in KKEE (−48.4 mV) compared to hNaV1.4 (−39.6 mV), whereas the Q(V) midpoint for each of the other mutations tested were unaffected (EEKK, −43.1 mV; KKQQ, −39.5 mV; KKRR, −41.5 mV; EEKK/KKEE, −38.7; Fig. 10 B).
FIGURE 10.

(A) ON and OFF gating currents measured from oocytes in the cut-open configuration in the presence of 1 μM TTX. Currents were evoked from a holding potential of −120 mV. Calibration: 5 ms; hNaV 1.4 1 μA, KKQQ 600 nA, KKRR 400 nA, EEKK 300 nA, KKEE 200 nA. Charge/voltage (B) and % charge immobilization (C) relations were obtained from experiments as shown in A. Charge immobilization was calculated as 1 − [IgOFF FAST/IgON FAST]. Values represent mean ± SE from 6–12 experiments.
The voltage dependence of charge immobilization is shown in Fig. 10 C. Boltzmann fits to the voltage dependence of charge immobilization indicated a left shift for KKQQ (−64.1 mV), KKEE (−65.1 mV), and EEKK (−57.8 mV) compared to hNaV1.4 (−50.5 mV), whereas EEKK/KKEE produced a right shift (−41.5 mV). The voltage dependence of charge immobilization in KKRR could not be fit to a Boltzmann distribution. At depolarized potentials, slightly less charge was immobilized in KKEE, KKQQ, and EEKK/KKEE compared to hNaV1.4 and <20% of the charge was immobilized in KKRR.
Since charge immobilization occurs concomitantly with the onset of fast inactivation, we were interested in determining kinetics of charge immobilization in the linker mutations, especially those that significantly affected the rate and voltage dependence of open-state fast inactivation. Onset of charge immobilization was determined using variable voltage and duration depolarizing pulses (Fig. 11 A). Time constants of charge immobilization onset were determined from exponential fits to the immobilization curves as shown in the examples for hNaV1.4 (Fig. 11 B) and KKEE (Fig. 11 C).
FIGURE 11.

Onset of gating charge immobilization. (A) Protocol used to obtain IgON and IgOFF gating currents in experiments shown below. (B, C) Plots of % charge immobilized in hNaV1.4 and linker mutation KKEE as a function of pulse duration. Charge immobilization was calculated as 1 − [IgOFF FAST/IgON FAST] in experiments in which the duration of the depolarizing step was varied from 0.2 ms to 15 ms, at depolarizing pulse voltages shown. Values represent the mean of 8–14 experiments in B and C.
Fig. 12 shows a comparison of time constants of ionic current decay obtained in the cut-open configuration to the onset of charge immobilization. These experiments revealed that the charge was immobilized in hNaV1.4 before fast inactivation. At voltages from −20 mV to 50 mV, charge immobilized with time constants on average 55% less than observed for open-state fast inactivation. EEKK and EEKK/KKEE also immobilized charge before fast inactivation. In contrast, similar kinetics for charge immobilization and fast inactivation were observed in mutations at K-1317, K-1318. In KKRR and KKEE the voltage dependencies of fast inactivation and of charge immobilization were shallow compared to those for hNaV1.4.
FIGURE 12.

Relationship of time constants of fast inactivation of ionic currents in the cut open configuration (open circles) to those for onset of charge immobilization (solid circles) after addition of 1 μM TTX.
These results confirmed the findings from ionic current measurements that suggest the cluster of charged amino acids near the inactivation “particle” affect voltage-dependent gating transitions associated with S4 movement. Linker charges do not appear to influence S4 movement via electrostatic interaction of voltage sensor charges, however. Charge-conserving mutations were equally effective in terms of disrupting charge movement and charge immobilization. In addition, the effect of EEKK/KKEE to restore voltage dependence of fast inactivation entry and charge immobilization onset compared to KKEE suggest that a local interaction in the DIII-DIV linker contributes to these effects.
Recovery of gating charge
The rate of charge remobilization is indicative of the time required for voltage sensors to return to their hyperpolarized-favored positions after immobilization. To determine whether the cluster of charged residues near the IFMT inactivating motif controls S4 remobilization, we measured the recovery of gating charge in hNaV1.4 and mutant channels with a double pulse protocol. We compared the initial IgON evoked with a 0-mV, 30-ms depolarization to recovering IgON elicited with a second depolarization after a variable voltage and duration interpulse.
Remobilization of the gating charge is shown in Fig. 13 in hNaV1.4 and linker mutations after an interpulse voltage of −80 mV. Time courses of charge recovery for hNaV1.4 and mutations of charged residues in the DIII-DIV linker are shown in Fig. 14. At −80 mV, 46.9% of the charge remobilized quickly in hNaV1.4, presumably reflecting the nonimmobilizable component of gating charge. For other mutations, that component was 50.4% (EEKK), 54.9% (KKEE), 61.3% (KKQQ), 66.6% (EEKK/KKEE), and 92.7% (KKRR). Recovery of the second, immobilizable component of the gating charge was analyzed with respect to time constant (τrecS). At −80 mV, the second component recovered with a time constant of 20.9 ms. For other mutations, recovery τrecS values were 14.3 ms (EEKK), 12.0 ms (KKEE), 23.8 ms (KKQQ), and 14.4 ms (EEKK/KKEE). In contrast, remobilization of the gating charge in KKRR was dominated by a single rate of recovery. These results confirm that the cluster of charges near the inactivation “particle” control the recovery of voltage sensors to their hyperpolarized-favored positions but that this control is allosteric rather than electrostatic.
FIGURE 13.

Remobilization of gating charge using a double pulse protocol. Initial IgON evoked with a 0-mV, 30-ms depolarization is shown along with recovering IgON elicited with a second 0-mV depolarization for 10 ms, after a variable voltage (−80 mV in the experiments shown here) and duration interpulse. Calibration for traces shown in (A) for IgON: 5 ms; hNaV1.4 500 nA, EEKK/KKEE 300 nA, EEKK 200 nA, KKRR 150 nA, KKRR 100 nA.
FIGURE 14.

Plots of recovery of IgON for hNaV1.4 and DIII-DIV linker charge mutations. TTX-blocked channels were depolarized to 0 mV for 30 ms, subjected to interpulse voltages for durations up to 50 ms, and charge remobilization determined from the response to second depolarization for 10 ms. Values represent the mean in 8–12 experiments.
DISCUSSION
This study was motivated by our earlier finding that effects of mutations at the negative cluster of charge EE-1314,15 on deactivation were dependent on alteration of charge (33). Specifically, charge reversal at this locus slowed the deactivating transition from the inactivated state. One interpretation from that study is that the positive charge added to the linker slows the return of the “inactivated” voltage sensors to the hyperpolarized position before channel recovery by an electrostatic repulsion between linker and voltage sensor charges. We hypothesized that native positive charge in the DIII-DIV linker might be the origin of charge immobilization of DIIIS4 or DIVS4 (11) and tested that hypothesis by comparing effects of charge reversing, neutralizing, and substituting mutations at KK-1317,18. We found that this cluster of positive charge in the linker affects gating charge and its immobilization and remobilization, but not as a consequence of electrostatic interaction with the voltage sensors. We also found that this cluster couples the voltage dependencies of activation, fast inactivation, and charge immobilization.
Role of linker charges in channel gating
Previous studies have shown that charge content in the DIII-DIV linker of sodium channels is not essential for fast inactivation (31–33,39). Instead, an IFMT motif flanked by clusters of positive and negative charge is requisite for fast inactivation (16–19,23). The most consistent effect reported for mutations that alter charge content in the DIII-DIV linker is modulation of open-state fast inactivation. In general, reduction of charge accelerates the entry of channels into the fast-inactivated state (31,39), whereas addition of positive charge slows the entry of channels into the fast-inactivated state (33). Thus, although fast inactivation is not directly voltage dependent, charge content of the DIII-DIV linker modulates the kinetics of fast inactivation, perhaps via interactions with the S4 voltage sensors.
In this study we found that charge neutralization at K-1317, K-1318 accelerated open-state fast inactivation. However, additional mutations at this residue to reverse or substitute charge (KKEE, KKRR) produced unexpected effects on fast inactivation. Each of these mutations slowed fast inactivation and uncoupled the voltage dependence between activation and fast inactivation. Mutations of a cluster of tyrosine residues adjacent to K-1317, K-1318 (18,40) or of nearby lysine residues (31) also reduce the voltage dependence of open-state fast inactivation. Our data are thus consistent with previous results showing that this region of the DIII-DIV linker comprises an important determinant of the mechanism by which activation is coupled to fast inactivation.
Each of the mutations tested in this study hyperpolarized the steady-state fast inactivation curve. The greatest effect was observed in KKQQ, which elicited a 13-mV hyperpolarization of midpoint. Stabilization of the fast-inactivated state by KKQQ was also exhibited by slower recovery and faster onset of closed-state and open-state fast inactivation. KKEE and KKRR also hyperpolarized the h∞ curve by ∼10 mV but depolarized the g(V) curve; these mutations thus uncoupled the voltage dependencies of the equilibrium parameters for activation and fast inactivation. In contrast to KKQQ, KKEE and KKRR slowed open-state fast inactivation. Therefore, their effects on steady-state fast inactivation are most likely dominated by a significant acceleration of closed-state fast inactivation. That effect may also explain the depolarizing effect of KKEE and KKRR on the g(V) curve by inactivating channels directly from the closed state.
Deactivation and charge immobilization
Deactivating transitions from the open and inactivated states are dissimilar in kinetics, such that the delay in the onset of recovery from fast inactivation is markedly slower than the decay in tail currents from open channels (38). A comparison of deactivation kinetics in DIVS4 mutations suggest that charge immobilized during fast inactivation dictates the kinetics of the two phases of recovery, deactivation, and unbinding of the IFMT motif (38,41). Recovery of channels is tightly coupled to remobilization of the DIVS4 voltage sensor (41), and effects of mutations on tail currents (open-state deactivation) predict effects of mutations on recovery delay (inactivated-state deactivation) in the absence of other gating perturbations (38). Thus, we have speculated that tail currents and recovery delay reflect deactivating transitions and that their kinetic disparity is a consequence, at least in part, of the immobilization of charge incurred with fast inactivation.
Interestingly, one of the mutations in this study dramatically affected recovery in a way that would not be predicted by its effect on remobilization of gating charge; although KKRR accelerated charge remobilization and recovery delay, this mutation paradoxically slowed recovery of channels from the fast-inactivated state. We interpret these effects as a partial uncoupling of the two steps of recovery: voltage sensor translocation and unbinding of the IFMT motif. Thus, deactivation from the inactivated state is a steeply voltage-dependent process that may drive an apparent voltage-dependent recovery, analogous to the coupling of activation and fast inactivation.
We also found a rapid remobilization of charge in certain linker mutations (KKQQ, EEKK/KKEE) that actually prolongs recovery delay. We speculate that effects of linker mutations at K-1317, K-1318 are twofold: they accelerate the movement of the S4 voltage sensors during remobilization of the gating charge but differentially influence the unbinding of the IFMT motif or other gating transitions (i.e., passage through closed states) to effect the return of channels to an immediately available state. Voltage sensor mutations, on the other hand, may affect charge remobilization but not IFMT binding, such that charge remobilization dictates deactivation and recovery predictably.
Mechanism for coupling of activation to fast inactivation
The gating particles proposed by Hodgkin and Huxley (42) drive independent permissiveness, or activation, and nonpermissiveness, or inactivation. Their gating scheme has been modified in light of studies that show that the voltage-dependent nature of fast inactivation is a consequence of coupling to activation (43–45), although it must be noted that other studies suggest that fast inactivation has at least a limited voltage sensitivity independent of activation (46,47). Structural determinants of the coupling between sodium channel activation and fast inactivation have been identified as the outermost arginine of the domain IV voltage sensor (27,28) and a pair of tyrosine residues flanking the IFMT motif (18,40). Our findings indicate that the KK-1317,18 cluster adjacent to these tyrosine residues also plays a role in the coupling between activation and fast inactivation. Further, we show that uncoupling of these processes with mutagenesis of these two lysine residues similarly reduces the voltage dependence of the onset of charge immobilization.
Several findings lend support to the idea that a local interaction of the linker is an important aspect of the coupling process. The quadruple mutations EEKK/KKEE and EEQQ/KKQQ “recoupled” the voltage dependencies of activation and fast inactivation. Second, the EEKK/KKEE mutation restored the voltage-dependent onset of charge immobilization. Finally, this mutation restored the wild-type choreography of charge immobilization and fast inactivation: that charge is immobilized before fast inactivation. Our findings suggest that a local interaction within the DII-DIV linker, mediated by the cluster of charged residues adjacent to the IFMT motif, is an important structural determinant of coupling between activation, fast inactivation, and charge immobilization.
We used the EEKK/KKEE mutation to test for a local electrostatic interaction as the determinant in the effects of mutations (specifically KKEE) on fast inactivation and charge immobilization. We expected additive effects of EEKK and KKEE in the quadruple mutation if these residues act independently, and a wild-type phenotype if these residues interact electrostatically, since this mutation swapped rather than altered charge. This mutation restored, albeit incompletely, the voltage dependencies of fast inactivation and onset of charge immobilization and restored the wild-type phenotype of charge immobilization before entry into fast inactivation. Therefore, we propose that electrostatics are important in the local structure of the channel and that this local structure is a key determinant in the coupling of activation to fast inactivation and charge immobilization.
An alternative but not exclusive hypothesis is that these positive charges K-1317, K-1318 interact with local tyrosine residues Y-1319, Y-1320 through cationic interaction with π orbital electrons (48). Such a cation-π interaction has been elegantly demonstrated in TTX-selective binding in NaV1.4 (49). In a separate study it was shown that mutations of the Y-1319, Y-1320 cluster removing aromaticity at this cluster “uncouple” activation to fast inactivation, whereas aromatic amino acid substitutions do not (40). The solution structure of this portion of the inactivation gate suggests a stable helical conformation (19). That structure is consistent with a putative cation-π interaction between tyrosine and lysine residues. Thus, mutations at K-1317, K-1318 could disrupt such an interaction. However, substitution of lysine for arginine at K-1317, K-1318 might be expected to strengthen such an interaction (48). Therefore, at present our results suggest only that a local, electrostatic interaction between negative and positive clusters proximal to the IFMT motif play an important role in the impact of the local conformation in the linker on fast inactivation and its coupling to the activation process.
Interaction of linker residues with voltage sensors
Our results raise interesting possibilities about the relationship of fast inactivation and charge immobilization. One interpretation is that since each process is similarly affected by mutagenesis, charge immobilization arises as a consequence of channel inactivation. In squid giant axon, the time constant of fast inactivation parallels the development of charge immobilization (29). However, we find that in skeletal muscle channels, charge is immobilized before fast inactivation. With mutagenesis at K-1317, K-1318, the kinetics of fast inactivation more closely parallel the development of charge immobilization. These results may be explained by domain-specific perturbation of voltage sensor movement by the linker mutations. Although both DIII and DIV voltage sensors are immobilized during the course of fast inactivation (11), immobilization in DIVS4 is independent of binding of the IFMT motif whereas binding is requisite for immobilization of DIIIS4 (50,51). We speculate that charge reversing or substituting mutations at K-1317, K-1318 restrict the movement of DIVS4 such that DIIIS4 movement is the rate-limiting substrate of charge immobilization after binding of the inactivation particle. Alternatively, the enhanced rate of closed-state fast inactivation in KKRR and KKEE may underlie their relative effects on fast inactivation and charge immobilization kinetics, if charge is not effectively immobilized when channels do not open before inactivating.
The exact molecular basis for charge immobilization is not certain, but our findings indicate that a direct electrostatic interaction between the positive charge cluster and charged residues in the voltage sensors is unlikely. Interestingly, in previous work we found that charge-altering mutations of central residues R-4 and R-5 in both DIIIS4 and DIVS4 produce significant effects on recovery delay that are based in structural alteration, rather than in charge reduction (52,53). Charge-altering mutations of central residues in DIVS4 profoundly affect charge immobilization (41,53). These findings lead us to speculate that charged residues in the DIII-DIV linker and in voltage sensors interact allosterically with each other or with a common determinant of charge immobilization.
Countercharges responsible for stabilization of DIVS4 have been proposed for charge immobilization of that voltage sensor (41). Interactions of residues in DIVS3 and DIVS4 have been experimentally verified (54), and mutations in DIVS3 affect recovery delay. A role for pore residues as countercharges for DIVS4 (55) may explain our finding that charge is immobilized more effectively during open-state fast inactivation than during closed-state fast inactivation.
Acknowledgments
The authors thank J. Abbruzzese (Utah State University) for comments on a draft of this manuscript and A Correa (University of California, Los Angeles) for help with the cut-open oocyte technique.
This work was supported by National Science Foundation RUI 023558 to J.R.G., National Institutes of Health R01 NS29204 to P.C.R., and National Institutes of Health P20 RR016454 INBRE to I.S.U.
Editor: Francisco Bezanilla.
References
- 1.Armstrong, C. M., and B. Hille. 1998. Voltage-gated ion channels and electrical excitability. Neuron. 20:371–380. [DOI] [PubMed] [Google Scholar]
- 2.Catterall, W. A. 2000. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 26:13–25. [DOI] [PubMed] [Google Scholar]
- 3.Noda, M., S. Shizimu, T. Tanabe, T. Takai, T. Kayano, T. Ikeda, H. Takahashi, Y. Nakayama, N. Kanaoka, N. Minamino, K. Kangawa, K. Matsuo, H. Raftery, M. Hirose, T. Inayama, H. Hayashida, T. Miyata, and S. Numa. 1984. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 312:121–127. [DOI] [PubMed] [Google Scholar]
- 4.Goldin, A. L., R. L. Barchi, J. H. Caldwell, F. Hofmann, J. R. Howe, J. C. Hunter, R. G. Kallen, G. Mandel, M. H. Meisler, Y. B. Netter, M. Noda, M. M. Tamkun, S. G. Waxman, J. N. Wood, and W. A. Catterall. 2000. Nomenclature of voltage-gated sodium channels. Neuron. 28:365–368. [DOI] [PubMed] [Google Scholar]
- 5.Yu, F. H., and W. A. Catterall. 2003. Overview of the voltage-gated sodium channel family. Genome Biol. 4:207.1–207.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang, N., and R. Horn. 1995. Evidence for voltage-dependent movement in sodium channels. Neuron. 15:213–216. [DOI] [PubMed] [Google Scholar]
- 7.Yang, N., A. L. George Jr., and R. Horn. 1996. Molecular basis of charge movement in sodium channels. Neuron. 16:213–218. [DOI] [PubMed] [Google Scholar]
- 8.Mitrovic, N., A. L. George Jr., and R. Horn. 1998. Independent versus coupled inactivation in sodium channels. Role of the domain 2 S4 segment. J. Gen. Physiol. 111:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horn, R., S. Ding, and H. J. Gruber. 2000. Immobilizing the moving parts of ion channels. J. Gen. Physiol. 116:461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cestele, S., V. Yarov-Yarovoy, Y. Qu, F. Sampieri, T. Scheuer, and W. A. Catterall. 2006. Structure and function of the voltage sensor of sodium channels probed by a β-scorpion toxin. J. Biol. Chem. 281:21332–21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha, A., P. C. Ruben, A. L. George Jr., E. Fujimoto, and F. Bezanilla. 1999. Voltage sensors in domains III and IV, but not in I and II, are immobilized by Na+ channel fast inactivation. Neuron. 22:73–87. [DOI] [PubMed] [Google Scholar]
- 12.Chanda, B., and F. Bezanilla. 2002. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 120:629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuhmer, W., F. Conti, H. Suzuki, X. Wang, M. Noda, N. Yahagi, H. Kubo, and S. Numa. 1989. Structural parts involved in activation and inactivation of the sodium channel. Nature. 339:597–604. [DOI] [PubMed] [Google Scholar]
- 14.Kontis, K. J., A. Rounaghi, and A. L. Goldin. 1997. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol. 110:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groome, J. R., E. Fujimoto, A. L. George Jr., and P. C. Ruben. 1999. Differential effects of homologous S4 mutations in human skeletal muscle sodium channels on deactivation gating from open and inactivated states. J. Physiol. 516:687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vassilev, P. M., T. Scheuer, and W. A. Catterall. 1988. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 241:1658–1661. [DOI] [PubMed] [Google Scholar]
- 17.West, J. W., D. E. Patton, T. Scheuer, Y. Wang, A. L. Goldin, and W. A. Catterall. 1992. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. USA. 89:10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kellenberger, S., J. W. West, W. A. Catterall, and T. Scheuer. 1997. Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. J. Gen. Physiol. 109:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohl, C. A., F. A. Boeckman, C. Baker, T. Scheuer, W. A. Catterall, and R. E. Klevit. 1999. Solution structure of the sodium channel inactivation gate. Biochemistry. 38:855–861. [DOI] [PubMed] [Google Scholar]
- 20.Tang, L., R. G. Kallen, and R. Horn. 1996. Role of an S4–S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J. Gen. Physiol. 108:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerche, H., W. Peter, R. Fleischhauer, U. Pika-Hartlaub, T. Malina, N. Mitrovic, and F. Lehmann-Horn. 1997. Role in fast inactivation of the IV/S4–S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J. Physiol. 505:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith, M. R., and A. L. Goldin. 1997. Interaction between the sodium channel inactivation linker and domain III S4–S5. Biophys. J. 73:1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filatov, G. N., T. P. Nguyen, S. D. Kraner, and R. L. Barchi. 1998. Inactivation and secondary structure in the D4/S4–5 region of the SkM1 sodium channel. J. Gen. Physiol. 111:703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPhee, J. C., D. S. Ragsdale, T. Scheuer, and W. A. Catterall. 1998. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel β-subunit in fast inactivation. J. Biol. Chem. 273:1121–1129. [DOI] [PubMed] [Google Scholar]
- 25.Popa, M. O., A. K. Alekov, S. Bail, F. Lehmann-Horn, and H. Lerche. 2004. Cooperative effect of S4–S5 loops in domains D3 and D4 on fast inactivation of the Na+ channel. J. Physiol. 561:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheets, M., J. W. Kyle, R. G. Kallen, and D. A. Hanck. 1999. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chahine, M., A. L. George Jr., M. Zhou, S. Ji, W. Sun, R. L. Barchi, and R. Horn. 1994. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 12:281–294. [DOI] [PubMed] [Google Scholar]
- 28.Chen, L.-Q., V. Santarelli, R. Horn, and R. G. Kallen. 1996. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 108:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong, C. M., and F. Bezanilla. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meves, H., and W. Vogel. 1977. Inactivation of the asymmetrical displacement current in giant axons of Loligo forbesi. J. Physiol. 267:377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patton, D. E., J. W. West, W. A. Catterall, and A. L. Goldin. 1992. Amino acid residues required for fast Na+ channel inactivation: charge neutralizations and deletions in the III–IV linker. Proc. Natl. Acad. Sci. USA. 89:10905–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller, J. R., M. K. Patel, J. E. John, J. P. Mounsey, and J. R. Moorman. 2000. Contributions of charged residues in a cytoplasmic linking region to Na channel gating. Biochim. Biophys. Acta. 1509:275–291. [DOI] [PubMed] [Google Scholar]
- 33.Groome, J. R., E. Fujimoto, and P. C. Ruben. 2003. Negative charges in the DIII-DIV linker of skeletal muscle Na+ channels regulate deactivation gating. J. Physiol. 548:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groome, J. R., J. L. Abbruzzese, E. Fujimoto, and P. C. Ruben. 2004. Charged residues in the DIII-DIV linker of hNaV1.4 couple charge immobilization to activation. Biophys. J. 611–Pos. (Abstr.)
- 35.Ho, H. N., H. D. Hunt, R. M. Morton, K. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 77:51–59. [DOI] [PubMed] [Google Scholar]
- 36.Featherstone, D. E., E. Fujimoto, and P. C. Ruben. 1998. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J. Physiol. 506:627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo, C.-C., and B. P. Bean. 1994. Na+ channels must deactivate to recover from inactivation. Neuron. 12:819–829. [DOI] [PubMed] [Google Scholar]
- 38.Groome, J. R., E. Fujimoto, and P. C. Ruben. 2000. The delay in recovery from fast inactivation in skeletal muscle sodium channels is deactivation. Cell. Mol. Neurobiol. 20:521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moorman, J. R., G. E. Kirsch, A. M. Brown, and R. H. Joho. 1990. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science. 250:688–691. [DOI] [PubMed] [Google Scholar]
- 40.O'Leary, M. E., L.-Q. Chen, R. G. Kallen, and R. Horn. 1995. A molecular link between activation and inactivation of sodium channels. J. Gen. Physiol. 106:641–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhn, F. J. P., and N. G. Greeff. 1999. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J. Gen. Physiol. 114:167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodgkin, A. L., and A. F. Huxley. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bezanilla, F., and C. M. Armstrong. 1977. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 70:549–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aldrich, R. W., and C. F. Stevens. 1987. Voltage-dependent gating of single sodium channels from mammalian neuroblastoma cells. J. Neurosci. 7:418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cota, G., and C. M. Armstrong. 1989. Sodium channel gating in clonal pituitary cells. The inactivation step is not voltage dependent. J. Gen. Physiol. 94:213–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horn, R., and C. A. Vandenberg. 1984. Statistical properties of single sodium channels. J. Gen. Physiol. 84:505–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stimers, J. R., F. Bezanilla, and R. E. Taylor. 1985. Sodium channel activation in the squid giant axon. Steady state properties. J. Gen. Physiol. 85:65–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallivan, J. P., and D. A. Dougherty. 1999. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA. 96:9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santarelli, V. P., A. L. Eastwood, D. A. Dougherty, R. Horn, and C. A. Ahern. 2007. A cation-pi interaction discriminates among sodium channels that are either sensitive or resistant to tetrodotoxin block. J. Biol. Chem. 282:8044–8051. [DOI] [PubMed] [Google Scholar]
- 50.Sheets, M. F., J. W. Kyle, and D. A. Hanck. 2000. The role of the putative inactivation lid in sodium channel gating current immobilization. J. Gen. Physiol. 115:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheets, M., and D. A. Hanck. 2005. Charge immobilization of the voltage sensor in domain IV is independent of sodium current inactivation. J. Physiol. 563:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51a.Groome, J. R., E. Fujimoto, L. Walter, and P. C. Ruben. 2002. Outer and central charged residues in DIVS4 of skeletal muscle sodium channels have differing roles in deactivation. Biophys. J. 82:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groome, J. R., H. M. Alexander, E. Fujimoto, M. Sherry, and D. Petty. 2007. Central charged residues in DIIIS4 regulate deactivation gating in skeletal muscle sodium channels. Cell. Mol. Neurobiol. 27:87–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruben, P. C., J. Cha, J. L. Abbruzzese, E. Fujimoto, and F. Bezanilla. 1999. DIVS4 mutations in rSkM1 and hSkM1 sodium channels uncouple charge immobilization from fast inactivation. Biophys. J. 76–194. (Abstr.)
- 54.Ji, S., A. L. George Jr., R. Horn, and R. L. Barchi. 1996. Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSkM1 sodium channel gating. J. Gen. Physiol. 107:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuhn, F. J. P., and N. G. Greeff. 2002. Mutation D384N alters recovery of the immobilized gating charge in rat brain IIA sodium channels. J. Membr. Biol. 185:145–155. [DOI] [PubMed] [Google Scholar]