

Abstract
Lipid bilayers are two-dimensional fluids. Here, the effect of monovalent ion concentration on the mixing, and consequently the organization, of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) bilayers has been examined. Epifluorescence microscopy was used to visualize the organization. Fluorescence recovery after photobleaching and attenuated total reflection-Fourier transform infrared spectroscopy were used to assess the fluidity of the lipids. At high ionic strength the DOPC and DOPA lipids appear uniformly mixed. Upon lowering the ionic strength, rapid separation is observed. The DOPA-rich regions appear fractal-like and exhibit hysteresis in their properties. The lipids freely exchange between the two regions. These experiments clearly demonstrate the significant effect that electrostatics can have on membrane organization.
INTRODUCTION
In cell membranes, lipids have two primary functions: to act as a semipermeable barrier and to provide a location for the integral membrane proteins to reside in. The extent to which lipids have other functions is a largely open question. One area of considerable interest regards whether lipids are in any way organized. In particular, “lipid rafts” have received considerable attention due to the possibility that they may concentrate important signaling proteins (1,2). Model system work clearly shows that ternary mixtures of saturated lipids, unsaturated lipids, and cholesterol separate to form liquid-ordered/liquid-disordered regions (3). This article will focus instead on the effect that electrostatics have on organization, an area that has been less well explored. Ion concentrations fluctuate in cells and may provide a means to concentrate membrane components and a mechanism to switch between two states of activity (4).
The effect of ion concentration on membrane organization has been examined by a number of groups using indirect methods, e.g., differential scanning calorimetry, pyrene excimer/monomer ratios, and Monte Carlo simulations (5–9), among others. The aim of this article is to examine the effect of ion concentration on the organization of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) bilayers with epifluorescence microscopy. Microscopy allows the direct visualization of organization, or lack thereof, and can thus provide critical information about the effect of electrostatics on organization.
Mixtures of phosphatidylcholine (PC) and phosphatidic acid (PA) were chosen for the following reasons: the pKa1 and pKa2 of PA are close to physiological conditions, PC is the most commonly occurring zwitterionic lipid, and PA can be created from PC by the action of phospholipase D. We further chose to work with PC/PA mixtures containing dioleoyl tails because these lipids should remain fluid (the physiologically relevant phase) at room temperature. Additionally, the pKas of DOPA in DOPC were recently determined: for 10 mol % DOPA in DOPC, pKa1 = 3.2, pKa2 = 7.9 (10).
In brief, previous experimental work on PC/PA mixtures has shown the following: Ionic strength and pH both affect the chain melting temperature of PA (5,6). The charge on PA is highly sensitive to changes in monovalent electrolyte concentration (8). When PA is mixed with PC, the charge on the PA headgroup affects the miscibility of the two components (7). In the particular case of DMPA/DPPC (1,2-dimyristoyl-sn-glycero-3-phosphate 1,2 dihexadecanoyl-sn-glycerol-3-phosphocholine) mixtures, at pH 4, there is indirect evidence that a fluid-fluid immiscibility occurs (7). The presence of this immiscibility is attributed to the reduction in repulsion between the PA molecules along with an increase in hydrogen-bonding between the PA lipids. In this article, direct observation of ion-induced reorganization of DOPC/DOPA bilayers will be shown. At high ionic strengths the lipids appear, to the level of resolution, uniformly mixed; but when the concentration of monovalent salt is decreased (75 mM or less), rapid separation occurs.
MATERIALS AND METHODS
Materials
Chloroform stock solutions of DOPC, DOPA, and 1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine (NBD-PC) were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. 2-(N-Morpholino)ethanesulfonic acid hydrate (MES hydrate), phosphate-buffered saline (PBS) tablets, and EDTA were purchased from Sigma Chemical (Poole, Dorset, UK). Potassium chloride (KCl) and potassium hydroxide (KOH) were purchased from Mallinckrodt Chemicals (Phillipsburg, NJ). Alexa Fluor 647 carboxylic acid, succinimidyl ester was purchased from Invitrogen (Carlsbad, CA). The Superdex 200 gel-filtration column was purchased from GE Healthcare Life Sciences, and the Microcon YM-100 centrifugal filter units were obtained from Millipore (Bedford, MA).
The buffer used for lipid rehydration and fusion was composed of 50 mM MES hydrate, 0.1 mM EDTA, and 250 mM KCl, adjusted to pH 5.0 with concentrated KOH. Fluid-fluid separation was initiated using an exchange buffer composed of 50 mM MES hydrate, 0.1 mM EDTA, and 0–100 mM KCl, pH 5.0.
Vesicle and supported lipid bilayer preparation
Large unilamellar vesicles (LUVs) were prepared by the extrusion method. Briefly, the chloroform-solvated lipids were mixed at the appropriate molar ratios, dried under nitrogen, and held under vacuum for 1 h. The dried lipids were rehydrated in the 50 mM MES hydrate, 0.1 mM EDTA, and 250 mM KCl buffer, pH 5.0. The lipid suspension was then extruded 21 times through a polycarbonate membrane with 50-nm pores. Following extrusion, the LUV solution was centrifuged for 5 min at 14,000 rpm (Eppendorf Minispin Plus, Westbury, NY). The extruded vesicles were stored at 20°C, shielded from light, and used within 1 day. Supported lipid bilayers were formed, by vesicle fusion, inside a 60-μl perfusion chamber (Invitrogen) on appropriately treated glass slides. The slides were washed in dilute ICN 7× detergent (VMR International, West Chester, PA), rinsed extensively in 18 MΩ water, and baked at 450°C for 4 h; slides were used within a day of preparation. After 5 min, excess vesicles were removed from the perfusion chamber using the same buffer used in vesicle preparation. The perfusion chamber was used to exchange the outside buffer solution; at least 1 ml of buffer was passed through the perfusion chamber to ensure complete exchange.
Imaging of the supported lipid bilayer
A Nikon (Tokyo, Japan) TE2000 fluorescence microscope equipped with either a Cascade 512B or a Cascade 650 charge-coupled device (CCD) camera (Roper Scientific, Tucson, AZ) was used to image the bilayers. An X-Cite 120 arc lamp (EXFO, Quebec, Canada) was used as a light source. The NBD and Alexa fluorophores were imaged using NBD and Alexa 647 filter sets, respectively (Chroma Technology, Rockingham, VT). Figs. 1, 6, 7, and 8 were acquired using a 100×, 1.30 numerical aperture (NA) objective. Figs. 2, 3, and 5 were acquired using a 40×, 1.30 NA objective.
FIGURE 1.

Epifluorescence images of a 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC bilayer. The bilayer was formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer. The ionic strength of the outside solution was then dropped to 0 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5, and images acquired every 100 ms (a–h). The images are contrasted to clearly show the light and dark regions. The difference in intensity between the two regions is ∼13% (this calculation excludes the bright rings). A 100×, 1.3 NA objective was used to acquire the images. The scale bar represents 10 μm.
FIGURE 6.

Epifluorescence images of bilayers containing (a) 0, (b) 10, (c) 20, (d) 30, and (e) 40 mol % DOPA in DOPC with 1 mol % NBD-PC. The bilayers were formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer. The ionic strength of the outside solution was then dropped to 0 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5. The scale bar represents 10 μm.
FIGURE 7.

Epifluorescence images of a 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC bilayer. The bilayer was formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer. After formation, the excess vesicles were rinsed away and the bilayer imaged (a); the ionic strength of the outside solution was then dropped to 0 mM KCl (b) and raised back up to 250 mM KCl (c). The scale bar represents 10 μm.
FIGURE 8.

Epifluorescence images of bilayers at pH 5 (a–c) and 7.4 (d–f). Bilayer images were taken before (a, d) and after (b, e) the addition of 1:20 protein/lipid. (c, f) Epifluorescence images of the adsorbed protein (α-synuclein labeled with Alexa 647). The bilayer was composed of 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC. The scale bar represents 10 μm.
FIGURE 2.

Epifluorescence images of a 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC bilayer. The bilayer was formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer. The ionic strength of the outside solution was then dropped to 0 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5, and images acquired every 150 ms (a–h). A 40×, 1.3 NA objective was used to acquire the images. The scale bar represents 20 μm.
FIGURE 3.

FRAP: The bilayer was composed of 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC. It was formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer and the outside solution was exchanged for one containing 25 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5. (a) Plot of fluorescence recovery as a function of time; an APD was used to collect the light; the laser power was 150 nW. (b) Epifluorescence image of the bilayer before bleaching, (c) immediately after bleaching, and (d) 20 min later. The bleached spot shows the location that the APD samples. The scale bar represents 10 μm.
FIGURE 5.

Epifluorescence images of a 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC bilayer. The bilayer was formed in a 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer. The ionic strength of the outside solution was then dropped to (a) 100 mM KCl, (b) 75 mM KCl, (c) 50 mM KCl, (d) 25 mM KCl, and (e) 0 mM KCl in 50 mM MES, 0.1 mM EDTA, pH 5. The images are of separate samples and were acquired after the separation was complete. The scale bar represents 50 μm.
Fluorescence recovery after photobleaching
The FRAP measurement system is discussed in detail elsewhere (11). Briefly, a Nikon TE2000 fluorescence microscope equipped with a 40× oil immersion objective, an NBD filter set (Chroma Technology), and a silicon avalanche photodiode (APD) single photon counting module (SPCM-AQR-16-FC, PerkinElmer, Vaudreuil, Quebec) was used to focus, collect, and count the emitted fluorescence. A 15-mW argon ion laser (488 nm; Melles Griot, Carlsbad, CA) was used to both bleach and monitor the lipid bilayer. To reduce further photobleaching of the fluorophore during the recovery period, the laser intensity was reduced 100,000-fold using a 5× (focal transmission of 1 × 105) neutral density filter (NE50B, Thorlabs, Newton, NJ). A LabVIEW program was used to acquire the counts from the APD, control the filter wheel, and trigger the shutter (Uniblitz; VA, Rochester, NY).
Attenuated total reflection-Fourier transform infrared spectroscopy
A Nicolet 470 Fourier transform infrared spectroscopy (FTIR) equipped with a mercury cadmium telluride type A detector was used to collect the spectra. A home-built attenuated total reflection (ATR) setup was used, as described in detail elsewhere (12). In brief, infrared light is sent out of the spectrometer and coupled into an ATR element created in-house (15 mm × 9 mm × 525 μm silicon wafer) at 45°. Before introducing the lipids, a background of the silicon ATR element and the buffer was collected. To form supported lipid bilayers, LUVs were injected into the custom-made Delrin flow-cell (20 μL) and allowed to incubate for 30 min. Once the bilayer had formed, buffer was flushed through the flow-cell to rinse away excess vesicles, and the sample spectrum was obtained and ratioed against the background spectra. For both the background and sample spectra, 1600 scans were signal averaged at a resolution of 4 cm−1 using Happ-Genzel apodization and zero filling.
Expression, purification, and labeling of α-synuclein
The protein is expressed in Escherichia coli and purified as described previously (13). The protein was preferentially labeled at the N-terminus: monomeric α-synuclein (0.56 mM in 225 μL PBS, pH 7.0) is mixed with Alexa Fluor 647 carboxylic acid, succinimidyl ester (0.25 mg in 25 μL dimethyl sulfoxide (DMSO)). The mixture is placed on a Microplate shaker at 800 rpm for 2 h. The protein solution is then loaded onto a Superdex 200 gel-filtration column and eluted in 20 mM HEPES, pH 7.4. The protein concentration and degree of labeling are determined using ultraviolet-visible absorbance measurements (the degree of labeling of the protein is typically 20%, mol/mol). The protein is then aliquoted and stored at −20°C. Before each experiment an aliquot is thawed and centrifuged through a Microcon YM-100 centrifugal filter unit with a molecular mass cutoff of 100 kDa to remove any aggregates. For binding experiments at pH 5, the concentrated protein eluted in HEPES, pH 7.4 is diluted 100-fold in MES buffer at pH 5 and used immediately.
RESULTS
Fig. 1 a shows a bilayer composed of 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC. The bilayer was formed by vesicle fusion in a 250 mM KCl, 50 mM MES, and 0.1 mM EDTA, pH 5.0 buffer. The bilayer is uniform and mobile. Upon exchanging the bulk solution with a buffer of 0 mM KCl, 50 mM MES, and 0.1 mM EDTA, pH 5.0, separation is observed (Fig. 1, b–h). The images are contrasted to clearly show the light and dark regions; the difference in intensity between the two regions, in these and subsequent images, is ∼13%. The NBD moiety is attached to the tail of a PC lipid. Also visible in these and subsequent images are rings (in lower resolution images the rings look like bright spots). As detailed elsewhere (14) these bilayers bend away from the surface to form caps when exposed to asymmetric screening environments. The caps appear as rings because of the lack of z-resolution.
To ensure that the separation is not light/heat induced, images were acquired at various locations on the bilayer where minimal light exposure occurred. The separation is observed uniformly over the entire bilayer.
In Fig. 1, only one nucleation site is visible; to examine the nucleation density we repeated the experiment in Fig. 1 with a lower magnification objective (Fig. 2). Examining many samples, under these conditions, we find that on average there are ∼3 nucleation sites per 40,000 μm2. By patterning the bilayers (15), it is possible to examine what happens when the area of the bilayer is smaller than the typical domains. In this case it is observed that there is at least one nucleation site per bilayer region (data not shown). This suggests that the number of nucleation sites is set by the speed with which the domains propagate, i.e., there are many possible nucleation sites but rapid propagation eliminates them.
To determine whether the lipids in both regions are mobile, fluorescence recovery after photobleaching (FRAP) experiments were performed. In Fig. 3 the recovery can be observed. The bilayer was formed as before, but in this case the bulk salt concentration was only dropped to 25 mM. This salt concentration was chosen because it results in domains that are of a similar size to the laser bleach spot. To assess whether there is exchange between the two regions, it is necessary to bleach an entire dark region (as compared with the bright region, there are ∼13% fewer NBD-PC lipids in the dark region). An image of the bilayer was acquired before bleaching (Fig. 3 b). The bilayer was then bleached to background levels in 1 s using an Argon ion laser operating at 15 mW. The laser power was attenuated by 100,000-fold to 150 nW, and the recovering fluorescence monitored using an APD (Fig. 3 a). At this laser power level it is possible to monitor the bilayer indefinitely without further bleaching the sample (11). The intensity was normalized by acquiring data before bleaching. To show the bleached area, the bilayer was bleached again with the laser and an image quickly acquired with a CCD camera (Fig. 3 c). The bleached spot shows the location that the APD samples. Another image was acquired ∼20 min later (Fig. 3 d). The bleach spot completely covers the dark region, and the recovery is nearly complete (≥95%); it is clear that there is exchange of lipids between the two regions.
The diffusion of the probe molecule will be different in the two regions due to compositional differences. Fitting the recovery curve precisely is therefore nontrivial. By determining the half-time to recovery, we can estimate the diffusion coefficient (16); doing so yields a value of ∼1 μm2/s, a value typical of fluid bilayers.
The C-H stretching region is sensitive to the lipid phase (17); therefore, we have also examined the bilayers using ATR-FTIR before and after the ionic strength was dropped. In Fig. 4 the C-H stretch region of a 30 mol % DOPA/69 mol % DOPC, 1 mol % NBD-PC bilayer when uniformly mixed (solid line) and separated (dashed line) is shown. There is a slight change in intensity between the two spectra; this is likely due to the removal of stray vesicles from the flow cell. The peak positions do not shift from one spectra to the other, indicating that the C-H groups are in a similar environment regardless of whether the lipids are uniformly mixed or not.
FIGURE 4.

ATR-FTIR: The bilayer was composed of 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC. Spectra were collected before (solid line) and after (dashed line) ionic strength induced separation. The ν(C–H) region is shown.
The area fraction of the dark regions should depend on the ionic strength. To determine if this is the case, bilayers were formed as in the previous figures. The bulk solution was then exchanged for a 50 mM MES, 0.1 mM EDTA, pH 5 buffer containing 0–100 mM KCl. In Fig. 5, it can be seen that visible separation does not occur until the KCl concentration drops to 75 mM; each image is of a different sample. The extent of the separation depends on the KCl concentration: the lower the concentration the more extensive the separation. It can also be observed that the nucleation density depends on the extent of the separation. In general the farther the separation propagates, the lower the nucleation density is (we know from real-time monitoring that when the separation is extensive, i.e., Fig. 5, d and e, there are few nucleation points).
The separation should also depend on the amount of DOPA. To confirm this, bilayers were formed as before except that the concentration of DOPA varied from 0–40 mol %; it was not possible to form bilayers containing 50 mol % or more DOPA. The bulk solution was exchanged for a 0 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer and the bilayers examined (Fig. 6). As can be seen it is necessary to have >10 mol % DOPA present for visible separation. Here again the nucleation density depends on the extent of the separation.
The reversibility of the separation was also examined. The patterns formed are fractal-like and suggest a nonequilibrium process. Bilayers were formed as before (Fig. 7 a), and the bulk solution was exchanged with a 0 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 buffer (Fig. 7 b). Then the bulk solution was returned to 250 mM KCl, 50 mM MES, 0.1 mM EDTA, pH 5 (Fig. 7 c). The rings disappear, as they should (14), but the separation remains. These samples have been monitored for 24 h with no change observed. Hysteresis in the properties of PC/PA bilayers has previously been observed (5,6). Consequently, the ionic strength was raised to 1 M and the pH was kept at 5 or adjusted to 7.4 or 12. The patterns were not observed to disappear under any of these conditions.
An increase in pH was not enough to disrupt the patterns, yet the charge on the PA should be increasing. To examine whether this is the case, we looked at the binding of α-synuclein to PC/PA membranes at two different pHs. In both cases the membranes were prepared as in Figs. 1–3. To increase the pH, the bulk solution was exchanged for one containing 50 mM HEPES, 0.1 mM EDTA, pH 7.4; no change was observed in the bilayer. α-Synuclein is well known to bind to negatively charged lipids (18,19) and can thus be used as a marker for the presence of negatively charged lipids; in these experiments, the α-synuclein was labeled on the N-terminus with Alexa 647. Fig. 8, a and b, shows a bilayer at pH 5 before and after the addition of 1:20 protein/lipid. The protein binds uniformly to the surface except where there are caps (Fig. 8 c). Fig. 8, d and e, shows a bilayer at pH 7.4 before and after the addition of 1:20 protein/lipid. Here, the protein prefers the fractal-like regions (Fig. 8 f). These results will be discussed in more detail below.
DISCUSSION
We have visualized ionic strength-dependent separation in DOPC/DOPA bilayers. In all of the samples shown, the marker was tail-labeled NBD-PC. If we replace the NBD-PC with head-labeled Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE; Invitrogen-Molecular Probes, Eugene, OR), the same separation patterns are observed, i.e., the Texas Red DHPE is depleted in the fractal-like regions. As mentioned in the Introduction, charged PA promotes mixing. However, when the PA is protonated the repulsion between the PA lipids is reduced and favorable hydrogen-bonds can form between the PA lipids, promoting separation of the uncharged PA lipids from the other lipids (7). Therefore, it is assumed that the dark regions are PA-rich and that the bright regions are PC-rich.
The fractal-like regions are not observed to move, at least not over the timescale of ∼1 day. From Fig. 3 we see that there is rapid exchange of lipids between the PC-rich and the PA-rich regions. So although the domains are immobile, the lipids are mobile and freely exchanging, ∼1 μm2/s. The gel-to-fluid phase transition temperature of DOPA− is −8°C (20), and upon protonation the Tm of PA lipids increases but only by a few degrees (5,21). Consequently, we expect that the DOPA remains fluid, regardless of the charge state. The lack of shift in the C-H stretching region in Fig. 4 supports the conclusion that the lipids in both regions are fluid.
As expected, the extent of the separation depends on both the ionic strength (Fig. 5) and the amount of PA present (Fig. 6). To examine this dependence further we have used the expressions derived by Ninham and Parsegian (22) to determine the percentage of uncharged DOPA as a function of monovalent salt concentration (see Appendix). Shown in Fig. 9 is a plot of the area fraction of the DOPA-rich regions (left axis) and the percentage of uncharged DOPA (right axis) as a function of KCl concentration. The area fraction values were determined by examining, over a large area, several bilayers at each ionic strength; conditions were the same as Fig. 5. The calculated values of percentage of uncharged DOPA and the experimental area fraction data show the same general trend with decreasing KCl, i.e., a very rapid rise. A certain amount of uncharged DOPA is needed. Fig. 6 suggests at least 10 mol %, before separation begins. Upon exceeding this threshold, the calculated values suggest that the area fraction should increase rapidly, and it does. At high KCl concentration the data are not expected to match exactly because the comparison is with area fraction, not amount of uncharged DOPA. Once all of the PA lipids are protonated the expression is no longer valid. Physically this means that the amount of uncharged PA plateaus; we have added a horizontal dotted line to guide the eye.
FIGURE 9.

Plot of area fraction of the fractal-like regions (left axis) and percentage uncharged DOPA (right axis) versus KCl concentration. The area fractions are experimentally determined (see Discussion for details). The mol % uncharged DOPA is calculated (see Appendix for details), H = 10−5 M; Z = 10−3.2 M; S = 266 Å2/ionizable group.
In the analysis of the previous paragraph we have assumed that one of the variables in the derived expression remains constant: S—the surface area per ionizable group. Globally, this is true; averaged over the whole surface there is the same ratio of PA/PC lipids. Locally, however, the PC-rich and the PA-rich regions have different surface areas per ionizable group. In Fig. 10, the percentage of uncharged DOPA as a function of KCl concentration is shown for different values of S. The bold line was calculated using S = 266 Å2/charge; this is our best estimate for a 30 mol % DOPA/69 mol % DOPC/1 mol % NBD-PC bilayer (see Appendix for details). When the bilayer separates, regions with higher (PC-rich) and lower (PA-rich) surface areas per ionizable group form. The PC-rich regions are to the left of the bold curve, whereas the PA-rich regions are to the right of the bold curve.
FIGURE 10.

Plot of percentage uncharged DOPA versus KCl concentration for varying values of S. Bold line, S = 266 Å2/ionizable group. Dashed lines, S = 600–100 Å2/ionizable group, varying in increments of 50 Å2/ionizable group.
It is possible that Fig. 10 explains one of the surprising observations of this work: that raising the ionic strength, even to 1 M, does not cause the bilayer to remix. If, in the PA-rich regions, the surface area per ionizable group is low, then even at 1 M not all of the DOPA will be charged. Consequently, the drive for the charged DOPA lipids to mix may be counterbalanced by the interactions between the neutral lipids. Unfortunately, fluorescence microscopy is not necessarily sensitive to compositional differences. The change in intensity between the dark and bright regions is ∼13% for all the images shown in this article. NBD-PC is, however, very different from DOPC and may not be entirely representative of the extent of the separation.
Increasing the pH should increase the number of negatively charged PA lipids. It would be expected that this would disrupt the patterns; yet it does not. In Fig. 8, it is observed that at pH 5 the α-synuclein does not have a preference for the PA-rich or PC-rich regions. This indicates that the charge density of the two regions is the same. Fig. 10 suggests that at pH 5 and low ionic strength the majority of the PA lipids are uncharged. At pH 7.4 the α-synuclein prefers the PA-rich regions, indicating that at least some of the PA lipids are charged. This shows that increasing pH increases the charge on the PA lipids, as it should; however why the patterns do not disappear is unclear at this time. The charge density of the PA-rich region increases at the cost of losing hydrogen-bond interactions between the PA lipids. This may shift the conditions for charging the membrane. At pH 7.4 the charge density has increased, but possibly not enough to disrupt the patterns. In the end, we were not able to find conditions that reversed the patterns. Whether this is because the pattern formation is irreversible or because there is a significant hysteresis and the necessary conditions have not been found is at this time unclear.
Fractal-like separation has been observed in two other systems that we are aware of: 1) rapid compression of DMPE monolayers (23), and 2) heating cationic-supported bilayers above the acyl chain melting temperature and cooling to room temperature (24,25). In both of these systems, the lipids in the fractal-like patterns are in the gel or condensed phase. Here, the lipids in the fractal-like pattern move rapidly. This raises the question of why the PA-rich regions do not move. The bilayers in this article are on solid supports; consequently, there is frictional coupling between the bilayer and the support (26). The viscosity of the water trapped between the bilayer and the solid support is orders of magnitude larger than bulk water—surprisingly lipids in supported bilayers continue to move at rates very close to those observed in free bilayers (27). The difference in free and supported bilayers becomes evident when domains are present. We have previously shown that it is possible to form circular liquid-ordered domains in bilayers on glass slides; similarly, the lipids are free to exchange but the domains themselves are immobile (27).
Frictional coupling explains why the patterns do not collectively move, but it does not explain why the patterns appear fractal-like rather than circular to minimize line tension. We have not attempted to calculate a fractal dimension. For an object to be a fractal it must appear the same on all length scales; in these experiments, only one length scale is accessible. Nevertheless we note that the patterns look very much like diffusion-limited aggregation (28,29). This raises the question of whether some nonbilayer structure is being formed, disks, for instance, which stick together. Optical microscopy does not afford the resolution to determine if other structures are forming. However, given that lipids readily exchange between the two regions (Fig. 3), we feel this is unlikely. AFM images of glass slides do not show any discernable patterns (27); nevertheless, it is possible that scratches or chemical heterogeneities in the glass serve as nucleation sites, trapping a small core of lipids.
At the moment we do not understand why the patterns are fractal-like; a few possible reasons/observations are discussed here: 1), The patterns form rapidly; in such instances fractal-like patterns are often observed (30). Perhaps, due to the solid supports the fractal-like patterns are trapped, and we are observing an intermediate structure. To determine if this is the case, experiments using unsupported bilayers are required; we are currently working on this. 2), Although much of the PA is neutralized, negatively charged PA is still present. If the PA− species like to sit at the interface this would promote the formation of extended structures (due to charge repulsion). The charged PA consequently would behave like a surfactant, lowering the line tension of the interface. 3), Our group has examined separation in numerous lipid mixtures using supported bilayers: the dominant features observed in these mixtures (PC/PG, PC/PE, PC/cholesterol) are circular domains. Regardless of what we do to the PC/PA mixtures—change the tail composition, add cholesterol, and/or add lysoPC (data not shown)—the separation appears fractal-like. In addition to changing the monovalent ionic strength, we can induce separation by changing the pH, concentration of divalent ions, and/or concentration of protein (data not shown); again, only fractal-like separation is observed. Although this does not rule out the solid support as a contributing factor, it strongly suggests that the properties of PA also contribute.
Switches are necessary in normal cell function, i.e., the activity of an enzyme, the assembly of a complex, and the binding affinity of a molecule. Here, we have, for the first time to our knowledge, visualized the switching of organization in a charged membrane. Evidence strongly suggests that a fluid-fluid immiscibility occurs. By concentrating the PA lipids, the properties of the PA-rich region change dramatically and are then largely insensitive to further environmental changes. This work shows how changing the environment can significantly alter local properties; it is interesting as a way to manipulate fluid membranes and may have biological relevance.
CONCLUSIONS
It has been shown that altering monovalent ionic strength can control the organization of DOPC/DOPA bilayers. Upon reducing the ionic strength, the bilayers separate into PC-rich and PA-rich regions. The PA-rich regions appear fractal-like and exhibit hysteresis in their formation and disappearance. This is the first work, to our knowledge, to visualize the effect of monovalent ions on the organization of fluid membranes.
Acknowledgments
Jennifer S. Hovis is a recipient of a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund. This work was also supported by National Institutes of Health grant R01 NS049221 (J.-C.R. and J.S.H.) and a grant from the Showalter Trust (J.S.H. and J.-C.R.).
APPENDIX
In their 1971 article, Ninham and Parsegian (22) theoretically examined the properties of charged surfaces in saline solutions. The analysis was done for two charged surfaces in solutions separated by a distance, b. In our experiments the solution above the bilayer is very large; we therefore used the expressions for b ≥ 1/κ. For this condition and the condition of only monovalent ions (no divalent ions), the fraction of surface groups dissociated, α, is given by
 |
(1) |
where H is the H+ concentration in the reservoir, Z the dissociation constant, 1/κ the screening length, n the reservoir anion concentration, and S the surface area per ionizable group. The θ functions θ3(0,q) and θ4(0,q) are defined by
 |
(2) |
To convert α into the percentage of uncharged DOPA we used the following expression:
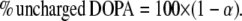 |
(3) |
To calculate the curve shown in Fig. 9, we used the following values: H = 10−5 M; Z = 10−3.2 M; S = 266 Å2/ionizable group; κ and n vary with the salt concentration. S was determined as follows: We assumed the radius of DOPC = 4.8 Å (31) and that because the dioleyl tails are so large they are the limiting factor on charge density and so the radius of DOPA = the radius of DOPC. We further assumed that the lipids were arranged in a hexagonal closed packed arrangement. If the lipids were further spread out, S would increase and the curve would shift down and to the left. If DOPA is smaller than DOPC, S would decrease and the curve would shift up and to the right. In Fig. 10 the same values of H and Z were used as in Fig. 9, and S was allowed to vary.
Editor: Enrico Gratton.
References
- 1.Brown, D. A., and E. London. 1998. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 164:103–114. [DOI] [PubMed] [Google Scholar]
- 2.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 3.Veatch, S. L., and S. L. Keller. 2002. Organization in lipid membranes containing cholesterol. Phys. Rev. Lett. 89:268101. [DOI] [PubMed] [Google Scholar]
- 4.Lipowsky, R., and E. Sackmann, editors. 1995. Handbook of Biological Physics. Elsevier Science, Amsterdam, The Netherlands.
- 5.Galla, H. J., and E. Sackmann. 1975. Chemically-induced phase separation in mixed vesicles containing phosphatidic-acid—optical study. J. Am. Chem. Soc. 97:4114–4120. [DOI] [PubMed] [Google Scholar]
- 6.Träuble, H., and H. Eibl. 1974. Electrostatic effects on lipid phase-transitions: membrane structure and ionic environment. Proc. Natl. Acad. Sci. USA. 71:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garidel, P., C. Johann, and A. Blume. 1997. Nonideal mixing and phase separation in phosphatidylcholine phosphatidic acid mixtures as a function of acyl chain length and pH. Biophys. J. 72:2196–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tokutomi, S., K. Ohki, and S. I. Ohnishi. 1980. Proton-induced phase-separation in phosphatidylserine-phosphatidylcholine membranes. Biochim. Biophys. Acta. 596:192–200. [DOI] [PubMed] [Google Scholar]
- 9.Hinderliter, A. K., J. Y. Huang, and G. W. Feigenson. 1994. Detection of phase-separation in fluid phosphatidylserine phosphatidylcholine mixtures. Biophys. J. 67:1906–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kooijman, E. E., K. M. Carter, E. G. van Laar, V. Chupin, K. N. J. Burger, and B. de Kruijff. 2005. What makes the bioactive lipids phosphatidic acid and lysophosphatidic acid so special? Biochemistry. 44:17007–17015. [DOI] [PubMed] [Google Scholar]
- 11.Seu, K. J., L. R. Cambrea, R. M. Everly, and J. S. Hovis. 2006. Influence of lipid chemistry on membrane fluidity: tail and headgroup interactions. Biophys. J. 91:3727–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hull, M. C., L. R. Cambrea, and J. S. Hovis. 2005. Infrared spectroscopy of fluid lipid bilayers. Anal. Chem. 77:6096–6099. [DOI] [PubMed] [Google Scholar]
- 13.Rochet, J. C., K. A. Conway, and P. T. Lansbury. 2000. Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 39:10619–10626. [DOI] [PubMed] [Google Scholar]
- 14.Cambrea, L. R., and J. S. Hovis. 2007. Formation of three-dimensional structures in supported lipid bilayers. Biophys. J. 92:3587–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boxer, S. G. 2000. Molecular transport and organization in supported lipid membranes. Curr. Opin. Chem. Biol. 4:704–709. [DOI] [PubMed] [Google Scholar]
- 16.Soumpasis, D. M. 1983. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 41:95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantsch, H. H., and R. N. A. H. Lewis. 1991. Phospholipid phase transitions in model and biological membranes as studied by infrared spectroscopy. Chem. Phys. Lipids. 57:213–226. [DOI] [PubMed] [Google Scholar]
- 18.Jo, E., J. McLaurin, C. M. Yip, P. St. George-Hyslop, and P. E. Fraser. 2000. α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275:34328–34334. [DOI] [PubMed] [Google Scholar]
- 19.Davidson, W. S., A. Jonas, D. F. Clayton, and J. M. George. 1998. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273:9443–9449. [DOI] [PubMed] [Google Scholar]
- 20.Silvius, J. R. 1982. Lipid-Protein Interactions. John Wiley & Sons, New York.
- 21.Blume, A., and H. Eibl. 1979. Influence of charge on bilayer membranes—calorimetric investigations of phosphatidic-acid bilayers. Biochim. Biophys. Acta. 558:13–21. [DOI] [PubMed] [Google Scholar]
- 22.Ninham, B. W., and V. A. Parsegian. 1971. Electrostatic potential between surfaces bearing ionizable groups in ionic equilibrium with physiologic saline solution. J. Theor. Biol. 31:405–428. [DOI] [PubMed] [Google Scholar]
- 23.Miller, A., W. Knoll, and H. Mohwald. 1986. Fractal growth of crystalline phospholipid domains in monomolecular layers. Phys. Rev. Lett. 56:2633–2636. [DOI] [PubMed] [Google Scholar]
- 24.McKiernan, A. E., T. V. Ratto, and M. L. Longo. 2000. Domain growth, shapes, and topology in cationic lipid bilayers on mica by fluorescence and atomic force microscopy. Biophys. J. 79:2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muresan, A. S., H. Diamant, and K. Y. C. Lee. 2001. Effect of temperature and composition on the formation of nanoscale compartments in phospholipid membranes. J. Am. Chem. Soc. 123:6951–6952. [DOI] [PubMed] [Google Scholar]
- 26.Evans, E., and E. Sackmann. 1988. Translational and rotational drag coefficients for a disk moving in a liquid membrane associated with a rigid substrate. J. Fluid Mech. 194:553–561. [Google Scholar]
- 27.Seu, K. J., A. P. Pandey, F. Haque, E. A. Proctor, A. E. Ribbe, and J. S. Hovis. 2007. Effect of surface treatment on diffusion and domain formation in supported lipid bilayers. Biophys. J. 92:2445–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witten, T. A., and L. M. Sander. 1981. Diffusion-limited aggregation, a kinetic critical phenomenon. Phys. Rev. Lett. 47:1400–1403. [Google Scholar]
- 29.Sander, L. M. 2000. Diffusion-limited aggregation: a kinetic critical phenomenon? Contemp. Phys. 41:203–218. [Google Scholar]
- 30.Ben-Jacob, E. 1993. From snowflake formation to growth of bacterial colonies. Part I. Diffusive patterning in azoic systems. Contemp. Phys. 34:247–273. [Google Scholar]
- 31.Kucerka, N., S. Tristram-Nagle, and J. F. Nagle. 2006. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208:193–202. [DOI] [PubMed] [Google Scholar]