

Abstract
We have investigated the kinetics of DNA hybridization to oligonucleotide arrays on high-capacity porous silica films that were deposited by two techniques. Films created by spin coating pure colloidal silica suspensions onto a substrate had pores of ∼23 nm, relatively low porosity (35%), and a surface area of 17 times flat glass (for a 0.3-μm film). In the second method, latex particles were codeposited with the silica by spin coating and then pyrolyzed, which resulted in larger pores (36 nm), higher porosity (65%), and higher surface area (26 times flat glass for a 0.3-μm film). As a result of these favorable properties, the templated silica hybridized more quickly and reached a higher adsorbed target density (11 vs. 8 times flat glass at 22°C) than the pure silica. Adsorption of DNA onto the high-capacity films is controlled by traditional adsorption and desorption coefficients, as well as by morphology factors and transient binding interactions between the target and the probes. To describe these effects, we have developed a model based on the analogy to diffusion of a reactant in a porous catalyst. Adsorption values (ka, kd, and K) measured on planar arrays for the same probe/target system provide the parameters for the model and also provide an internally consistent comparison for the stability of the transient complexes. The interpretation of the model takes into account factors not previously considered for hybridization in three-dimensional films, including the potential effects of heterogeneous probe populations, partial probe/target complexes during diffusion, and non-1:1 binding structures. The transient complexes are much less stable than full duplexes (binding constants for full duplexes higher by three orders of magnitude or more), which may be a result of the unique probe density and distribution that is characteristic of the photolithographically patterned arrays. The behavior at 22°C is described well by the predictive equations for morphology, whereas the behavior at 45°C deviates from expectations and suggests that more complex phenomena may be occurring in that temperature regime.
INTRODUCTION
An important goal for the advancement of DNA array technology is to increase the signal strength, which would have two major benefits. First, an enhanced signal would assist with the analysis of dilute or weakly binding samples (such as samples with high AT content) and thereby increase the array sensitivity. Furthermore, signal amplification could allow reduction of the feature size without sacrificing signal intensity, which would enable more information to be encoded on a single substrate. To enable greater signal intensity, three-dimensional substrates are receiving increasing attention, as reviewed by Dufva (1).
A wide variety of routes for fabricating high-capacity substrates have been investigated. Organic materials offer a great degree of flexibility for developing very high-capacity materials, varying the physical and chemical properties of the matrix, and using a wide variety of synthesis routes. A pioneering approach that has been very successful is the use of polymer gel layers (2–6), primarily based on polyacrylamide. Other organic gel films that have been used include sugar polyacrylate hydrogels (7), agarose (8,9), electropolymerized polypyrrole (10,11), and plasma-polymerized allylamine (12). Organic films have also been formed by adsorption of polymers (13,14) and polyelectrolyte multilayers (15). DNA-based (16) and phosphorous-based dendrimers (17) have also been used to increase the available probes for hybridization. Stillman et al. reported the measurement of similar binding affinities on planar glass and in nitrocellulose membranes (18). Other membrane materials used include microporous polyamide-6 (19). Membrane approaches (both organic and inorganic) are reviewed by Jones (20). To increase hybridization kinetics, polymer gels functionalized with DNA have also been immobilized in microchannels (21,22). Molecular recognition in high-capacity organic films has also extended beyond DNA hybridization to include antibody/antigen binding (23,24) and glycogen interactions (25).
Inorganic materials provide a complementary approach, offering advantages of mechanical rigidity and inertness to chemicals during processing. Porous silicon has received a great deal of attention because of the ideally ordered porous structure (26–31), as is the case for porous alumina surface layers (32). The mechanical rigidity of inorganic materials also allows them to be used in “flow-through” modes, such as porous silicon/silica (33–35), porous alumina (36,37), and glass microchannels (38,39). Three-dimensional layers have also been formed by the adsorption of oligonucleotide-covered gold nanoparticles (40). However, these various routes for fabrication of inorganic supports have not been optimized for high-resolution photolithographic patterning and are not favorable due to the optical scattering that results from the large scale of the features, opaqueness (in the case of silicon and aluminum), and/or reflectivity (in the case of gold nanoparticles).
Hybridization kinetics on high-capacity supports are a more complex phenomenon than on flat glass because mass transfer and nonuniformity within the film must be considered. Although a variety of high-capacity platforms have been investigated experimentally, theoretical modeling and investigation of the fundamental parameters governing the performance has received less attention (2). Livshits and Mirzabekov proposed and tested the theory of “retardation”, whereby targets being “washed out” of a polymer gel on which DNA probes have been immobilized are impeded by repeated association and dissociation with the probes in the matrix (41). They went on to propose that the diffusion of targets into the matrix during hybridization is controlled by the same type of interactions (4). This theory has been further developed by Sorokin et al. (2,3).
In previous work, we have discussed the use of colloidal silica films as substrates for high-capacity DNA arrays (42,43). Films created via this method are attractive for photolithographic array fabrication because they are robust and easily processed and the small pore size does not result in optical scattering. In this report we compare and contrast the hybridization kinetics of films created with two deposition techniques. In the first, the silica is spin coated onto a substrate, followed by a low temperature thermal treatment, and the pores in the film are formed by the natural voids between the solid particles. In the second, latex is codeposited with the silica colloid and then removed at high temperature, leaving behind large voids of a controlled size. The resulting “templated” films have larger pore size, porosity, and surface area than films created from pure silica deposition (42).
In this work, we use the analogy to diffusion of reactants in catalyst beds to analyze the diffusion of target oligonucleotides into arrays on the porous silica films. The goal of the analysis is to develop an understanding of two key aspects of the hybridization behavior: 1), the “thermodynamic” retardation effects and how they relate to the model proposed by Sorokin et al. (2,3) and Livshits et al. (4), and 2), the morphology-dependent components of the diffusivity.
This research builds on previous studies of binding kinetics on planar photolithographically patterned arrays (44). These studies provide values for fundamental adsorption parameters (ka, kd, and K) that are used in the analysis on the porous films. Furthermore, the studies were conducted with arrays patterned on a chemically and mechanically similar supporting surface (glass), which provides consistency between the systems. A model was presented to interpret the contribution of non-1:1 probe/target structures. It has been proposed by Levicky and Horgan that the diversity of experimental observations even on planar glass arrays could reflect in part the formation of structures more complex than one-to-one hybridization, such as target bridging and hybridization to multiple probes (45). In this research our goal is to extend the investigation of photolithographically patterned arrays to the third dimension and in the process to consider the potential effects of the unique probe density and distribution, non-1:1 structures, and the nature of the interactions a target experiences as it diffuses into the substrate.
MATERIALS AND METHODS
Preparation of surfaces and probe layer
The deposition of pure colloidal silica films (43) and films “templated” with polymer latex (42) have been previously described. For the pure colloidal silica films, ZL silica (Nissan Chemicals, Tokyo, Japan; 65 ± 16 nm) was used. Unless stated otherwise, films were spin coated with 20 wt % suspensions at 2500 rpm. The films were then heated to 350°C at 10°C/min, held for 4 h, and cooled to room temperature at 20°C/min. The final film thickness was ∼0.3 μm.
For the templated films, referred to as “T-S50”, a 2:1 mixture of latex/silica (8% vol solids) consisting of S50 silica (Nissan Chemicals, 16 ± 5 nm) and sulfate-modified latex (IDC, Eugene, OR; 20 ± 3 nm) was spun at 1000 rpm. To remove the latex, the films were heated to 400°C at 2°C/min, held for 4 h, and cooled to room temperature at 20°C/min, resulting in films also 0.3-μm thick. The flat glass and porous silica surfaces were cleaned and silanated and the probe layer was synthesized using MeNPOC chemistry as described by McGall et al. (46).
Hybridization kinetics
The probe arrays are a variant of those used by Glazer et al. (44) and Forman et al. (47) with 100 μm features and only repeating sets of 20-mer perfect match (20 PM) and one-base-mismatch (20 MM) probes. The sequence was (3′) AGG TCT TCT GGT CTC CTT TA (5′), with the 3′ end attached to the surface (43). This sequence was chosen to have approximately equal AT and GC content and was designed to minimize self-complementary target/target and probe/probe interactions. The effect of a mismatch located at the center position of each probe is evaluated. The results reported are the average of ∼20 independent features per substrate and a minimum of two substrates per condition. Intrasubstrate variability in hybridization signal intensity was low (≤5%), whereas intersubstrate variability was typically higher (≤20%).
Hybridization time courses were observed as described in Glazer et al. (44). Briefly, fluorescein-labeled 20-mer oligonucleotide target (perfect match to 20-mer probe) diluted in MES buffer (containing 0.1 M 2-[N-morpholino]ethanesulfonic acid (Sigma, St. Louis, MO), 0.89 M NaCl, and 0.03 M NaOH) at a controlled temperature was continuously circulated through a flow cell (also temperature controlled) containing the array, and scans were taken at regular time intervals. At this total ionic strength of ∼1 M electrostatic interactions are localized to a Debye length of ∼3 Å (48). All data are background corrected by subtracting the signal from a region of the sample with no probe synthesis (average of 20 independent features per substrate). A high flow rate of ∼80 mL/min was used to minimize the depletion layer in solution. For the flat glass surface, this flow rate was sufficient to supply target at a rate that minimized the depletion layer in solution, so that the adsorption kinetics would be indicative of the fundamental interface parameters (44). For the porous glass surfaces, the impact of diffusion within the surface is one of the key topics that will be discussed in subsequent sections.
Development of the model for diffusion and hybridization in porous silica films
In this section, we develop a model for hybridization kinetics based on the analogy between the porous silica films and catalyst beds. The model takes into account two primary factors: 1), retardation caused by probe/target interactions as the target penetrates the film, and 2), limitations caused by the film morphology.
Modeling approach
Adsorption in the porous system is described by the schematic shown in Fig. 1. In the bulk solution (from y = h to y = b), the concentration of free target in solution (Cs) is described by Eq. 1 (49).
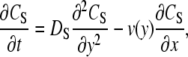 |
(1) |
where Ds is the target diffusivity in free solution (cm2/s). v is the fluid velocity (m/s) and is described by Eq. 2 (49).
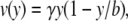 |
(2) |
where γ is the wall shear rate (1/s) and b is the thickness of the flow cell. As explained in the Appendix, mass transfer to the film and the gradients in the solution concentration are approximated based on laminar flow and boundary layer theory.
FIGURE 1.

Schematic representation of hybridization in the flow cell. Buffer enters the cell with target concentration CO. A thin depletion layer (δ) forms near the porous layer, across which target must diffuse. The concentration drop is much sharper in the porous layer, where the diffusion of the target is impeded by repeated association/dissociation with the probes. The image is scanned from the “backside”, with the emitted light being collected by a PMT. Note that the drawing is not to scale, and that b, the cell thickness, is 1 mm and h, the layer thickness, is typically 0.3–0.8 μm.
Within the porous layer (from y = 0 to y = h), the solution concentration and the concentration of adsorbed target (A) are described (4) by coupled Eqs. 3 and 4.
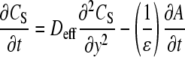 |
(3) |
and
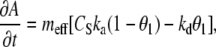 |
(4) |
where Deff is the “effective” diffusivity of the target in the porous film, which will be discussed in detail in the following sections. In Eq. 3, ɛ is the porosity, and the factor of ɛ−1 is included to normalize for the fact that the fluid volume in the layer depends on the porosity (i.e., adsorption of target in a low porosity film has a greater effect on Cs within the layer). meff is the “effective” volume density (M) of adsorption sites for full duplexes and is determined by Eq. 5.
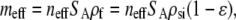 |
(5) |
where SA is the specific surface area of the colloidal silica (m2/g), ρsi is the density of pure silica (2.2 g/cm3), and ρf is the density of the porous film. neff is the effective density of sites that can form full duplexes (per unit of surface area) and differs from nT, the total density of probes on the surface, which includes both full-length and truncated probes, as will be discussed in the following sections. ka and kd are the adsorption and desorption coefficients, with units M−1s−1 and s−1, respectively. The determination of these values is discussed thoroughly in our article on planar glass substrates, and the values in Table 1 have been taken directly from that research (44). These values agree reasonably well with parameters measured by other authors on arrays on which intact probes have been immobilized. Based on the variability in the data (standard deviation) and the fit of the linear regression, the error in the ka values in Table 1 ranges 14%–16% (maximum for 20 PM, 45°C), and the error in kd ranges 17%–22% (maximum for 20 MM, 45°C).
TABLE 1.
Summary of ka and kd values for the probe sets at 22°C and 45°C (44)
|
ka (M−1sec−1 × 10−4)
|
kd (sec−1 × 104)
|
K(M−1 × 10−8)
|
||||
|---|---|---|---|---|---|---|
| Temperature | Match | Mismatch | Match | Mismatch | Match | Mismatch |
| 22°C | 6.4 | 6.2 | 0.50 | 1.2 | 13 | 5.2 |
| 45°C | 19 | 21 | 12 | 29 | 1.6 | 0.72 |
The boundary conditions are as follows: 1), the solution reaches the bulk concentration and is fixed at a distance δ from the surface, and 2), there is no flux at the boundary of the cell or at the boundary with the underlying glass substrate. These conditions are described by Eqs. 6 and 7, respectively.
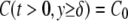 |
(6) |
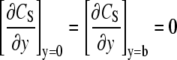 |
(7) |
Equation 6 is based on the approximation that the solution concentration is fixed beyond the initial boundary layer at the surface that is due to the no-slip condition. δ is solved in the Appendix and will be used as an approximation for the numerical model. The initial condition is that the concentration throughout the flow cell is zero, as described by Eq. 8.
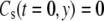 |
(8) |
To solve this system of equations, we developed a numerical model with a custom-written C++ routine.
Model for retarded diffusion in polymer gels
Extensive work and modeling has been performed on the diffusion of target molecules into and out of polymer gels on which DNA probes have been immobilized (2–4,41). This work provides an excellent basis for comparing and contrasting the hybridization behavior in our system and addresses the dependencies on the binding constant and the concentrations of both free and immobilized oligonucleotides. For diffusion in a nonfunctionalized gel, the characteristic time of diffusion is defined by Eq. 9 (2). In contrast, for a gel that has been functionalized with DNA probes, the characteristic diffusion time will depend on whether the binding is “strong” (Eq. 10) or “weak” (Eq. 11) (2).
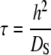 |
(9) |
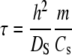 |
(10) |
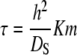 |
(11) |
Strong binding occurs when K is large and therefore KCs ≫ 1. Because the surface adsorbs a significant amount of the target, Cs becomes the key limiting factor of the hybridization. Conversely when K is small and KCs ≪ 1, the target will repeatedly associate and dissociate with the surface with frequency and duration governed by Km.
Modeling diffusion of oligonucleotides in porous silica films
The porous silica layer is similar to a very “thin” catalyst bed, and the mechanism of transport is most similar to “pore diffusion”, in which transport occurs primarily within the fluid phase of a porous particle (50). A solute molecule transported by pore diffusion may attach to the sorbent and detach many times along its path. Based on morphology considerations (i.e., binding effects are not included), the diffusion coefficient for pore diffusion in a typical porous catalyst can be estimated (51) by Eqs. 12 and 13.
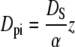 |
(12) |
and
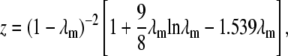 |
(13) |
where Dpi is the effective diffusivity in a porous catalyst due to morphology effects and α is the tortuosity factor, which quantifies the actual distance a molecule must travel between any two given points in the film relative to a straight line, and can be estimated by α = 1/ɛ (52). The factor z accounts for the restriction in diffusivity due to the pore size, and λm is the ratio of the size of the molecule to the size of the pore (rm/rp).
For pore diffusion control, the effective diffusivity is also strongly influenced by association and dissociation of the molecule with the surface of interest (50). For the case of the DNA arrays, we assume this interaction is dominated by binding to the surface-bound probes, as the signal in the probe regions is much greater than the background. Estimation of nT is complicated because the stepwise synthesis yield is 92%–94% (46), so the probe population will be a mixture of full-length and truncated probes. For the purpose of analyzing the pore diffusion, we begin with the assumption that the density of sites for interaction (nT) is not necessarily the same as the density of the sites that adsorb targets into stable duplexes (neff). In other words, the diffusing target can interact transiently with both full-length and truncated probes. As we have done for the estimation of ka, kd, and K (44), we approximate the target's interaction with the surface as a linear isotherm, or equivalently as a Langmuir surface at very low surface coverage (i.e., all adsorbing molecules interact only with an adsorption site and not with each other (53)). This assumption is based on the observation on flat glass that the full probe population is much greater than the portion that adsorbs targets into stable duplexes (44). The implications of these assumptions and the sensitivity of the analysis are discussed fully in the section “Physical interpretation of the R values and the transient complexes”. Finally, under this set of assumptions, the diffusion in the porous film is described by Eq. 14 (50).
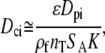 |
(14) |
where Dci is the effective diffusivity for pore diffusion. Combining Eqs. 12–14, we can estimate the overall effective diffusivity (Deff,si) in the porous layer, as shown in Eq. 15,
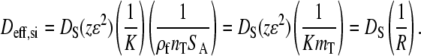 |
(15) |
The diffusivity is a product of morphology-dependent parameters and the product KmT, where mT is the total volume density of full-length and truncated probes that may differ from meff. KmT is analogous to the derivation for weak binding by Sorokin et al. (2). KmT acts as a “partition factor”, or approximately the ratio of the time a given target spends bound to the surface versus diffusing in solution. R is the overall retardation factor, which is equal to the ratio of the diffusivity in free solution to the retarded diffusivity within the porous layer. In the following sections, these equations will be used to analyze the overall retardation rate and to isolate the contributions of morphology and thermodynamic factors.
The equations above describe the primary approach used in this research. As part of the research on planar glass (44), a model was developed that described an “overshoot” phenomenon, and this model will also be applied to the porous glass films. However, the approach we have taken also has limitations. As the overshoot on planar glass demonstrates, structures other than 1:1 probe/target binding are possible on the photolithographically patterned arrays. Other types of structures of this nature may occur during the hybridization and are not accounted for explicitly in the analysis. Additionally, a significant contribution to the understanding of DNA arrays in recent years has been the modeling of electrostatic contributions (54–57). However, as discussed in the article on planar glass, given the complexity of the potential binding interactions on the photolithographically patterned surface, a direct application of the electrostatics model is not possible.
RESULTS
In this section, we first present the basic properties of the porous silica films. The hybridization kinetics on both ZL and T-S50 silica under a variety of conditions, including variations in film thickness, temperature, and solution concentration, are then discussed. Finally, we apply the retardation model to extract overall retardation values for each set of conditions.
Properties of porous silica substrates
Porous silica films were characterized with the techniques described previously (43), including ellipsometry (refractive index and thickness), profilometry (thickness), nitrogen adsorption (surface area, pore size), and scanning electron microscopy (SEM) (imaging). Fig. 2 shows SEM images of the surfaces, and the surface properties of the two films are compared in Table 2. The templated films simultaneously have higher pore size (rp), porosity (ɛ), and surface area than the ZL films, a morphology that should have advantages for mass transfer.
FIGURE 2.

SEM images of S50, “templated” S50 (T-S50), and ZL silica surfaces. The T-S50 silica has higher surface area, pore size, and porosity than the pure ZL silica.
TABLE 2.
Comparison of the properties of pure ZL and T-S50 films for 0.3 μm surfaces
| Property/Surface | Pure ZL Silica | T-S50 Silica |
|---|---|---|
| Porosity (ɛ) | 35% ± 5% | 65% ± 5% |
| Pore size (rp) | 23 ± 2 nm | 36 ± 4 nm |
| Matrix particle size | 65 ± 16 nm | 16 ± 5 nm |
| Surface area multiple (relative to flat) | 17 ± 3 | 26 ± 4 |
| Surface area accessed by synthesis* (relative to flat) | 14 ± 3 | 20 ± 4 |
| Relative synthesis yield (“RSY”; relative to flat glass) | 1.1 ± 0.1 | 1.2 ± 0.1 |
Silanated surface accessed by hydroxyl staining assay (43).
The efficiency of synthesis on the pure colloidal silica films has been evaluated previously (43), and the same techniques were used to characterize the efficiency on the T-S50 surfaces. Evaluation of the surface cleanliness and ability to support array synthesis were achieved with fluorescent staining (46) and HPLC-based (43) assays. Residual noncovalently bound fluorescein was negligible on all surfaces. Additionally, the relative synthesis yield of a probe on the porous surfaces was equivalent to that of flat glass, indicating that the porous nature of the films did not interfere with the step-wise synthesis reactions. Given this evidence, we concluded that the pyrolysis procedure resulted in a surface condition that is amenable to oligonucleotide synthesis without further postpyrolysis processing.
Hybridization on ZL silica films
Fig. 3 shows the hybridization behavior of a 0.3-μm ZL silica film at 22°C and 100 nM target concentration. For comparison to hybridization on flat glass, Fig. 4 is included by permission from the authors (44). The signal amplification ratio (relative to flat glass) of eightfold was consistent for both probe sets. Previously, we have shown that this signal amplification ratio can achieve up to ∼70% of the ratio of the surface area to flat glass (43). To fit the retardation model to the hybridization data, we must fit two key parameters: 1), the “shock transition zone” (STZ), and 2), the overall retardation value.
FIGURE 3.

Hybridization time course of 0.3-μm layer of ZL silica (22°C, 100 nM). The adsorbed target density is eightfold higher than the signal on flat glass for both probe sets (see comparison to Fig. 4). Note that the line is shown only as a guide to the eye. All hybridization data shown are the average of a minimum of 20 independent features.
FIGURE 4.

Hybridization time course on flat glass array at 100 nM, 22°C (reproduced by permission from Glazer et al. (44)).
Shock transition zone
For a relatively thin layer, a key assumption is how much of the outermost portion of the porous layer is “open” and is therefore at the same concentration as the neighboring solution. Numerical modeling shows that the best fit of the retardation model occurs when ∼0.15 μm, or about half of the layer, is set to the same concentration as the solution. This open portion of the top surface is analogous to an STZ that is often observed at the outermost portion of catalyst pellets (58). However, for catalyst beds, which have much higher adsorption capacity, this is often a steady-state phenomenon, whereas in the porous silica films the available sites in the outer portion rapidly saturate. The observed STZ is equivalent to the thickness of 2–3 stacked ZL silica particles, which implies that the top of the surface is more loosely packed than the deeper portion of the film.
Overall retardation values
Fig. 5 a shows the hybridization curves (20 PM, 22°C, 100 nM) for three thicknesses of ZL silica (0.3, 0.5, and 0.8 μm, created by deposition of 20, 30, and 42 wt % solutions, respectively), and Fig. 5 b shows the same data for the 20 MM. In Fig. 5 a, the best fit of the retardation model corresponds to an average R value of 4500 (4500, 4500, and 4400 for the 0.3, 0.5, and 0.8 micron films, respectively). More variability was typically observed in the thin films at short times than in the thick films. We attribute this to the presence of the STZ, which accounts for half or more of the thickness of the thin films; and therefore small differences in this zone can greatly influence the overall time to saturation. For the 20 MM in Fig. 5 b, the model corresponds to an average R value of 3800 (4000, 3800, and 3600 for the 0.3, 0.5, and 0.8 micron films, respectively). The R values for both the PM and MM probe sets are relatively independent of film thickness, and therefore the retardation model is able to represent the hybridization behavior.
FIGURE 5.

(a) Hybridization time course (20 PM, 22°C, 100 nM) on ZL silica layers 0.3, 0.5, and 0.8 μm thick and the best fit (—) of the retardation model, which corresponds to an average R = 4500 (4500, 4500, and 4400 for 0.3, 0.5, and 0.8 μm, respectively). The model fits the long diffusion-limited regime more closely than the portion where the surface approaches saturation. (b) Hybridization time course (20 MM, 22°C, 100 nM) on ZL silica layers 0.3-, 0.5-, and 0.8-μm thick, and the best fit (—) of the retardation model, which corresponds to an average R = 3800 (4000, 3800, and 3600 for 0.3, 0.5, and 0.8 μm, respectively).
As shown in Fig. 5, a and b, the model is most effective at simulating the long approach to equilibrium although there is slight deviation in the region where the film approaches saturation. This deviation may be due to the fact that we have approximated the adsorption as simple Langmuir behavior up to this point and have not taken into account the slight “overshoot” behavior (see Fig. 3). On flat glass distinct overshoot behavior was observed for 18-mer and 16-mer probe sets, on which the adsorbed target density reached a higher value before declining to the final plateau (44). In the section “Overshoot behavior on high-capacity ZL and T-S50 surfaces” the potential contributors to overshoot behavior on the porous silica films are discussed. Additionally, the assumption has been made that Dci does not change as the surface coverage increases because of the large population of truncated, unoccupied probes. If the actual portion of the population that interacts with the target is much smaller, then this assumption may not hold true and could contribute to deviation of the model near saturation. The sensitivity to assumptions about the probe population is discussed further in the section “Physical interpretation of the R values and the transient complexes”.
The increase in time to equilibrium with increased film thickness suggests a diffusion-limited process, and the long diffusion-limited regime is clearly evident in Fig. 6, where we plot the 0.5 micron data versus t0.5. Early in the time course, the signal increases rapidly as the STZ saturates. After saturation of the top surface, the target must diffuse deeper into the film to find available sites, resulting in a long regime that is linear versus t0.5, as is expected for diffusion-limited adsorption (49). Finally, as the film approaches saturation, the slope drops off due to the low availability of sites.
FIGURE 6.

Description of the regimes during hybridization on ZL silica films (0.5 micron, 20 PM, 22°C, 100 nM).
For the 0.5 and 0.8 μm films, we note a decrease in the signal that occurs after ∼7–10 h (or later), although no changes were observed in the thickness of the silica film itself. Similarly, abrupt changes in the signal on the planar glass films were often observed at this time. This effect is likely attributed to film degradation resulting from the extremely high flow rate, which is 15–20 times faster than that used for conventional assays, for which no such degradation is observed (44).
Hybridization on T-S50 surfaces
Fig. 7 shows the hybridization of the T-S50 surface (0.3 μm, 22°C, 100 nM). In comparison to a ZL surface of similar thickness (see Fig. 3), the T-S50 reaches a higher adsorbed target density at the plateau (1.3–1.5 times greater than ZL silica), hybridizes more quickly, and has a distinct overshoot that is observed in both probe sets. Overall the hybridization is 11 times higher than flat glass for a 0.3-μm film and shows that the templated surface has several advantages as a substrate for high-capacity DNA arrays.
FIGURE 7.

Hybridization time course on 0.3-μm layer of T-S50 silica (22°C, 100 nM). In contrast to the ZL surface, the T-S50 shows much more distinct overshoot behavior. R = 1400 for both probe sets, as shown by the models (— for 20 PM and — for 20 MM). The overshoot is not due to nonspecific adsorption, as the background is constant over the time during which the overshoot occurs.
The best fit of the retardation model for the 20 PM probe set corresponds to R = 1400, and under these conditions, retardation is 4500/1400 ≈ 3 times greater on the ZL silica. The STZ equals 0.06 μm, which corresponds to two to three of the matrix (S50) particles. The model fits the initial adsorption period (up to ∼70% of the maximum) equally well regardless of whether we consider the maximum (top of the overshoot) or the plateau capacity, which affirms that the overshoot does not cause misinterpretation of the data. The overshoot is not due to nonspecific adsorption of the target to the porous silica, as all signals have been corrected by subtracting the background signal from a region with no probe synthesis. The background was constant over the same timespan that the overshoot occurs in the probe regions, as shown in Fig. 7.
Effects of temperature and concentration on hybridization
Effect of temperature
Fig. 8 shows the effect of elevating the temperature to 45°C for the T-S50 and ZL silica surfaces (0.3 μm, 20 PM, 100 nM). By comparison to data under the same conditions on flat glass (44), we observe that the amplification ratios on the ZL and T-S50 silica are ∼6 and 8, respectively, or slightly lower than the amplification at 22°C. One contributing factor to this difference could be the secondary rise in adsorption observed on the flat glass arrays, which is not observed on the porous arrays. If these types of substrates are used in practical DNA arrays in the future, it may be possible to reach greater amplification after further optimization of the hybridization and washing conditions. The hybridization overshoot is very clear in the T-S50 but is not evident in ZL, although both surfaces approach their maxima at similar rates. The ZL surface is fit by R = 800 and the T-S50 is fit by a similar value of R = 700, which contrasts the behavior at 22°C, where retardation on the ZL silica is about three times greater.
FIGURE 8.

Hybridization time course at 45°C for 0.3-μm layers of ZL and T-S50 silica (20 PM, 100 nM). In contrast to the behavior at 22°C, both surfaces hybridize at similar rates, in this case with R = 800 for ZL and R = 700 for T-S50.
Effect of concentration
Fig. 9 shows the behavior of the 20 PM at 22°C and 10 nM target concentration. The 0.3-μm ZL film corresponds to R = 4400, which is nearly identical to the value at 100 nM. The best fit for the T-S50 is for R = 2000, which is slightly higher than observed at 100 nM but still less than half of the value for the ZL silica. For the T-S50, the signal shows instability developing at ∼500 min, making interpretation of the overshoot at extended times more difficult. Fig. 10 shows that for the ZL silica the retardation factor is independent of the solution concentration at 45°C, with R = 800, as was the case for 100 nM. For T-S50 the value is slightly higher than at 100 nM, with R = 1100.
FIGURE 9.

Hybridization time course at 10 nM on 0.3-μm layers of ZL and T-S50 silica (20 PM, 22°C). The best fit (—) of the T-S50 is given by R = 2000, whereas the ZL silica is twofold slower, at R = 4400.
FIGURE 10.

Hybridization time course at 10 nM and 45°C on 0.3-μm layers of ZL and T-S50 silica (20 PM, 45°C). The best fits (—) of the ZL and T-S50 are given by R = 800 and 1100, respectively.
Table 3 shows a summary of the R values for all of the conditions observed on the 0.3 micron films. Error values are determined from the variability (standard deviation) in the data and the accuracy of the fit by the model equations. At 22°C, the ratios of RZL/RT-S50 range 2.2–3.2 and are slightly higher at 100 nM than at 10 nM. At 45°C the range is 0.5–1.1, and again the ratios are slightly higher at 100 nM than at 10 nM. The temperature dependence is also evident from the ratios of R22°C/R45°C in Table 3, which range 5.5–8.3 for ZL and 1.8–2.0 for T-S50. Based on this analysis, it appears that the behavior at 22°C and 45°C is governed by different factors. In the following section the retardation model will be further developed, and the potential contributors to this behavior will be considered.
TABLE 3.
Summary of R values on 0.3 micron silica films
|
R
|
RZL/RT-S50
|
R22°C/R45°C
|
||||||
|---|---|---|---|---|---|---|---|---|
| Surface | T | Cs (nM) | 20 PM | 20 MM | 20 PM | 20 MM | 20 PM | 20 MM |
| ZL | T 22°C | 100 | 4500 ± 400 | 4000 ± 400 | 3.2 | 2.9 | 5.6 | 6.7 |
| T-S50 | 1400 ± 300 | 1400 ± 300 | 2.0 | 2.0 | ||||
| ZL | 10 | 4400 ± 500 | 5000 ± 500 | 2.2 | 2.5 | 5.5 | 8.3 | |
| T-S50 | 2000 ± 400 | 2000 ± 400 | 1.8 | 1.8 | ||||
| ZL | T 45°C | 100 | 800 ± 200 | 600 ± 100 | 1.1 | 0.9 | ||
| T-S50 | 700 ± 200 | 700 ± 200 | ||||||
| ZL | 10 | 800 ± 200 | 600 ± 200 | 0.7 | 0.5 | |||
| T-S50 | 1100 ± 100 | 1100 ± 300 | ||||||
A key factor for the practical application of DNA arrays is the development of the discrimination ratio (MM/PM). As hybridization assays on photolithographically patterned arrays are often performed at slightly elevated temperatures and low concentrations, we consider, for example, the data at 45°C and 10 nM. The flat glass, ZL silica, and T-S50 silica achieved similar ratios of 0.36 to 0.40, 0.39 to 0.43, and 0.41 to 0.45, respectively. The ratios were achieved in 15–20 min on flat glass and in much longer times of 200–300 and 300–400 min on ZL and T-S50 silica, respectively. If high-capacity silica arrays are used in the future for practical applications of DNA arrays, it may be possible to improve the speed and selectivity of the arrays by further optimizing the hybridization and washing conditions for the high-capacity substrates.
DISCUSSION
In this section, we first analyze the trends in the overall retardation values versus the surface type, temperature, and solution concentration. From these values, we then extract the morphology dependence of the hybridization kinetics. Finally, we are able to relate the retardation values to the nature of the transient probe/target duplexes that form as the target penetrates into the porous film.
Analysis of trends in overall retardation (R values)
Behavior at 22°C
To compare the retardation values on the ZL and T-S50 surfaces to theoretical expectations, we begin by examining the effective diffusivities using Eq. 15. To estimate the z factors, we must first evaluate λm. For a single-stranded target molecule diffusing in solution, rm can be estimated as the radius of gyration (rg) of the random coil in free solution. Previously it was estimated that rg for the 20-mer target under the conditions used in this study is ∼2 nm (43), and the target is at most 10% of the size of the pore (see Table 2). By substitution into Eq. 13, z = 0.7 for ZL and 0.8 for T-S50, and we observe that for pores that are at least an order of magnitude larger than the target, the pore size plays a relatively minor role in the diffusivity.
We then combine the z factors, the porosity values from Table 2, the surface area of pure ZL and S50 silica (35 and 104 m2/g, respectively, from Table 2), and take the ratio [Deff]T-S50/[Deff]ZL, which gives the estimate that the T-S50 should hybridize ∼2.3 times faster than ZL. The increased porosity of the T-S50 enhances the rate but competes with increased retardation due to the higher surface area (and therefore probe concentration) of the small matrix silica. This ratio is very similar to the range of RZL/RT-S50 at 22°C shown in Table 3. This is an important finding and indicates that when the hybridization kinetics are “normalized” for the surface morphology, the nature of the transient complexes that form as the target diffuses through the ZL and T-S50 surfaces are of similar nature.
Behavior at 45°C
Table 3 shows that at 45°C, the overall R values for the ZL and T-S50 films are similar. Surprisingly, retardation on the T-S50 substrate is slightly greater than the ZL in some cases. Based on the morphology predictions and the behavior at 22°C, this is an unexpected result. One possibility is that at low retardation values the film thickness becomes less significant when compared to the diffusion rate of the target, and therefore the morphology predictions become less reliable. Even single catalyst pellets are typically orders of magnitude greater than the layer thickness, such as 10 μm or more, and a packed bed reactor may be many orders of magnitude greater.
As a qualitative comparison, we can take the maximum retardation coefficient of ∼4500 (ZL, 22°C, 100 nM, 20 PM), and using the relationship x = (Dt)0.5 and the layer thickness of 0.3 μm, we estimate that even with retarded diffusion a target molecule can traverse the layer in ∼5 s. For the minimum observed retardation value of ∼600 (ZL, 45°C, 100 nM, 20 MM), the same path takes only ∼0.7 s, and the target spends negligible time in the layer. Correspondingly, the ratio of RZL/RT-S50 changes from a maximum of ∼3 to <1, and the predictive equations based on morphology may no longer be valid. As the thickness of the porous layer becomes small with respect to the characteristic diffusion length, it may be that the impact of the porous morphology diminishes as well.
Given these considerations, an alternative interpretation of the behavior at 45°C can be made by neglecting the morphology-dependent parameters in Eq. 15, which can be accomplished by setting zɛ2 = 1. Note that the 1 − ɛ factor is left in the calculation of ρf, as this parameter affects the concentration of probes. Given this set of assumptions, the predicted ratio of RZL/RT-S50 is proportional to mZL/mT-S50 = [(1 − ɛ)SA]ZL/[(1 − ɛ)SA]T-S50 = 0.6, and by this rationale the T-S50 may actually respond more slowly than the ZL. This ratio agrees well with the range of RZL/RT-S50 at 45°C in Table 3. As the influence of the morphology diminishes, it appears the hybridization is dominated increasingly by the probe concentration.
This analysis implies that the morphology predictions are less applicable at 45°C than at 22°C, and to further test this possibility, a more quantitative approach is needed. One method is to compare the actual rate of hybridization to what would be expected if there were no diffusion limitations (59). The maximum rate can be estimated by evaluating Eq. 4 uncoupled from Eq. 5, which can be accomplished with the mathematical model developed for the coupled analysis. For this analysis we evaluate the hybridization for θ ≤ 0.75, which takes into account the STZ and the majority of the diffusion-limited regime, before the final interface-limited regime dominates (see Fig. 6). The results are shown in Table 4 for the 100 nM experiments.
TABLE 4.
Comparison of maximum theoretical versus observed hybridization rates (100 nM)
| Surface | Temperature | Actual time to reach 75% of plateau (min) | Theoretical time to reach 75% of plateau (min) | Actual/theoretical |
|---|---|---|---|---|
| ZL | 22°C | 43 | 3.6 | 12 |
| T-S50 | 22°C | 21 | 3.6 | 6 |
| ZL | 45°C | 11 | 1.3 | 8 |
| T-S50 | 45°C | 8 | 1.3 | 6 |
This analysis is useful for comparing the surfaces and temperatures. The ZL surface is slightly more limited by diffusion than the T-S50 at both temperatures and is affected significantly by the change in temperature. The T-S50 on the other hand shows similar ratios at both temperatures. However, overall the ratios are not significantly different, which suggests that these surfaces are similarly affected by diffusion, and the model equations should have the same applicability and limitations.
It is therefore possible that our approach, which assumes the morphology and thermodynamic factors can be considered independently, may not completely describe the system, especially at 45°C. A type of a phenomenon beyond consideration of our model would be if the morphology and thermodynamics have more complex interactions. For example, as the temperature increases and the transient interactions become less stable, pathways may open in the ZL that were not accessible at lower temperatures. The pores in the ZL are ∼23 nM. Although in free solution the target length is estimated as a 2-nm coil, an extended 20-mer duplex can span up to 7 nm (43). If two duplexes were extended from opposite surfaces, an extended target could nearly bridge them. In the T-S50 where the pores are larger, a subtle difference of this nature may have less of an effect. However, given the variability in the pore size and distribution, the complex probe distribution, and even potential target/target binding, an explicit model of this sort of phenomenon is beyond the scope of our research.
Physical interpretation of the R values and the transient complexes
Factors governing the transient complexes
As shown in Eq. 15, the overall retardation factor should be equal to KmT/zɛ2. In this section we compare and contrast this model to that proposed for polymer gels and evaluate the implications for the types of interactions occurring on the arrays. In the analyses by Sorokin et al. (2,3), the characteristic diffusion time observed was proportional to m/Cs for strong and Km for weak binding, with strong binding occurring for KCs ≫1 and weak for KCs ≪ 1. For the full-length probes, binding constants on the porous glass surfaces can be approximated by the values determined on the flat glass surface (see Table 1). KCs would range from 130 (20 PM, 22°C, 100 nM) to 0.7 (20 MM, 45°C, 10 nM) and therefore should play a factor for many of the conditions and will have some influence even at the lowest end of this range.
However, despite these large values of KCs, the binding observed in this study appears to correspond much more closely to the model for weak binding and in particular the dependence on Km. Based on Table 3, the retardation does not change significantly over a 10-fold change in concentration, whereas the Sorokin model for strong binding would predict a significant inverse dependence on the solution concentration. The T-S50 has a slight increase in R with decreasing concentration but much less significantly than expected from the Sorokin model and only slightly above the uncertainty of the measurements. Lower concentrations were not tested because of instability that can develop in the films at very long hybridization times due to the very high flow rate used in this study.
Additionally, the R values and the ratios of R22°C/R45°C change much more significantly with temperature than with concentration, suggesting a correlation to the K value of the probe set. For example the ratio of ZL silica (20 PM, 100 nM) at 22°C vs. 45°C is 5.6, which is similar to the ratio of the K values of 8.1 (see Table 1). The ratio for the T-S50 under these conditions is only 2.0, and as noted above this may be attributed to the limitations of the catalyst equations for lower retardation values.
To understand why the behavior appears to fit the weak binding model even for significant values of KCs, we must further examine the probe distribution on the photolithographically patterned arrays. An important distinction exists between polymer gel arrays on which intact full-length probes have been immobilized. Given that the stepwise synthesis yield cited in the literature is 92%–94% (46), the probe distribution will consist of both full-length and truncated probes. The density of sites for probe synthesis is 70–90 pmol/cm2 (42). The minimum number of full-length probes is therefore 70 pmol/cm2 × (0.92)20 = 13 pmol/cm2. The number of probes that achieve hybridization at equilibrium is ∼1 pmol/cm2 on flat glass arrays (44). In our previous work we have discussed how limitations on planar arrays are likely a result of both electrostatic limitations, which have been discussed by other authors (54–56,60), as well as steric influences (44). Therefore, because the adsorbed target density is much less than the full population of full-length and truncated probes, the population that the target can interact with does not change significantly as the surface approaches saturation. The assumption behind the strong binding model is that Cs determines the equilibrium concentration of free probes available to interact with the target. As a result the binding is better represented by the weak binding approximation of proportionality to Km. In the following sections we evaluate the range of possible scenarios that contribute to the effective K and m values the target experiences.
The work by Sorokin et al. (2,3) provides the basis for other comparisons to their results. Their most recent work addresses the factors that contributed to the rate of hybridization and discrimination between perfect match and mismatch probes. Higher probe concentration and lower porosity contribute to longer hybridization times. In our current work these variables are not independent, as the T-S50 has both higher porosity and higher probe concentration (due to higher surface area). However, our interpretations are consistent with their observations. At 22°C, the higher porosity of the T-S50 appears to contribute to faster hybridization. Conversely, at 45°C it appears the higher probe concentration in the T-S50 may be the dominant factor, and kinetics are similar or slower than on the ZL. With regard to discrimination, their work suggests that there should be a significant difference in the characteristic times for the PM and MM probes due to differences in K. However, our results in Table 3 show very similar retardation values for PM and MM under a given set of conditions. Again, it is likely that transient interactions with the truncations define the retardation rate, rather than Kfull length. For the polymer system the discrimination was inversely proportional to the fluorescence intensity. This finding does not hold true for the ZL and T-S50, which have very similar ratios although the adsorbed target on the T-S50 is greater.
Scenarios for Ktransient and mT for the transient complexes
Based on the assumption that the retardation has a component proportional to KmT, we can analyze the various scenarios that could contribute to this product. Since there is uncertainty in both the value of K for the transient complexes (i.e., how many basepairs form?) and the value of m (with which part of the probe population do the targets interact?), we must consider these factors separately. A range of possible scenarios is shown in Table 4
To vary mT, we vary nT so that the probe population can be compared directly to results on planar glass (by normalizing for the surface area). We first consider scenarios for Ktransient, which could have a maximum value of Kfull length. In this case, nT would be five to six orders of magnitude less than the full probe population of 70–90 pmol/cm2 (see the section “Physical interpretation of the R values and the transient complexes”). On the planar glass films, it was observed that ≤1 pmol/cm2 of DNA hybridized, or ∼1% of the probes, which is therefore three to four orders greater. It therefore is unlikely that Ktransient is equal to Kfull length.
At the other extreme is the case where nT = 90 pmol/cm2, and the resulting values of Ktransient are five to six orders of magnitude lower than Ksurface. If we use the behavior on planar glass as a basis and set nT = 0.9 pmol/cm2, then Ktransient is still three to four orders of magnitude lower than Ksurface. Finally, it has been observed that ka does not vary greatly for short duplexes, and if the assumption is made that ka,transient = ka,full length and nT = 0.9 pmol/cm2, then kd,transient is less stable than kd,surface by the same factor as the ratio of K values for that scenario in Table 4.
What is significant about this analysis is that the data set on planar glass allows an internally consistent comparison for the porous films and does not rely on comparisons to solution-based values (see discussion below) or other DNA array systems. A rigorous analysis of this type for the same probe/target system on both a planar and porous system has not been undertaken in the literature. The use of chemically and mechanically similar supports, glass and silica, maximizes the consistency of the comparison. For example, the different environments on planar glass and in a polymer gel make this comparison more difficult. The comparison of the planar values to the transient complexes makes clear that the latter are much less stable and therefore consist of fewer basepairs. In the following section we further explore the number of basepairs involved.
Estimation of nbp in the transient complexes
To tie the values of Ktransient to a number of basepairs (nbp), free energies of nucleation and free energy change per basepair reaction are needed. However, this fundamental knowledge is not available on DNA arrays in general (45) and for the photolithographically patterned arrays in particular. Forman et al. (47) observed a range of probe lengths, but values for ka and kd were not derived for each. In the article on planar glass (44), only a small range of probe lengths was considered.
We therefore consider parameters measured for solution-based hybridization. The comparison of the stability of surface-bound duplexes versus solution phase has been a subject of extensive research, such as summarized in Levicky (45). In most cases the surface-bound duplexes are less stable than their solution-based counterparts. If we assume that basepairing is the dominant probe/target interaction mechanism, then we can use the nearest-neighbor approach of Breslauer et al. (61), who determined the ΔH and ΔS for the 10 possible basepairing reactions. However, if we use this approach, the stability of the 20 PM at 22°C would be expected from only 10.3 bases and 13.3 at 45°C. Therefore, by using solution-based values, we are essentially calculating the lower limit for the number of basepairs in the transient complexes.
Peterson et al. observed that targets have more rapid hybridization kinetics when they are complementary to the upper portion of a probe than the portion near the surface (60), and we therefore estimate the number of bases and strength of interaction by excluding the basepairs nearest to the surface. These K values can then be combined with a range of potential probe densities to estimate the expected retardation, and in Fig. 11 these estimates are overlaid with the average values observed for the ZL silica (0.3 micron surface, 20 PM, 100 nM, 22°C). Over a very wide range of assumptions about the site density, the nbp varies from 6.6 to 8.8. When all data on the 0.3 micron surface is considered for both probe sets, the range is only slightly greater at 6.5–8.8 bases. The same analysis on T-S50 yields the identical range of 6.5–8.8.
FIGURE 11.

Calculation of the expected retardation of the diffusing target based on nbp and the portion of the probe population (nT) that may interact with the target. For the observed retardation of the ZL silica (20 PM, 100 nM, 22°C), the resulting estimate of nbp corresponds to 6.6–8.8 bases. The diagonals have been determined by estimating the retardation contributed by each of the basepairs in the surface-bound duplex. Since each basepair does not contribute the same degree of retardation (i.e., G/C pairs have stronger effect than A/T), the diagonal lines are not purely linear on the exponential scale.
When this same analysis is performed at 45°C the range is 7.9–11.1 for ZL, and similarly it is 8.3–11.7 for T-S50. However, as discussed previously, morphology effects play a more significant role for higher values of overall retardation, such as at 22°C. If we repeat the analysis where zɛ2 = 1 (see the section “Analysis of trends in overall retardation (R values)”), then the new estimates of nbp (9.0–12.3 for ZL and 8.8–12.2 for T-S50) are in slightly better agreement. However, because of the exponential dependence on nbp, correlation to specific ranges of bases using solution-based values is unreliable. This analysis reemphasizes the significance of the comparison to the planar arrays, and an interesting comparison is to the K values for 16-mers observed on planar glass (44). At 22°C, the 16-mers are only threefold less stable than the 20-mers, and at 45°C they are fivefold lower. In comparison to the data in Table 5, this implies that the transient complexes may consist of well less than 16 basepairs.
TABLE 5.
Scenario analysis for Ktransient and nT on 0.3 μm films
| 22°C/100 nM
|
22°C/10 nM
|
45°C/100 nM
|
45°C/10 nM
|
|||||
|---|---|---|---|---|---|---|---|---|
| ZL silica | 20 PM | 20 MM | 20 PM | 20 MM | 20 PM | 20 MM | 20 PM | 20 MM |
| nT/ntotal probe populiation if Ktransient = Ksurface | 6.5 E − 06 | 1.3 E − 05 | 6.6 E − 06 | 1.9 E − 05 | 9.8 E − 06 | 1.6 E − 05 | 9.8 E − 06 | 1.6 E − 05 |
| Ktransient/Ksurface if nT = 0.9 pmol/cm2 | 6.5 E − 04 | 1.3 E − 03 | 6.6 E − 04 | 1.9 E − 03 | 9.8 E − 04 | 1.6 E − 03 | 9.8 E − 04 | 1.6 E − 03 |
| Ktransient/Ksurface if nT = 90 pmol/cm2 | 6.5 E − 06 | 1.3 E − 05 | 6.6 E − 06 | 1.9 E − 05 | 9.8 E − 06 | 1.6 E − 05 | 9.8 E − 06 | 1.6 E − 05 |
| T-S50 silica | 20 PM | 20 MM | 20 PM | 20 MM | 20 PM | 20 MM | 20 PM | 20 MM |
| nT/ntotal probe populiation if Ktransient = Ksurface | 4.8 E − 06 | 1.2 E − 05 | 6.8 E − 06 | 1.7 E − 05 | 1.9 E − 05 | 4.3 E − 05 | 3.0 E − 05 | 6.8 E − 05 |
| Ktransient/Ksurface if nT = 0.9 pmol/cm2 | 4.8 E − 04 | 1.2 E − 03 | 6.8 E − 04 | 1.7 E − 03 | 1.9 E − 03 | 4.3 E − 03 | 3.0 E − 04 | 6.8 E − 03 |
| Ktransient/Ksurface if nT = 90 pmol/cm2 | 4.8 E − 06 | 1.2 E − 05 | 6.8 E − 06 | 1.7 E − 05 | 1.9 E − 05 | 4.3 E − 05 | 3.0 E − 05 | 6.8 E − 05 |
Binding scenarios that lead to the transient complexes
To further develop a physical interpretation of the transient complexes, we must consider the photolithographic synthesis process and the implications for the probe length distribution and possible binding stoichiometries (i.e., the number of targets bound per probe). The minimum spacing between probes (i.e., full-length and/or truncations) is <2 nm (43). This spacing is shorter than the length of an extended 20-mer duplex, which may reach up to nearly 7 nm (43), and therefore it is possible that a single target may have interactions with multiple probes.
If this scenario occurs, the target will eventually proceed to either fully zipper (62,63) with one of the probes or to desorb and continue diffusing, as shown in Fig. 12. It follows that retardation is the result when the second case occurs. Since the total probe population with which the target can interact (∼90 pmol/cm2) is about two orders of magnitude greater than the probes that eventually hybridize (<1 pmol/cm2), it is reasonable to suggest that these types of transient interactions are much more prevalent than full hybridization.
FIGURE 12.

Scenario for formation of metastable complexes, which then leads to either the target desorbing and continuing to diffuse (“retardation”) or the formation of a stable duplex with a full-length probe. Because of the close spacing of full-length and truncated probes, photolithographic arrays may be prone to these types of complexes. Based on the density of stable duplexes on flat glass (∼1 pmol/cm2) versus the total density of full-length and truncated probes (70–90 pmol/cm2), the retardation scenario will be much more prevalent.
The lower limits of the ranges observed at both 22°C and 45°C (seven and eight basepairs, respectively) imply the formation of complexes with targets adsorbed to multiple probes, as it is unlikely that this many bases could form between a single probe/target combination without the combination fully zippering. This type of behavior may be more prominent on photolithographically patterned arrays than arrays where full-length, intact probes have been immobilized at lower densities.
Overshoot behavior on high-capacity ZL and T-S50 surfaces
In the study on flat glass substrates (44), we proposed a model that explains how the overshoot on the planar glass substrates can be attributed to the formation and decay of “oversaturated” complexes, a phenomenon that was observed on 16- and 18-mer probes, but not 20-mers. On the porous glass a large overshoot is observed on the T-S50 20-mers under several conditions, and only a slight overshoot is observed on the ZL (22°C, 100 nM). It is therefore evident that in addition to a dependency on the probe set, the overshoot is also influenced by the film morphology.
When the parameters determined in the planar glass study (from the 18-mer probe set) are applied to the 20-mer data on the porous surfaces, the predicted overshoot is less than observed on the ZL silica and much less than observed on the T-S50. It is likely that a full analysis of the overshoot behavior on the high-capacity substrates would require a more complex model that fully takes into account the implications of the pore size distribution. For example, it is possible that the smallest pores in the film have disproportionate impacts on the diffusion. In small pores, the diffusion is restricted excessively due both to geometric factors as well as to an increased local concentration of probes with which the target can interact. These effects are expected to be even more pronounced in the T-S50 glass, which has a bimodal distribution of pores (i.e., pores are formed both by the large voids left behind by pyrolyzed latex, as well as the small natural voids between the matrix silica).
This hypothesis agrees well with the data presented in Figs. 3 and 7, where the T-S50 film shows much larger overshoots than the ZL. The majority of hybridization will occur quickly as the target takes the path of least resistance through the large voids. Diffusion into the small pores will slow the reaction in the crevices of the film, and targets that enter these small pores will also be slow to wash out, which could explain the surface-dependent component of the large overshoot observed in the T-S50 films. Small pores in the ZL silica (due to polydispersity) most likely contribute by the same mechanism to the overshoot observed on those films, although this effect is much smaller than on the T-S50.
In addition to the morphology, the overshoot is also a function of concentration. Figs. 8 and 10 show that at 45°C the overshoot disappears as the concentration decreases from 100 to 10 nM. As is shown in Fig. 10 of the article on planar glass, when the concentration is high (such as 100 nM) binding occurs rapidly, the decay of the secondary complexes occurs over the same period, and the overshoot is observed. In contrast, at 10 nM the binding occurs over a long time and the decay of the secondary complexes does not cause a noticeable deviation from monotonic behavior.
This behavior also implies that the overshoot is a function of flow rate, as the effect of a lower flow rate would be to expand the depletion layer and slow the diffusion of target to the surface (see the Appendix), which would have a similar effect to lowering the concentration. However because of the instability of films at long times (see the section “Hybridization on ZL silica films”), an optimization by lowering the flow rate (which would lengthen hybridization time) and increasing the concentration would require significant trial and error and was beyond the scope of this research.
CONCLUSION
Porous silica films provide a high-capacity support that is effective for obtaining greater adsorbed target densities on photolithographically patterned arrays. The properties of the film can be modulated easily with use of different matrix silica, concentrations, spin speeds, and templating with latex. Discrimination on the arrays appears similar to planar glass, suggesting that the porous matrix does not interfere with the relative stabilities of the probe sets. This similarity to binding on planar glass, as well as use of the same chemical processes, can help facilitate the use of these types of surfaces and may avoid the necessity for reinterpretation of the body of data developed on planar arrays.
We have developed a theoretical model that explores the governing factors on the porous silica films. Adsorption parameters measured on planar glass provide key input parameters for the model, and the use of similar surfaces maximizes the consistency of the comparison. The adsorption is characterized by retarded diffusion due to both morphology factors and transient interactions between the diffusing target and the probes. The transient complexes are much less stable than full duplexes, with the latter having binding constants greater by three orders of magnitude or more. These short-lived complexes may be a result of the unique nature of the photolithographically patterned system, in which the total density of full-length and truncated probes greatly outnumbers the density of hybridized duplexes, and the proximity of probes that may facilitate non-1:1 binding structures (44). The behavior at 22°C is fit well by the predictive equations for morphology. The behavior at 45°C does not match the expectations based on morphology and suggests that either the behavior is dominated by thermodynamics or that there are more complex interactions between morphology and thermodynamics than captured in the model.
The models and interpretations developed can help to broaden the understanding of DNA hybridization in three-dimensional surface environments. The existing models and interpretations for polymer gel arrays provided a starting point for the analysis, but they do not discuss the potential impacts of factors such as heterogeneous probe populations, partial complexes forming during diffusion, or binding of a target to more than one probe. In future studies, it would be valuable to compare the kinetics on the photolithographic system to those observed for immobilized arrays on polymer gels, on which the probe population is nearly homogeneous, and to examine how much of the difference in the kinetic behavior is due to morphology factors versus the probe spacing and heterogeneity. Additionally, it should be noted that only one probe/target sequence, probe synthesis chemistry, and probe distribution have been addressed in this study, and additional studies are needed to compare other systems.
As the drive to increase the signal output from arrays continues, so will the demand for optimization of binding and discrimination in three-dimensional volumes. The substrates and models described in this report can provide a step toward that goal. Beyond DNA arrays, these types of substrates and models can also find use for high-capacity molecular recognition of other molecules, such as in the rapidly growing field of proteomics.
Acknowledgments
The authors thank David Stern, Audrey Suseno, Paul Bury, Nineveh Parker, Michael Savage, and Paul Ciccolella of Affymetrix and Larry Bailey of Stanford University. Nathan Wilson of Stanford University provided assistance in the development of the C++ routine for analysis of the kinetics data.
In addition to Affymetrix, funding was provided by the Advanced Technology Program (ATP; sponsored by the Department of Commerce, administered by the National Institute of Standards and Technology), the Center for Polymer Interfaces and Macromolecular Assembly (CPIMA; sponsored by the National Science Foundation), and the Graduate Research Fellowship Program (GRF; sponsored by the National Science Foundation).
APPENDIX: MASS TRANSFER FROM THE FLOW STREAM
For the numerical simulation of the adsorption process, we refer to the analysis of the boundary layer in a laminar flow arrangement (59,64). The mass transfer coefficient (kc) is calculated with Eq. A1.
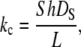 |
(A1) |
where Ds is the diffusivity of the target in free solution (∼10−6 cm2/s), and L is the chamber length (1.25 cm). Sh is the Sherwood number and is calculated by Eq. A2 (64).
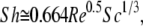 |
(A2) |
where Re is the Reynolds's number, which is calculated by Eqs. A3 and A4.
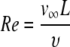 |
(A3) |
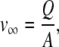 |
(A4) |
where v∞ is the fluid flow velocity (m/s), Q is the volumetric flow rate (∼80 mL/min), A is the cross sectional area of the flow cell (1.25 cm × 0.1 cm = 0.125 cm2), and ν is the kinematic viscosity (∼10−6 m2/s). Sc is the Schmidt number, defined by Eq. A5.
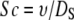 |
(A5) |
The equations are solved to give v∞ = 11 cm/s, Re ≈ 1400 (indicative of laminar flow), and kc = 4 × 10−6 m/s. These values give a depletion layer (δ) with thickness that is estimated by the relation δ = Ds/kc = 20 μm.
For the numerical solution of the system of mass transfer and adsorption equations (Eqs. 1–4), we have simplified the model by assuming constant bulk solution concentration outside of the boundary layer and then solving for the concentration gradients within the boundary layer and the porous film. The R values were not highly sensitive to the estimate of the boundary layer thickness (within a reasonable variation of the thickness). This finding is not surprising given that diffusion within the layer is two to three orders of magnitude slower than free solution and is therefore the limiting factor in the hybridization kinetics.
Editor: Jonathan B. Chaires.
References
- 1.Dufva, M. 2005. Fabrication of high quality microarrays. Biomol. Eng. 22:173–184. [DOI] [PubMed] [Google Scholar]
- 2.Sorokin, N. V., V. R. Chechetkin, M. A. Livshits, V. A. Vasiliskov, A. Y. Turygin, and A. D. Mirzabekov. 2003. Kinetics of hybridization on the oligonucleotide microchips with gel pads. J. Biomol. Struct. Dyn. 21:279–288. [DOI] [PubMed] [Google Scholar]
- 3.Sorokin, N. V., V. R. Chechetkin, M. A. Livshits, S. V. Pan'kov, M. Y. Donnikov, D. A. Gryadunov, S. A. Lapa, and A. S. Zasedatelev. 2005. Discrimination between perfect and mismatched duplexes with oligonucleotide gel microchips: role of thermodynamic and kinetic effects during hybridization. J. Biomol. Struct. Dyn. 22:725–734. [DOI] [PubMed] [Google Scholar]
- 4.Livshits, M. A., and A. D. Mirzabekov. 1996. Theoretical analysis of the kinetics of DNA hybridization with gel-immobilized oligonucleotides. Biophys. J. 71:2795–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubina, A. Y., S. V. Pan'kov, E. I. Dementieva, D. N. Pen'kov, A. V. Butygin, V. A. Vasiliskov, A. V. Chudinov, A. L. Mikheikin, V. M. Mikhailovich, and A. D. Mirzabekov. 2004. Hydrogel drop microchips with immobilized DNA: properties and methods for large-scale production. Anal. Biochem. 325:92–106. [DOI] [PubMed] [Google Scholar]
- 6.Rehman, F. N., M. Audeh, E. S. Abrams, P. W. Hammond, M. Kenney, and T. C. Boles. 1999. Immobilization of acrylamide-modified oligonucleotides by co-polymerization. Nucleic Acids Res. 27:649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soto, C. M., C. H. Patterson, P. T. Charles, B. D. Martin, and M. S. Spector. 2005. Immobilization and hybridization of DNA in a sugar polyacrylate hydrogel. Biotechnol. Bioeng. 92:934–942. [DOI] [PubMed] [Google Scholar]
- 8.Koch, C. A., P. C. H. Li, and R. S. Utkhede. 2005. Evaluation of thin films of agarose on glass for hybridization of DNA to identify plant pathogens with microarray technology. Anal. Biochem. 342:93–102. [DOI] [PubMed] [Google Scholar]
- 9.Heller, M. J., A. H. Forster, and E. Tu. 2000. Active microelectronic chip devices which utilize controlled electrophoretic fields for multiplex DNA hybridization and other genomic applications. Electrophoresis. 21:157–164. [DOI] [PubMed] [Google Scholar]
- 10.Lassalle, N., E. Vieil, J. P. Correia, and L. M. Abrantes. 2001. Study of DNA hybridization on polypyrrole grafted with oligonucleotides by photocurrent spectroscopy. Biosens. Bioelectron. 16:295–303. [DOI] [PubMed] [Google Scholar]
- 11.Livache, T., H. Bazin, P. Caillat, and A. Roget. 1998. Electroconducting polymers for the construction of DNA or peptide arrays on silicon chips. Biosens. Bioelectron. 13:629–634. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, Z., W. Knoll, R. Foerch, R. Holcomb, and D. Roitman. 2005. DNA hybridization on plasma-polymerized allylamine. Macromolecules. 38:1271–1276. [Google Scholar]
- 13.Pirri, G., F. Damin, M. Chiari, E. Bontempi, and L. E. Depero. 2004. Characterization of a polymeric adsorbed coating for DNA microarray glass slides. Anal. Chem. 76:1352–1358. [DOI] [PubMed] [Google Scholar]
- 14.Consolandi, C., B. Castiglioni, R. Bordoni, E. Busti, C. Battaglia, L. R. Bernardi, and G. De Bellis. 2002. Two efficient polymeric chemical platforms for oligonucleotide microarray preparation. Nucleos. Nucleot. Nucl. 21:561–580. [DOI] [PubMed] [Google Scholar]
- 15.Zhou, X., L. Wu, and J. Zhou. 2004. Fabrication of DNA microarrays on nanoengineered polymeric ultrathin film prepared by self-assembly of polyelectrolyte multilayers. Langmuir. 20:8877–8885. [DOI] [PubMed] [Google Scholar]
- 16.Wang, J., M. Jiang, T. W. Nilsen, and R. C. Getts. 1998. Dendritic nucleic acid probes for DNA biosensors. J. Am. Chem. Soc. 120:8281–8282. [Google Scholar]
- 17.Trevisiol, E., V. Le Berre-Anton, J. Leclaire, G. Pratviel, A.-M. Caminade, J.-P. Majoral, J. M. Francois, and B. Meunier. 2003. Dendrislides, dendrichips: a simple chemical functionalization of glass slides with phosphorous dendrimers as an effective means for the preparation of biochips. New J. Chem. 27:1713–1719. [Google Scholar]
- 18.Stillman, B. A., and J. L. Tonkinson. 2001. Expression microarray hybridization kinetics depend on length of the immobilized DNA but are independent of immobilization substrate. Anal. Biochem. 295:149–157. [DOI] [PubMed] [Google Scholar]
- 19.Tang, J., N. He, L. Nie, P. Xiao, and H. Chen. 2004. Hydrolysis of microporous polyamide-6 membranes as substrate for in situ synthesis of oligonucleotides. Surf. Sci. 550:26–34. [Google Scholar]
- 20.Jones, K. D. 2001. Membrane immobilization of nucleic acids: Part 1: Substrates. IVD Technology. July/August:50–55.
- 21.Olsen, K. G., D. J. Ross, and M. J. Tarlov. 2002. Immobilization of DNA hydrogel plugs in microfluidic channels. Anal. Chem. 74:1436–1441. [DOI] [PubMed] [Google Scholar]
- 22.Zangmeister, R. A., and M. J. Tarlov. 2003. UV graft polymerization of polyacrylamide hydrogel plugs in microfluidic channels. Langmuir. 19:6901–6904. [Google Scholar]
- 23.Rubina, A. Y., V. I. Dyukova, E. I. Dementieva, A. A. Stomakhin, V. A. Nesmeyanov, E. V. Grishin, and A. S. Zasedatelev. 2005. Quantitative immunoassay of biotoxins on hydrogel-based protein microchips. Anal. Biochem. 340:317–329. [DOI] [PubMed] [Google Scholar]
- 24.Charles, P. T., E. R. Goldman, J. G. Rangasammy, C. L. Schauer, M.-S. Chen, and C. R. Taitt. 2004. Fabrication and characterization of 3D hydrogel microarrays to measure antigenicity and antibody functionality for biosensor applications. Biosens. Bioelectron. 20:753–764. [DOI] [PubMed] [Google Scholar]
- 25.Dyukova, V. I., E. I. Dementieva, D. A. Zubtsov, O. E. Galanina, N. V. Bovin, and A. Y. Rubina. 2005. Hydrogel glycan mircoarrays. Anal. Biochem. 347:94–105. [DOI] [PubMed] [Google Scholar]
- 26.Lin, V. S.-Y., K. Motesharei, K.-P. S. Dancil, M. J. Sailor, and M. R. Ghadiri. 1997. A porous silicon-based optical interferometric biosensor. Science. 278:840–843. [DOI] [PubMed] [Google Scholar]
- 27.Lie, L. H., S. N. Patole, A. R. Pike, L. C. Ryder, B. A. Connolly, A. D. Ward, E. M. Tuite, A. Houlton, and B. R. Horrocks. 2003. Immobilisation and synthesis of DNA on Si(111), nanocrystalline porous silicon and silicon nanoparticles. Faraday Discuss. 125:235–249. [DOI] [PubMed] [Google Scholar]
- 28.Archer, M., and P. M. Fauchet. 2003. Electrical sensing of DNA hybridization in porous silicon layers. Phys. Stat. Sol. (A). 198:503–507. [Google Scholar]
- 29.Archer, M., M. Christophersen, and P. M. Fauchet. 2004. Macroporous silicon electrical sensor for DNA hybridization detection. Biomed. Microdevices. 6:203–211. [DOI] [PubMed] [Google Scholar]
- 30.Chan, S., P. M. Fauchet, Y. Li, L. J. Rothberg, and B. L. Miller. 2000. Porous silicon microcavities for biosensing applications. Phys. Stat. Sol. (A). 18:541–546. [Google Scholar]
- 31.Bessueille, F., V. Dugas, V. Vikulov, J. P. Cloarec, E. Souteyrand, and J. R. Martin. 2005. Assessment of porous silicon substrate for well-characterised sensitive DNA chip implement. Biosens. Bioelectron. 21:908–916. [DOI] [PubMed] [Google Scholar]
- 32.Pan, S., and L. J. Rothberg. 2003. Interferometric sensing of biomolecular binding using nanoporous aluminum oxide templates. Nano Lett. 3:811–814. [Google Scholar]
- 33.Kessler, N., O. Ferraris, K. Palmer, W. Marsh, and A. Steel. 2004. Use of the DNA flow-thru chip, a three-dimensional biochip, for typing and subtyping of influenza viruses. J. Clin. Microbiol. 42:2173–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer, M., R. Philip, H. E. Matthai, E. Eastman, and A. J. O'Beirne. 2003. Using 4-D diagnostic tools for genetic analysis. IVD Technology. July:47–53.
- 35.Beattie, K. L., W. G. Beattie, L. Meng, S. L. Turner, R. Coral-Vazquez, D. D. Smith, P. M. McIntyre, and D. D. Dao. 1995. Advances in genosensor research. Clin. Chem. 41:700–706. [PubMed] [Google Scholar]
- 36.Matsumoto, F., K. Nishio, and H. Masuda. 2004. Flow-through-type DNA array based on ideally ordered anodic porous alumina substrate. Adv. Mater. 16(23–24):2105–2108. [Google Scholar]
- 37.Matsumoto, F., M. Harada, K. Nishio, and H. Masuda. 2005. Nanometer-scale patterning of DNA in controlled intervals on a gold-disk array fabricated using ideally ordered anodic porous alumina. Adv. Mater. 17:1609–1612. [Google Scholar]
- 38.Benoit, V., A. Steel, M. Torres, Y.-Y. Yu, H. Yang, and J. Cooper. 2001. Evaluation of three-dimensional microchannel glass biochips for multiplexed nucleic acid fluorescence hybridization assays. Anal. Chem. 73:2412–2420. [DOI] [PubMed] [Google Scholar]
- 39.Steel, A., M. Torres, J. Hartwell, Y. Yu, N. Ting, G. Hoke, and H. Yang. 2000. The flow-thru chiptm: a three-dimensional biochip platform. In Microarray Biochip Technology. M. Schena, editor. Eaton Publishing, Natwich, MA. 87–117.
- 40.Han, S., J. Lin, M. Satjapipat, A. J. Baca, and F. Zhou. 2001. A three-dimensional heterogeneous DNA sensing surface formed by attaching oligodeoxynucleotide-capped gold nanoparticles onto a gold-coated quartz crystal. Chem. Commun. 7:609–610. [Google Scholar]
- 41.Livshits, M. A., V. L. Florentiev, and A. D. Mirzabekov. 1994. Dissociation of duplexes formed by hybridization of DNA with gel-immobilized oligonucleotides. J. Biomol. Struct. Dyn. 11:783–795. [DOI] [PubMed] [Google Scholar]
- 42.Glazer, M., C. Frank, J. Lussi, J. Fidanza, and G. McGall. 2001. Colloidal silica films as substrates for high density DNA arrays. In Organic/Inorganic Hybrid Materials—2000 (Materials Research Society Proceedings, vol. 628). R. M. Laine, C. Sanchez, C. J. Brinker, and E. Gianellis, editors. Materials Research Society, Warrendale, PA. CC10.4.1–CC10.4.6.
- 43.Glazer, M., J. Fidanza, G. McGall, and C. Frank. 2001. Colloidal silica films for high-capacity DNA probe arrays. Chem. Mater. 13:4773–4782. [Google Scholar]
- 44.Glazer, M., J. A. Fidanza, G. H. McGall, M. O. Trulson, J. E. Forman, A. Suseno, and C. W. Frank. 2006. Kinetics of oligonucleotide hybridization to photolithographically patterned DNA arrays. Anal. Biochem. 358:225–238. [DOI] [PubMed] [Google Scholar]
- 45.Levicky, R., and A. Horgan. 2005. Physicochemical perspectives on DNA microarray and biosensor technologies. Trends Biotechnol. 23:143–149. [DOI] [PubMed] [Google Scholar]
- 46.McGall, G. H., A. D. Barone, M. Diggelman, S. P. A. Fodor, E. Gentalen, and N. Ngo. 1997. The efficiency of light-directed synthesis of DNA arrays on glass substrates. J. Am. Chem. Soc. 119:5081–5090. [Google Scholar]
- 47.Forman, J. E., I. D. Walton, D. Stern, R. P. Rava, and M. O. Trulson. 1998. Thermodynamics of duplex formation and mismatch discrimination on photolithographically synthesized oligonucleotide arrays. In Molecular Modeling of Nucleic Acids. N. B. Leonitis and J. SantaLucia Jr., editors. American Chemical Society, Washington, DC. 206–228.
- 48.Israelachvili, J. 1992. Intermolecular and Surface Forces. Academic Press, New York. 238.
- 49.Andrade, J. D. 1985. Principles of protein adsorption. In Surface and Interfacial Aspects of Biomedical Polymers: Protein Adsorption, 2nd ed. J. D. Andrade, editor. Plenum Press, New York. 29–35.
- 50.LeVan, M. D., G. Carta, and C. M. Yon. 1997. Adsorption and ion exchange. In Perry's Chemical Engineers' Handbook, 7th ed. R. H. Perry, C. H. Chilton, and D. W. Green, editors. McGraw-Hill, New York. 19–28.
- 51.Brenner, H., and L. J. Gaydos. 1977. The constrained Brownian movement of spherical particles in cylindrical pores of comparable radius: models of the diffusive and convective transport of solute molecules in membranes and porous media. J. Colloid Interf. Sci. 58:312–356. [Google Scholar]
- 52.Wakao, N., and J. M. Smith. 1962. Diffusion in catalyst pellets. Chem. Eng. Sci. 17:825–834. [Google Scholar]
- 53.Adamson, A., and A. P. Gast. 1997. Physical Chemistry of Surfaces. John Wiley & Sons, New York. 395.
- 54.Vainrub, A., and M. B. Pettitt. 2002. Coulomb blockage of hybridization in two-dimensional DNA arrays. Phys. Rev. E. 66:041905-1–041905-4. [DOI] [PubMed] [Google Scholar]
- 55.Vainrub, A., and M. B. Pettitt. 2003. Sensitive quantitative nucleic acid detection using oligonucleotide microarrays. J. Am. Chem. Soc. 125:7798–7799. [DOI] [PubMed] [Google Scholar]
- 56.Vainrub, A., and M. B. Pettitt. 2004. Theoretical aspects of genomic variation screening using DNA microarrays. Biopolymers. 73:614–620. [DOI] [PubMed] [Google Scholar]
- 57.Halperin, A., A. Buhot, and E. B. Zhulina. 2004. Sensitivity, specificity, and the hybridization isotherms of DNA chips. Biophys. J. 86:718–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Do, D. D. 1998. Adsorption Analysis: Equilibria and Kinetics. Imperial College Press, London. Ch. 9.
- 59.Fogler, H. S. 1992. Elements of Chemical Reaction Engineering. Prentice Hall, Englewood Cliffs, NJ. 543–571.
- 60.Peterson, A. W., L. K. Wolf, and R. M. Georgiadis. 2002. Hybridization of mismatched or partially matched DNA at surfaces. J. Am. Chem. Soc. 124:14601–14607. [DOI] [PubMed] [Google Scholar]
- 61.Breslauer, K. J., R. Frank, H. Blocker, and L. A. Marky. 1986. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA. 83:3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cantor, C. R., and P. R. Schimmel. 1980. The Behavior of Biological Macromolecules. W. H. Freeman, San Francisco. Ch. 23.
- 63.Saenger, W. 1984. Principles of Nucleic Acid Structure. Springer-Verlag, New York. 142–143.
- 64.Mills, A. F. 1999. Basic Heat and Mass Transfer. Prentice Hall, Upper Saddle River, NJ. Ch. 4.