

Abstract
In the cytochrome c-551 family, the heme 17-propionate caboxylate group is always hydrogen bonded to an invariant Trp-56 and conserved residues (His and Arg mainly, Lys occasionally) at position 47. The mutation of His-47 to Ala-47 for Pseudomas stutzeri ZoBell cytochrome c-551 removes this otherwise invariant hydrogen bond. The solution structure of ferrous H47A has been solved based on NMR-derived constraints. Results indicate that the mutant has very similar main chain folding compared to wild-type. However, less efficient packing of residues in the mutant surrounding the heme propionates leads to more solvent exposure for both propionate groups, which may account for decreased stability of the mutant. The mutant has a reduction potential different from wild-type, and furthermore, the pH dependence of this potential is not the same as for wild-type. The structure of the mutant suggests that these changes are related to the loss of the residue-47 propionate hydrogen bond and the loss of charge on the side chain of residue 47.
INTRODUCTION
Cytochrome c-551 is a class I cytochrome c (1) widely distributed among prokaryotes. Although smaller (∼82 amino acids) than its eukaryotic analog mitochondrial cytochrome c (∼103 amino acids), it has similar protein folding and redox potential (∼250 mV). Common structural features include Met/His heme axial ligation, partial burying of the heme propionate groups, and a network of hydrogen bonds to the heme propionates by invariant Trp and Arg/His/Lys residues. In the cytochrome c-551 family, residue 56 is the invariant Trp, and residue 47 always is a hydrogen donor group. For example, residue 47 is Arg in cytochrome c-551 from Pseudomonas aeruginosa but His in cytochrome c-551 from Pseudomonas stutzeri and Pseudomonas mendocina. In rarer cases such as the homolog from Nitrosomonas europae, the donor is Lys. This conserved hydrogen bond may participate in heme redox potential modulation (2). The effect of residue 47 on protein stability and redox potential was studied by mutating His-47 to Ala-47 for Pseudomonas stutzeri substrain ZoBell cytochrome c-551 (3). The solution structure of H47A has now been determined by NMR and is reported herein. The effect of residue 56 was probed earlier through Trp-56 mutations to either Tyr (W56Y) or Phe (W56F) for Pseudomonas aeruginosa cytochrome c-551 (4). Mutant structures were proposed in these cases based on molecular dynamics calculations without experimental constraints using the coordinates from the known wild-type crystal structure. The H47A ferrocytochrome structure to be presented was determined by NMR experimental restraints starting from random coil geometry and hence allows for unbiased conclusions regarding structure.
MATERIALS AND METHODS
The H47A mutant protein was expressed as described previously (3). The prior purification procedure yielded cytochrome c-551 free from other contaminating proteins, but it did contain peptidoglycan fragments released during the freeze-thaw steps that obscured important protein NMR resonances. Hence, the purification procedure was amended. Solid ammonium sulfate was added to the protein released by the freeze-thaw cycles to a final concentration of 2.0 M, and salt-induced precipitates were removed by centrifugation. The sample was applied to a pre-equilibrated (2.0 M ammonium sulfate, 100 mM ammonium bicarbonate) Pharmacia Octyl Sepharose 4 Fast Flow hydrophobic interaction chromatography (HIC) column (1 × 5 cm). The column was washed with 100 mM ammonium bicarbonate buffers containing 2.0 M and 1.5 M ammonium sulfate (50 ml each) to remove impurities, and then the red cytochrome was eluted with 100 mM ammonium bicarbonate buffer containing 1.0 M ammonium sulfate. To reduce the sample volume, solid ammonium sulfate was added to the eluent until the concentration reached 2 M. The mixture was then loaded onto a second HIC column (1 × 2.5 cm). After the column had been washed with 20 ml 2.0 M ammonium sulfate containing 100 mM ammonium bicarbonate buffer, the protein was eluted with deionized water in a very tight band. The HIC-concentrated sample was chromatographed on a Pharmacia G-25 Sephadex gel filtration column (1 × 25 cm equilibrated with 10 mM Tris HCl, pH 8.0, and eluted with the same). The eluent was loaded onto a DEAE-Sephacel anion exchange column (1 × 7 cm equilibrated with the Tris buffer above) and then washed consecutively with 100 ml of Tris buffer, 50 ml of Tris buffer containing 50 mM NaCl, then finally Tris buffer containing 100 mM NaCl, which eluted the red cytochrome. The volume of the eluent was reduced by an HIC concentration column as explained above. Finally, the concentrated sample was chromatographed on a G-50 Sephadex gel filtration column (3.5 × 46 cm) eluted with 100 mM ammonium bicarbonate. Cytochrome fractions were collected and lyophilized.
NMR samples were prepared as previously described (5). TOCSY, NOESY, and DQF-COSY NMR experiments were performed on the Bruker AVANCE 600 spectrometer with standard pulse programs (6–8) at temperatures in the range of 298 K to 320 K. Variable temperature spectra were recorded to resolve cases of overlap at a single temperature. All ferrocytochrome chemical shifts to be reported correspond to spectra at 298 K in 50 mM phosphate buffer containing sodium dithionite at ∼10 mM at pH 7.0 to ensure complete reduction. All spectra were processed with NMRPipe (9), and peaks were inventoried by NMRView (10). The effects of pH on the NMR spectra of the mutant cytochrome were measured at 298 K in 50 mM phosphate buffer containing internal 3-(trimethylsilyl)-tetradeutero sodium propionate (commonly known as TSP) as a shift standard of 0 ppm adjusted to the appropriate pH with either HCl or NaOH as measured with a glass electrode. The range was limited to 5 to 9 by the stability of the mutant protein. The chemical shifts of resolved heme methyl resonances were measured in wide-sweep one-dimensional spectra, and the error limits on shift measurements were conservatively estimated to be ±0.02 ppm. Heme propionate resonances and select aromatic side chain resonances were not resolvable in one-dimensional spectra, so their chemical shifts were measured from appropriate cross peaks in TOCSY or NOESY spectra in the dimension of highest digital resolution. These cross peaks represent broad scalar-coupled multiplets, and the error limits were estimated based on the peak width and digital resolution.
Main chain and side chain assignments of resolved resonances were made by now-standard techniques. Although there turned out to be considerable chemical shift similarity between the mutant and wild-type, all assignments for the mutant were made independently and confirmed by scalar and dipolar connectivity. Protein structure calculations were performed with the ARIA 2.0 ALPHA (11–14) and the CNS 1.1 (15) software suites starting from an extended random coil structure and data at 298 K. Default ARIA/CNS parameters were used in most cases. The frequency window size was 0.02 ppm in the direct proton dimension and 0.04 ppm in the indirect proton dimension. The ARIA calculation comprised nine iterations, and the 7 best structures of the 20 calculated in any iteration were chosen as models for the next iteration except in iteration 8, where 10 of 50 were used for analysis. For partial assignments, the maximum number of contributions was 20. The ambiguity cutoffs following the default scheme for nine iterations were 1.0, 0.9999, 0.999, 0.99, 0.98, 0.96, 0.93, 0.9, and 0.8 Å. For the violation analysis, violation thresholds were held constant at 0.5 Å in all iterations, but violation tolerances were varied following two schemes. Calculation for early runs used the default scheme, 1000.0, 5.0, 3.0, 1.0, 1.0, 1.0, 0.1, 0.1, and 0.1 Å, whereas calculation for later runs used tighter violation tolerances, 5.0, 3.0, 1.0, 0.5, 0.2, 0.1, 0.1, 0.1, and 0.1 Å. The CNS structure simulation used torsion angle dynamics at slower cooling rates (cool1, 10,000; cool2, 8000) to improve convergence levels for the structures. CNS analysis after the last iteration was enabled, and PROCHECK (16) analysis was performed.
RESULTS AND DISCUSSION
Chemical shift assignments and experimental constraints have been deposited with the Biological Magnetic Resonance Data Bank as accession number 7296. Three-dimensional atomic coordinates, chemical shifts, and experimental constraints have been deposited with the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) as accession number 2I8F. Assignment results showed that the ferrocytochrome H47A mutant had chemical shifts similar to the wild-type (17). However, distinctive chemical shift differences were observed for the residues near 47, indicating that it is the mutation site. Critical facts for Ala-47 assignment were: 1), the wild-type H-47 imidazole ring proton HE1 (8.70 ppm) and HD2 (7.81 ppm) cross peaks were missing in TOCSY spectra; 2), the assigned mutant 47 residue had an alanine-type spin-coupling network; 3), sequential NOE connections in the amide region (NH47-NH48) and aliphatic region (NH47-αH44, NH47-αH46, NH47-βH46, NH48-αH47, and NH48-βH47) were consistent with the assignments.
Backbone NH and αH chemical shift differences between H47A and wild-type are plotted versus residue number in Fig. 1. The similarity is very strong. The average chemical shift deviations from that of the wild-type are 0.142 ppm (backbone NHs) and 0.065 ppm (CαHs), which is the first strong indication that the overall protein folding is similar. Residues near position 47 (43–44, 47–48, and 51–53) show much bigger (±0.56 ppm at maximum) chemical shift deviations, indicating changes in these regions. Chemical shifts of H47A heme protons are generally very close to that of the wild-type, with an average deviation of 0.03 ppm. Larger changes in chemical shift (up to ±0.2 ppm) are observed for both propionate groups, indicating that the H47A mutation disturbed the local environments of the two propionates (Fig. 2).
FIGURE 1.

Chemical shift differences of backbone NHs (dHN) and CαHs (dHA) between H47A and wild-type. Gaps in the dHN line are result from prolines.
FIGURE 2.

Schematic of the heme c prosthetic group with substituents numbered according to IUB-IUPAC nomenclature for tetrapyrroles. The additional small decimal numbers are the chemical shift differences, H47A minus wild-type, for heme protons.
The ARIA/CNS structure calculation was performed with 67 dihedral angles and 1524 distance restraints, including manually assigned NOEs, NOEs assigned by ARIA, and H-bond restraints (Table 1). At the end of the final ARIA run, the 10 best structures of the 50 calculated were further refined in a shell of water to prevent any side-chain packing artifacts caused by the simplified treatment of nonbonded forces and missing solvent contact in earlier calculations. The average root mean-square deviation (RMSD) in position between the 10 structures and their mean structure for all residues is 0.47 ± 0.07 Å for backbone atoms and 0.75 ± 0.07 Å for all heavy atoms (Table 1). The relatively higher backbone RMSDs for residues 36–39, 57–58 (Fig. 3) are caused by a smaller number of experimental constraints. Spectral overlap was not a significant problem for these resonances, so this suggests possible higher flexibility in these regions, which is reasonable because both are within less structured loops located on the protein surface. Based on the protein backbone ψ-φ angle Ramachandran plot, among the nonglycine and nonproline residues, 83.3% have backbone torsion angles in the most favored region, 16.7% in an additionally allowed region, and 0% in generously allowed or disallowed regions (Table 1).
TABLE 1.
Experimental restraints and structure statistics
| ARIA NOEs | |
|---|---|
| Total | 1414 |
| Unambiguous* | 1105 |
| Ambiguous | 309 |
| H bonds | 30 |
| Backbone φ angles | 40 |
| Proline ω angles | 7 |
| Side-chain χ angles | 20 |
| Ensemble RMSD (Å) | |
| Secondary structure (backbone) | 0.38 ± 0.06 |
| Secondary structure (heavy) | 0.64 ± 0.05 |
| Backbone (all residues) | 0.47 ± 0.07 |
| Heavy atoms (all residues) | 0.75 ± 0.08 |
| RMSD (experimental restraints) | |
| NOEs (Å) | 0.031 ± 0.004 |
| H-bonds (Å) | 0.012 ± 0.005 |
| Dihedral angles (deg) | 0.03 ± 0.01 |
| RMSD (covalent geometry) | |
| Bonds (Å) | 0.00423 ± 0.00004 |
| Angles (deg) | 0.791 ± 0.007 |
| Impropers (deg) | 1.12 ± 0.08 |
| Ramchandran space | |
| Most favored region | 83.3% |
| Additionally allowed | 16.7% |
| Generously allowed | 0 |
| Disallowed | 0 |
Subcategories on NOEs include 493 intraresidue; 240 sequential; 94 short range (2 ≤ |i-j| ≤ 3); 46 medium range (4 ≤ |i-j| ≤ 5); and 232 long range (|i-j| ≥ 5).
FIGURE 3.

Atomic root mean-squared deviations (Å) of the 10 best-simulated annealing structures about the mean structure for backbone atoms (upper panel) and all heavy atoms (lower panel).
As indicated by their chemical shift similarity, the H47A backbone fold resembles that of the wild-type. The secondary structure of H47A contains five α-helices at residues 3–9, 13–15, 27–33, 40–50, and 68–79, corresponding to α-helix regions in the wild-type. A ribbon diagram overlay of both proteins reveals that the superposition in α-helix regions is very good, whereas in loop regions modest displacements are possible (Fig. 4).
FIGURE 4.

Overlay of the stereo diagrams of the main chain ribbons for H47A (black) and wild-type (gray).
Major structural differences were observed for regions around the two heme propionates. The carboxylate of 17-propionate is located in a pocket formed by residues 30, 34, 43–44, 47, 52–53, and 55–56 (Fig. 5). Replacing the bulkier His-47 imidazole group with the smaller Ala-47 methyl group creates a slight “van der Waals vacuum”, which allows residues 52 and 53 directly next to it to shift closer to the Ala side chain. The smaller van der Waals radii in combination with the lack of a hydrogen bond to the 47 side chain also shift the 17-propionate carboxylate and the hydrogen-bonded Trp-56 toward Ala-47. This causes Leu-44, Val-55, Val-30, and Lys-33 side chains to swing away from the propionate center. The net effect is that in H47A, residues surrounding the 17-propionate are packed less tightly, which results in a bigger solvent exposure of the propionate.
FIGURE 5.

Overlay of the stereo diagrams showing the side chains within 6 Å of the heme propionate 17 CG atom for H47A (black) and wild-type (gray).
The shift toward Ala-47 of residues 52–53 and 56 in turn disturbs the residue packing around the 13-propionate. In H47A and the wild-type, the 13-propionate carboxylate is buried in a U-shaped loop formed by residues 52–61, with residues Gln-53 and Pro-58, and Ile-59 at the opening of the “U”, residues Gly-54, Val-55, Trp-56, and Gly-57 as two arms of the U, and residues Pro-60 and Met-61 bent back toward the heme iron center (Fig. 6). In H47A, Gln-53 is shifted toward the 17-propionate and therefore away from the 13-propionate. The displacement of Trp-56 swings Gly-57, Pro-58, and Ile-59 also away from the 13-propionate. As a result, the “U” shape has a much broader opening, and the13-propionate is exposed to solvent to a greater extent, as in the case of the 17-propionate. The increased solvent exposure of the 17-propionate was quantified by a solvent-accessible surface area calculation performed with the GETAREA 1.1 software package (18). In wild-type, the surface-accessible areas for a water probe radius of 1.4 Å for heme carbonyl oxygens were 0.38 Å2 and 0.77 Å2 for the 17-propionate, and in H47A, the corresponding areas were 2.44 Å2 and 8.07 Å2.
FIGURE 6.

Overlay of the stereo diagrams showing the side chains within 6 Å of the heme propionate 13 CG atom for H47A (black) and wild-type (gray).
The less efficient packing of residues around the heme propionates may account for the decreased acidic stability of H47A reported previously (3). His-47 in wild-type is positively charged under acidic conditions (2). The loss of a positive charge for residue 47 at pH <5.0 could decrease the electrostatic balance of the surrounding residues and destabilize the mutant protein.
Cytochromes of the c-551 family have a characteristic dependence of redox potential on pH. Fig. 7 (adopted from Milller et al. (3)) displays this behavior for wild-type and the H47A mutant. The salient differences are that the mutant has a lower redox potential than wild-type until pH ∼8.8, and the change with pH for the mutant is confined to a narrower range than for wild-type.
FIGURE 7.

Comparison of the pH dependence of the standard reduction potential for H47A (squares) and wild-type (diamonds) (data obtained from Miller et al. (3)). The smooth solid curves for each data set represent fitting the data with an equation whereby the pH dependence results from to a single ionization event that has different pKa values in the reduced and oxidized states as given by Leitch et al. (19) with the parameters given by Miller et al. (3). The dashed curve for H47A represents fitting the potential to simple Henderson-Hasselbalch behavior with a single pKa of 6.4 as described in the text and Supplemental Information. The dashed curve has been displaced downward by 3 mV from the best fit to emphasize how it parallels the two-pKa equation. Without the displacement, the two curves are almost superimposed.
The pH dependence of the redox potential has previously been attributed to a single ionization event that has a different pKa in the oxidized (pKox) and reduced (pKred) states (19). This could still be a contributing factor, and the redox potential profile of the mutant was originally fit to this model with pKox of 6.7 ± 0.3 and pKred of 6.2 ± 0.1 (3). However, the close correspondence of these values to each other, the new three-dimensional information for the mutant, and a reappraisal of the shape of the mutant profile have suggested a new proposal to explain the mutant versus wild-type redox differences. The new proposal does not necessarily invalidate the Leitch et al. model because in the mutant there can still be slight differences in a pKox and a pKred, but it does provide a different prospective to view mutant versus wild-type changes. Fig. 8 schematically depicts this perspective.
FIGURE 8.

Schematic depicting the net charge on the ferric heme core and the net charge in the vicinity of the 17-propionate as a function of pH range for H47A and wild-type.
It has been shown by prior NMR studies that the 17-propionate has a pKa that can be around neutrality or lower, whereas His-47 hydrogen bonded to this carbonyl can have a pKa ≈ 8 (2). Therefore, at pH values less than ∼6.5, the propionate and the histidine will both be protonated, hence bearing charges of zero and +1, respectively, and leading to a local net charge for this cluster of +1. This is called state 1 of the cytochrome in Fig. 8. Between pH values of 6.5 to ∼8, the propionate is deprotonated and bears a negative charge while the histidine is still mainly +1, leading to a net local charge of zero (state 2). Above pH ∼8, the deprotonated histidine leads to a net local charge of −1 (state 3). There are no distinct boundaries between these states because of partial protonation equilibria, but the state names highlight the limiting behavior.
For the standard reduction half reaction,
 |
there is a standard midpoint reduction potential, 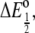 which is the value plotted in Fig. 7. Associated with the half-potential is a free energy change
which is the value plotted in Fig. 7. Associated with the half-potential is a free energy change 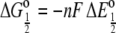 where n is the number of electrons transferred, i.e., one in this case, and F is Faraday's constant. Because
where n is the number of electrons transferred, i.e., one in this case, and F is Faraday's constant. Because 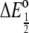 is more positive for the mutant, the free energy is more negative, and this means the reduction reaction is more favorable. This can also be taken as indicating that the oxidized state of the mutant is destabilized to a greater extent relative to the reduced state. When considering what might be the structural basis for this difference, it is important to also consider that there are little to no folding structural differences between redox states in the c-551 family. This was first shown by x-ray crystallography (20) and more recently by NMR (21–24), whereby it was shown that pseudocontact shifts of resonances throughout the polypeptide in the oxidized state were consistent with a single ferric iron g-tensor and the geometry of the reduced-state polypeptide.
is more positive for the mutant, the free energy is more negative, and this means the reduction reaction is more favorable. This can also be taken as indicating that the oxidized state of the mutant is destabilized to a greater extent relative to the reduced state. When considering what might be the structural basis for this difference, it is important to also consider that there are little to no folding structural differences between redox states in the c-551 family. This was first shown by x-ray crystallography (20) and more recently by NMR (21–24), whereby it was shown that pseudocontact shifts of resonances throughout the polypeptide in the oxidized state were consistent with a single ferric iron g-tensor and the geometry of the reduced-state polypeptide.
The net formal charge on a ferric heme core is +1 because the tetrapyrrole is formally −2 after loss of the two inner pyrrole hydrogens, and correspondingly, a ferrous heme is formally 0. Fig. 8 postulates that the ferric heme in state 1 of wild-type is destabilized versus a net neutral ferrous heme as a result of charge interaction with the 17-propionate cluster. So the redox potential is highest (i.e., reduction is most favored) in this state. In state 2 the net charge in the propionate cluster is zero, and so the ferric heme is more stable with respect to ferrous heme, and the redox potential drops, whereas in state 3, when the cluster has a net negative charge, the ferric heme becomes even more favorable compared to ferrous. In the mutant without an ionizable His-47, state 1 already has a net neutral cluster, so the redox potential is lower. State 2 is still more favorable because of the net negative cluster, but the transition occurs more steeply with pH because ionization of the propionate occurs at lower pH. There are no further ionization events possible for the mutant, and so its redox potential is independent of pH until it finally merges with the potential of wild-type, reflecting a common charge state for the 17-propionate cluster.
NMR spectra were collected as a function of pH in an attempt to gain additional insight into the pH dependence of the redox potential similar to previous studies on wild-type cytochromes c-551 (2,19). In the simplest of cases, resonances close to the ionizable 17-propionate might be expected to change their chemical shifts in a way that parallels the redox potential behavior shown in Fig. 7. However, this does not necessarily have to be the case because a multitude of factors can affect the chemical shifts that have no effect on redox potential. Fig. 9 shows the results for the heme 181-methyl adjacent to the 17-propionate in the ferric form. The large paramagnetic shift of this resonance is highly sensitive to local environmental changes, and it showed the largest pH-dependent shift changes. In the Supplementary Material section, the pH dependence for other resonances associated with the heme propionates is presented. Data are limited on the acidic side to pH 5 because the mutant is not as stable as wild-type and denatures below this pH. The midpoint for the pH jump for the mutant redox potential, or the transition pKa, is around pH 6.3. Neither the heme 181-methyl nor any other resonance close to the heme propionates shows a similar pKa. The general features were the same for all resonances that showed pH-dependent shifts greater than the error in recording the shift. There was a main shift either up or down beginning around pH 7. The data for the 181-methyl show a small wiggle between pH 6.3 and 7, close to but larger than the error bars of the measurements. This feature was observed in some but not all of the resonances followed. It may be experimental error, or it may be reflecting some other ionization event in the protein.
FIGURE 9.

Chemical shifts of the mutant H47A ferricytochrome heme 181-methyl resonance (solid squares) as a function of pH. The error bars are smaller than the data point symbols on this scale. The solid line represents hypothetical behavior of a heme methyl resonance whose chemical shift is affected by the ionization of both the 17-propionate and the 13-propionate with further conditions described in the text.
The lack of parallelism in the pH dependences between ferric resonances and redox potential can result from multiple causes, but one simple hypothesis can be discussed. The 17-propionate had been called the inner propionate in the cytochrome c-551 family, and the 13-propionate has been called the outer, because in the wild-type structure 17 is buried in the protein interior and 13 is close to the surface. The scheme of Fig. 8 proposes that the ionization of buried 17 with its elaborate hydrogen-bonding network is influencing the redox potential, but, potential is independent of the ionization state of the more exposed 13. Ionization of 13, however, could have a major impact on nearby chemical shifts, as big or bigger in magnitude as ionization of 17. If the pKa values of these events are close, the composite pH dependence can be dominated by the effect of the 13 ionization. In Supplementary Material there is presented a calculated hypothetical case whereby the pKa of 17 is at 6.3 and the pKa of 13 is at 4.4, a more typical weak acid value. Ionization of 17 causes a hypothetical jump of 0.92 ppm for a hypothetical heme methyl resonance at 15.5 ppm, whereas ionization of 13 causes a 1.88-ppm jump. The resulting combined effect is a weighted average dominated by the 13 behavior that shows an apparent pKa ≈ 5 and little to no visual evidence of the 17 ionization. The final composite profile truncated at pH 5 is plotted as the solid curve in Fig. 9. It should be stressed that this simulation is by no means unique. There are too many adjustable parameters (direction of the jump, magnitude of the jump, and pKa for each propionate, plus the possibility that other ionizations perturb resonances) to ensure a unique fit.
It is reasonable that the 13-propionate could cause a larger perturbation on the chemical shift of ferric heme resonances. These resonances have a large contact shift contribution. It has been noted multiple times in the literature (see Timkovich et al. (25) and citations therein to earlier work) that unpaired electron spin density is distributed asymmetrically over ferric hemes in cytochromes of the c-type, and it has been proposed that the pyrrole with highest spin density could most readily accommodate an electron transferring into the cytochrome. The 121-methyl has the largest contact shift of all heme methyls in H47A and wild-type, indicating that spin density is maximized on this pyrrole. Hence, 13-propionate ionization as a perturbation can have a larger effect on shifts, even though its effect on redox potential is minimal.
In wild-type Pseudomonas aeruginosa cytochrome c-551, the pKa of the inner 17-propionate has been assigned to be around 7.2, and the outer 13-propionate was assigned a pKa of 3.1 (2). This was rationalized on the basis that burying the inner propionate in the protein hydrophobic interior made it more difficult to ionize to a net negative charge, whereas the outer propionate became easier to ionize to help stabilize the net formal charge on ferric heme, as shown in Fig. 8. Structure results for H47A show that the 17-propionate is now more solvent exposed, and hence, it is reasonable that its pKa could shift to lower values. The pKa of 13 could then move up, perhaps also because of the rearranged solvent exposure or perhaps because some of the burden of stabilizing the ferric heme has been assumed by 17.
Fig. 10 displays the effect of pH on the chemical shift of the W-56 indole NH in the ferrous state. This is the proton hydrogen bonded to the carbonyl of the 17-propionate. This resonance showed the largest effect by pH in the ferrous state. Other resonances close to the heme showed only modest shifts of 0.03 ppm or less and therefore could not be reliably used to monitor pKa values. The pH effect in this redox state closely parallels the behavior of the midpoint potential. The most accurate transition midpoint is in doubt because data could not be obtained below pH 5, but the best fit to the data using a single pKa was at 5.9 rather than at 6.4. As discussed for the ferric case and demonstrated in Supplementary Material, a composite pH profile resulting from two ionizations can skew the apparent pKa. If the 13-propionate ionization in the ferrous state produces an effect of the same sign on the W-56 resonances but of smaller magnitude and on the order of a pKa of 4.4, then the composite curve would shift below an apparent pKa of 6.4.
FIGURE 10.

Chemical shifts of the mutant H47A ferrocytochrome W-56 indole NH resonance (solid squares) as a function of pH. The error bars are smaller than the data point symbols on this scale. The solid line represents hypothetical behavior of a resonance whose chemical shift is affected by a single Henderson-Hasselbalch–type ionization with a pKa of 5.9.
NMR is a powerful technique to follow a single site in a complex macromolecule, but it does have its limitations. There is currently no a priori method of predicting how sensitive a given nearby proton may be to an ionization event or even of predicting which direction a resonance may shift. The pH-dependent shifts observed in the ferric state have been interpreted as mostly caused by ionization of the 13-propionate with a pKa well below 5, whereas the shifts observed in the ferrous state have been interpreted as mostly related to the 17-propionate with a pKa ≈ 6.4. Nothing in the data precludes the possibility that the pKa values might be slightly different in each redox state, but the data are still consistent with the proposal of Fig. 8.
Invariant hydrogen bonds to the heme propionates have long been recognized as a unique structural feature in the entire c-type cytochrome family. The three-dimensional structure of the H47A mutant of c-551 affords insight into the contribution one such bond makes to the final structure and suggests a simple model for explaining the pH dependence of its redox potential.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Editor: Heinrich Roder.
References
- 1.Ambler, R. P. 1991. Sequence variability in bacterial cytochromes c. Biochim. Biophys. Acta. 1058:42–47. [DOI] [PubMed] [Google Scholar]
- 2.Cai, M., and R. Timkovich. 1992. Ionization of the heme porpionate substituents in Pseudomonad cytochromes c-551. FEBS Lett. 311:213–216. [DOI] [PubMed] [Google Scholar]
- 3.Miller, G. T., D. Q. Mackay, M. S. Standley, S. I. Fields, W. M. Clary, and R. Timkovich. 2003. Expression of Pseudomonas stutzeri Zobell cytochrome c-551 and its H47A variant in Escherichia coli. Protein Express. Purif. 29:244–251. [DOI] [PubMed] [Google Scholar]
- 4.Cutruzzola, F., I. Ciabatti, G. Roll, S. Falcinelli, M. Arese, G. Ranghino, A. Anselmino, E. Zennaro, and M. C. Silvestrini. 1997. Expression and characterization of Pseudomonas aeruginosa c-551 and two site-directed mutants: role of tryptophan 56 in the modulation of redox properties. Biochem. J. 322:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timkovich, R., D. Bergmann, D. M. Arciero, and A. B. Hooper. 1998. Primary sequence and solution structure for ferrocytochrome c-552 from Nitrosomonas europaea. Biophys. J. 75:1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaka, A. J., C. J. Lee, and A. Pines. 1988. Iterative schemes for bilinear operators. Application to spin decoupling. J. Magn. Reson. 77:274–293. [Google Scholar]
- 7.Hwang, T. L., and A. J. Shaka. 1995. Water suppression that works. excitation sculpting using arbitrary waveforms and pulsed field gradients. J. Magn. Reson. Ser A. 112:275–279. [Google Scholar]
- 8.Derome, A., and M. Williamson. 1990. 2D homonuclear shift correlation phase sensitive using TPPI with double quantum filter phase cycle. J. Magn. Reson. 88:177–185. [Google Scholar]
- 9.Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 10.Johnson, B. A., and R. A. Blevins. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 4:603–614. [DOI] [PubMed] [Google Scholar]
- 11.Nilges, M. 1995. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 245:645–660. [DOI] [PubMed] [Google Scholar]
- 12.Nilges, M., M. J. Macias, S. I. O'Donoghue, and H. Oschkinat. 1997. Automated NOESY interpretation with ambiguous distance restraints: the refined NMR solution structure of the pleckstrin homology domain from β-spectrin. J. Mol. Biol. 269:408–422. [DOI] [PubMed] [Google Scholar]
- 13.Nilges, M., and S. I. O'Donoghue. 1998. Ambiguous NOEs and automated NOE assignment. Prog. Nucl. Magn. Reson. Spectros. 32:107–139. [Google Scholar]
- 14.Habeck, M., W. Rieping, J. P. Linge, and M. Nilges. 2004. NOE assignment with ARIA 2.0. Methods Mol. Biol. 278:379–402. [DOI] [PubMed] [Google Scholar]
- 15.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Nilges, et al. 1998. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. D54:905–921. [DOI] [PubMed] [Google Scholar]
- 16.Laskowiski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- 17.Cai, M., and R. Timkovich. 1994. The solution conformation of cytochrome c-551 from P. stutzeri ZoBell determined by NMR. Biophys. J. 67:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraczkiewicz, R., and W. Braun. 1998. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 19:319–333. [Google Scholar]
- 19.Leitch, F. A., G. R. Moore, and G. W. Pettigrew. 1984. Structural basis for the variation of pH-dependent redox potentials of Pseudomonas cytochromes c-551. Biochemistry. 23:1831–1838. [DOI] [PubMed] [Google Scholar]
- 20.Matsuura, Y., T. Takano, and R. E. Dickerson. 1982. Structure of cytochrome c551 from P. aeruginosa refined at 1.6 A resolution, and comparison of the two redox forms. J. Mol. Biol. 156:389–409. [DOI] [PubMed] [Google Scholar]
- 21.Timkovich, R., and M. Cai. 1993. Investigation of the structure of oxidized Pseudomonas aeruginosa cyt c-551 by NMR: Comparison of observed and paramagnetic shifts and calculated pseudocontact shifts. Biochemistry. 32:11516–11523. [DOI] [PubMed] [Google Scholar]
- 22.Cai, M., and R. Timkovich. 1999. Solution conformation of ferricytochrome c-551 from Pseudomonas stutzeri substrain ZoBell. Biochem. Biophys. Res. Commun. 254:675–678. [DOI] [PubMed] [Google Scholar]
- 23.Tachiiri, N., H. Hemmi, S. J. Takayama, H. Mita, J. Hasegawa, Y. Sambongi, and Y. Yamamoto. 2004. Effects of axial methionine coordination on the in-plane asymmetry of the heme electronic structure of cytochrome c. J. Biol. Inorg. Chem. 9:733–742. [DOI] [PubMed] [Google Scholar]
- 24.Zhong, L., X. Wen, T. M. Rabinowitz, B. S. Russell, E. F. Karan, and K. L. Bren. 2004. Heme axial methionine fluxionality in Hydrogenobacter thermophilus cytochrome c552. Proc. Natl. Acad. Sci. USA. 101:8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timkovich, R., M. Cai, D. M. Arciero, and A. B. Hooper. 1994. Characteristics of the paramagnetic 1H NMR spectra of the ferricytochrome c-551 family. Eur. J. Biochem. 226:159–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.