

Abstract
The development of high resolution, high speed imaging techniques allows the study of dynamical processes in biological systems. Lateral resolution improvement of up to a factor of 2 has been achieved using structured illumination. In a total internal reflection fluorescence microscope, an evanescence excitation field is formed as light is total internally reflected at an interface between a high and a low index medium. The <100 nm penetration depth of evanescence field ensures a thin excitation region resulting in low background fluorescence. We present even higher resolution wide-field biological imaging by use of standing wave total internal reflection fluorescence (SW-TIRF). Evanescent standing wave (SW) illumination is used to generate a sinusoidal high spatial frequency fringe pattern on specimen for lateral resolution enhancement. To prevent thermal drift of the SW, novel detection and estimation of the SW phase with real-time feedback control is devised for the stabilization and control of the fringe phase. SW-TIRF is a wide-field superresolution technique with resolution better than a fifth of emission wavelength or ∼100 nm lateral resolution. We demonstrate the performance of the SW-TIRF microscopy using one- and two-directional SW illumination with a biological sample of cellular actin cytoskeleton of mouse fibroblast cells as well as single semiconductor nanocrystal molecules. The results confirm the superior resolution of SW-TIRF in addition to the merit of a high signal/background ratio from TIRF microscopy.
INTRODUCTION
Historical review of TIRF microscopy and superresolution microscopy
In the modern era of biological research, fluorescence microscopy is a powerful tool to visualize specific biomolecules to which fluorescent dyes can be selectively bound. Total internal reflection fluorescence (TIRF) microscopy recently has emerged as the method of choice to probe cellular processes near the basal plasma membrane of adherent cells by illuminating a very thin region on the order of 100 nm (1,2). The evanescent wave intensity decays exponentially from the interface, and this near-field excitation volume allows intrinsic optical sectioning to less than one-fifth of the excitation wavelength while keeping wide-field (WF) imaging capability. Although conventional WF fluorescence excitation generates out-of-focus emission, the selective excitation of TIRF removes the out-of-focus noise and reduces photobleaching of fluorophores outside the focal plane and is thus ideal for single-molecule imaging (3,4). On the other hand, TIRF does not allow deeper imaging into the interior of cells, and the lateral resolution of TIRF is identical to that of standard WF imaging.
The quest to image structural and functional biological information using far-field microscopy at high resolution has been hindered by the diffraction limit of light. The Abbe diffraction limit originates from the wave nature of light and depends on the wavelength and the numerical aperture (NA) of an objective lens. When an object that is substantially smaller than the diffraction limit is imaged by a microscope, its image would be significantly broadened compared to the original object. The intensity distribution of this effective point object is defined as the point spread function (PSF). The commonly accepted Rayleigh criterion defines the resolution as the distance between two point objects when one PSF falls on the first zero point of the other PSF, which can be barely distinguished in incoherent imaging. Also Sparrow suggested a resolution criterion when the gradient of the summed profile is zero or no dip at the midpoint (5). These resolution definitions are roughly equal to the full width at half-maximum (FWHM) of the PSF. With typical visible light emission and oil-immersion objectives, optical resolution is over 200 nm.
The challenge of achieving superresolution in optical microscopy beyond the diffraction limit has been overcome in practice only during the last couple of decades. In particular, extending lateral or transverse resolution has been exemplified with several techniques such as i) stimulated emission depletion (STED) microscopy, ii) saturated structured illumination microscopy (SSIM), iii) solid immersion lens (SIL), iv) structured illumination microscopy (SIM), and v) harmonic excitation light microscopy (HELM).
STED has achieved the highest far-field optical resolution of <30 nm using nonlinear photon-induced saturation depletion of the excited state in the outer regions of the excitation PSF. However, this technique suffers from relatively slow speed due to the point scanning nature (6–8). SSIM is almost an inverse version of STED, using WF mode providing comparable superresolution to STED. Photobleaching in SSIM is particularly a challenge under saturating light intensities (9). SIL microscopy takes advantage of high refractive index material replacing immersion oil, utilizing the evanescence field of SIL for near-field imaging, which also requires point scanning (10). In contrast to the point scanning methods, SIM or HELM use WF camera detection, allowing faster image acquisition by encoding either the diffraction grating illumination structure or the standing wave (SW) illumination. This contains high frequency patterned illumination onto specimen, providing up to a factor of 2 lateral resolution enhancement (11,12–14). To decode the high frequency information, only several images need to be taken with phase shift, and general lateral resolution enhancement requires rotation of this pattern.
Resolution enhancement by use of evanescent standing wave
Even higher lateral resolution in WF mode can be achieved by a combination of SW illumination and TIRF microscopy (15,16). Evanescent SW keeps the SW spacing narrower due to a higher refractive index of the substrate resulting in enhanced resolution. The SW total internal reflection fluorescence (SW-TIRF) microscope has been recently implemented (14,17).
Since the SW modulation is inherently sinusoidal, to reconstruct the high resolution in the direction of the SW, a minimum of three image acquisitions at different phases of the SW are required. Since phase shift is in principle fast, SW-TIRF does not necessarily increase the total image acquisition time compared to conventional WF imaging.
The lateral resolution mostly depends on the evanescent SW fringe period, which is proportional to the excitation wavelength in the substrate. As with other high resolution techniques based on interference, there exists a side-band artifact. This artifact can be easily removed by linear deconvolution if the emission PSF is narrow enough to keep the side-band lower than 30% (18). Thus this effectively enhances the resolution much more than twofold, as demonstrated in this article.
The advances in SW-TIRF microscopy achieved in this work include: 1), the first (to our knowledge) superresolution WF imaging of single molecular and biological specimens with 2D SW-TIRF images; 2), a numerical analysis of SW-TIRF microscopy PSF to explore the effect of more than two SW directions; and 3), a demonstration that a feedback control stabilization of the evanescent SW phase ensures that this technique is sufficiently robust for routine biomedical imaging.
THEORY
Point-spread function engineering with SW-TIRF microscopy
For one WF image with structured excitation modulation onto the specimen, the fluorescence image can be described as
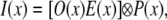 |
(1) |
and the final SW-TIRF PSF enhanced in the x-direction is given by
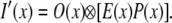 |
(2) |
where the reconstructed image 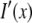 is the convolution of the fluorophore concentration distribution in the object
is the convolution of the fluorophore concentration distribution in the object 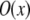 with
with 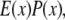 which is the multiplication of the conventional PSF
which is the multiplication of the conventional PSF 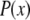 and the structured excitation intensity
and the structured excitation intensity 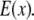 This final image with narrowed PSF is obtained by the weighted sum of several intermediate images. The excitation
This final image with narrowed PSF is obtained by the weighted sum of several intermediate images. The excitation 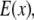 which carries the high spatial frequency component of structured illumination, has been transferred into the PSF to generate an effective PSF,
which carries the high spatial frequency component of structured illumination, has been transferred into the PSF to generate an effective PSF, 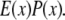 The detailed mathematical formulation of one-directional SW-TIRF (1D SW-TIRF) image reconstruction can be found in So et al. (15).
The detailed mathematical formulation of one-directional SW-TIRF (1D SW-TIRF) image reconstruction can be found in So et al. (15).
The excitation intensity profile from evanescent SW above the surface of a high refractive index substrate can be approximately described as
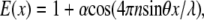 |
(3) |
where 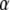 is the contrast of the SW, n is the refractive index of substrate, θ is the incident angle of the excitation beam at the interface, and λ is the vacuum excitation wavelength. This formulation gives lateral resolution enhancement in the direction of the SW. The extension of this 1D SW-TIRF theory to uniform lateral resolution enhancement can be achieved by superimposing the finite number of rotational SW-TIRF images (17).
is the contrast of the SW, n is the refractive index of substrate, θ is the incident angle of the excitation beam at the interface, and λ is the vacuum excitation wavelength. This formulation gives lateral resolution enhancement in the direction of the SW. The extension of this 1D SW-TIRF theory to uniform lateral resolution enhancement can be achieved by superimposing the finite number of rotational SW-TIRF images (17).
SW-TIRF image reconstruction
To generate a 1D SW-TIRF image, three WF images are required to be taken for each phase of the interfering SW excitation changed by 120° while the specimen remains the same. The phase difference of 120° is chosen to provide the highest signal/noise ratio (15). The superposition of the appropriately weighted images gives a high resolution image in the SW direction.
For two-dimensional SW-TIRF, finite equipolar angles are chosen and the corresponding 1D SW-TIRF is superposed, requiring a total of 3N WF images (N = number of equipolar angles). In this article, we used two-directional SW-TIRF (2D SW-TIRF) for demonstration. The numerical simulation in the following section shows that the anisotropy of effective PSF decreases to <10% for two orthogonal directions. In addition, the images are recorded with a camera with square pixels; this choice simplifies the image analysis.
Numerical simulation
As mentioned in the Theory section, general lateral resolution enhancement can be obtained by the superposition of the enhanced images in several directions. To determine how many directional images are required to get axially symmetric PSF in a practical sense, numerical simulation has been performed using a MATLAB program (MathWorks, Natick, MA) and described in Fig. 1. For N number of directional scan, uniform angular separation was used and only the profiles of the thinnest PSF FWHM and the thickest PSF FWHM direction were presented since all the other directional profiles lies between these two. Fig. 1 A shows the shape of lateral PSFs with corresponding contour plots for N = 0, 1, 2, 3, and 4. For N = 0, the PSF is the same as conventional TIRF or wide-field (WF) PSF. For N = 1, the thinnest PSF FWHM is achieved in the direction of SW propagation, resulting in ∼230% narrower PSF in the SW direction whereas the PSF FWHM in the perpendicular direction remains the same as the conventional TIRF (or N = 0 case). For N = 2, the difference of PSF FWHM in the thinnest and the thickest directions is <10% and thus it is reasonably isotropic. Also it is noticeable that the two directional PSF profiles virtually converge as N increases. Though N = 3 or 4 would be better in terms of the isotropic shape of PSF, the 2D SW-TIRF will be used for the experimental demonstration since using more directions will lengthen the total exposure time and compromise fast speed imaging. Future implementations of SW-TIRF will address this limitation. The simulated PSF FWHM of TIRF is 199 nm with the actual experimental parameter values. In actual measurement, the best measured PSF FWHM was ∼260 nm, which corresponds to that of NA 1.1 from numerical simulation. A similar result was observed in the literature (13,19) and will be further discussed in the Discussion. The MATLAB code for 2D SW-TIRF numerical simulation will be made available through the Supplementary Material online.
FIGURE 1.

(A) Effective PSF simulation of 2D SW-TIRF for directions N = 0, 1, 2, 3, and 4. N = 0 corresponds to the conventional TIRF. Anisotropy of PSF decreases as N increases (N: number of scan directions). (B) Comparison of the PSF profile in the thinnest and the thickest directions. All the other cross sectional profiles lie between these two profiles for corresponding N. Simulation condition: NA = 1.45, α = 1.0, 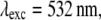
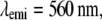 n = 1.52,
n = 1.52, 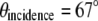 (or fringe period = 190 nm).
(or fringe period = 190 nm).
MATERIALS AND METHODS
Experimental design
Optical setup
The design of the TIRF microscope with SW excitation is depicted in Fig. 2. There are two ways to generate the TIRF system. One is prism launched and the other is objective launched. Prismless or objective-launched TIRF allows easy access to the specimen from the other side, which is particularly suitable for biological specimens in cell culture dishes, and thus is chosen in our setup (2). The excitation light from a laser (532 nm, Verdi-10, Coherent, Santa Clara, CA) is delivered to the setup via a single mode fiber through a fiber coupler (PAF-X-11-532-PC fiber coupler, OFR, Caldwell, NJ). Isolation of the laser source from the optical setup is necessary to minimize the effect of mechanical vibration of laser cooling. The fiber-delivered beam comes out of a fiber collimator and enters through a 50:50 beam splitter. One beam is reflected from a retroreflecting mirror attached with PZT (piezo-actuated transducer) to vary the optical path length for controlling the SW interference fringe phase. The other beam passes through a combination of a half-wave plate and a linear polarizer to match the intensity of two final beams entering into the objective lens. Each beam is coupled into a single-mode polarization-preserving optical fiber (Oz Optics, Ottawa, Canada), and the end fiber tips are housed in mirror mounts placed on XYZ-translators. Precise orientation and position control of the fiber tips are required with 5 degrees of freedom (X, Y, Z, yaw, and pitch) to place the corresponding excitation foci at the back aperture of the objective.
FIGURE 2.

Schematic of the objective-launched SW-TIRF microscope. Note that the diagram is very simplified (see the text).
The separation of two fiber tips with simple translation determines the final incidence angle onto a coverslip. The divergent beams emitted from these fiber tips are collimated by a collimation lens (f = 200 mm) and focused down by a tube lens (f = 125 mm). To keep our TIRF illumination in the s-polarization state, a linear polarizer is put in between the collimation lens and the tube lens (not shown in the diagram). The excitation beams enter a modified epiillumination light path of an inverted microscope (Olympus IX-71, Tokyo, Japan) through its side port and are reflected by a dichroic mirror (z532dc, Chroma, Rockingham, VT) and focused at the back aperture of a high NA objective lens (Olympus, Plan Apo 60×, NA 1.45). The collimated beams out of the objective enter the specimen/coverslip interface at a supercritical angle with beam diameter of several hundred micrometers. The incident angle is set at 67° ± 0.5°, which is above the critical angle of 62° assuming the index of specimen as 1.38 and that of coverslip as 1.52. By simply translating the excitation fiber launcher position, the setup can systematically vary the incidence angle over a range of 41.2°–72.5°, which makes it easy to switch imaging modes between SW fluorescence microscopy to SW-TIRF. The fluorescence emission is collected by the same objective and transmitted through the dichroic mirror along the emission path.
An additional barrier filter (HQ545LP, Chroma) is used to further attenuate the scattered excitation light. The emission out of the bottom port is expanded 16 times by relay optics and is focused onto a 12-bit intensified charge-coupled device (iCCD) camera (Pentamax, Princeton Instrument (now Roper Scientific), Trenton, NJ) with a field of view of 12 × 12 μm2. The residual excitation beams leaked through the back of the dichroic mirror exit through a hole bored at the back wall of the dichroic mirror cube. The residue excitation beams were expanded to form an interference pattern on a complementary metal oxide semiconductor (CMOS) sensor for SW phase detection.
With the current design, conventional TIRF images can be simply obtained by blocking one beam and conventional WF images can be taken by locating one beam at the center of the optical axis while blocking the other beam. This will be used for the comparison of different imaging modes.
Calibration of evanescent standing wave fringe period
As mentioned in the above section, the direct imaging measurement of the evanescent SW period is not trivial, due to its subdiffraction limited size. Instead, a thin uniform layer of fluorescent sample was prepared and SW emission was imaged by gradually increasing the incident angles from a subcritical angle, where SW can be imaged, to a supercritical angle where the SW cannot be imaged due to the resolution limit. This actual measurement of imaging SW fringes was matched with theoretical calculation from geometry within 2.5% and could be extrapolated to the setup angle of 67° ± 0.5°. We further need to calibrate the pixel size of the CCD camera. The pixel resolution was determined to be 23.9 nm by imaging a Ronchi ruling with known spacing (Edmund Optics, Barrington, NJ).
System stability and feedback control of the standing wave phase
The SW-TIRF system is basically an interferometer. Due to the thermal expansion and mechanical instability of the excitation beam path, the phase of the SW can drift over time (∼100°/min) and degrade the quality of final reconstructed image. Thus it is required to use closed loop feedback control of the SW phase (Fig. 3). Since the evanescent SW spacing (∼190 nm) on the coverslip is below the resolution of the imaging system (∼260 nm), an indirect way of phase estimation was devised. This is done by generating another alternative interference pattern onto a CMOS camera (S9227, Hamamatsu, Bridgewater, NJ) with ∼10× magnification relay optics at the back of the dichroic mirror mount. From the output of the CMOS array, an algorithm is implemented to calculate the fringe period and estimate the SW phase in real time. Assuming the Nyquist limit is satisfied and the alternating current component of the normalized fringe pattern is described by 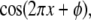 where
where 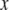 is the position along the CMOS array, the fringe phase,φ, can be obtained from
is the position along the CMOS array, the fringe phase,φ, can be obtained from 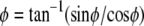 after forming and solving the matrix equation below.
after forming and solving the matrix equation below.
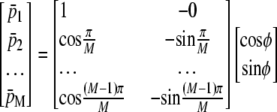 |
(4) |
FIGURE 3.

SW phase control with closed loop feedback controller. (A) Schematic diagram of the SW phase closed loop control unit. The embedded microcontroller monitors the output phase from the CMOS detector and this estimated phase is used to control input in real time (∼70 Hz). (B) Typical fringe pattern on CMOS image sensor. The invisible evanescent SW on the sample can be indirectly monitored in this alternative way. (C) SW fringe phase measurement with a feedback loop open (left) and closed (right). Reference angle: 120° and measurement duration: 0.2–45 s. The standard deviation of the fluctuation is <3°.
In our setup, the CMOS array has 512 pixels with size of 6.4 μm. Here M, which is approximately equal to six pixels, denotes the number of pixels corresponding to one period of fringes, which can be estimated by Fourier transform and taking the lowest but closest integer of the pixel values. 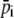 is the average intensity of the ith pixel in a period. Since 50 fringes can be recorded by the CMOS array, 50 values were averaged to provide a single average pixel value.
is the average intensity of the ith pixel in a period. Since 50 fringes can be recorded by the CMOS array, 50 values were averaged to provide a single average pixel value.
The SW phase drift was removed by a feedback control system utilizing an embedded microprocessor (SBC0486, Micro/sys, Montrose, CA) driving the PZT-driven retroreflector. The feedback signal is provided by the estimated phase of the interference pattern on the CMOS. A typical proportional, integrative, and derivative control scheme was used and the gains were tuned using the Zigler-Nichols method (20). The drift of the phase could be minimized by covering most of the beam path and attaching optical fibers on the rigid part. The fluctuation was reduced by isolating any mechanically moving sources from the optical table except a cooling fan inside the iCCD.
Data acquisition and image reconstruction
The raw images from the iCCD camera were stored using a desktop computer with Windows 98, running the WinView (Roper Scientific, Trenton, NJ) program. The typical exposure time for each raw image was ∼0.1– 0.2 s with total excitation power entering into the objective <10 mW. To minimize the photobleaching of fluorophore, a mechanical shutter was used to restrict the illumination during the image acquisition. Nonfluorescent immersion oil (Olympus, n = 1.516 at 23°C) was used. Each image was subtracted by the background image of a sample without fluorophore under the same condition. The image acquisition was synchronized with the real-time feedback control of the SW fringe phase by the embedded microprocessor using custom-written software in the C programming language (Turbo C++ version 3.0). Postimage reconstruction and digital image analysis was performed on an IBM (Armonk, NY) Thinkpad laptop computer (T40) using MATLAB software. The electronic postprocessing takes a few seconds, and potentially real-time reconstruction would be feasible with the development of computing technology.
To generate a 2D SW-TIRF image, the specimen was mechanically rotated by 90° between the recordings of two 1D SW-TIRF image sets that were subsequently combined. The registration of two-directional enhanced images was performed by using cross correlation of two images to find the relative shift of images before superposition. However, care needs to be taken since this cross correlation may not work well when the overall intensity distribution of each image is not the same. The mechanical sample rotation may be avoided in the future by the use of multiple directional excitation (13).
Fluorescent polystyrene spheres and semiconductor nanocrystals (quantum dots)
Fluorescent polystyrene microspheres (F-8792, Molecular Probes, Eugene, OR), with a nominal diameter of 44 nm and peak emission at 560 nm, were sonicated for several minutes and were loaded onto a cover slide followed by covering with a standard coverslip (22 × 22 mm2, No. 1.5 thickness). The coverslip was sealed with nail polish to prevent the evaporation of water. Initially most beads were moving due to Brownian motion. After overnight, most beads are attached on the coverslip and ready for imaging. The number concentration of microspheres was adjusted to get ∼10–30 beads in the microscope field of view (12 × 12 μm2).
The sample preparation of quantum dots was similar to that of polystyrene microspheres except using quantum dots (Qdot565 streptavidin conjugated, Invitrogen, Carlsbad, CA). It was recognized that the quantum dots are prone to lose their signal if stored in diluted solution. In addition, their blinking nature was adverse to the SW-TIRF technique based on the quantitative signal of intermediate images.
Cell culture and fluorescent labeling of the actin cytoskeleton
Fibroblasts were grown in standard 100 mm × 20 mm cell culture dishes (Corning, VWR, West Chester, PA) in Dulbecco's modified Eagle's medium (Cellgro, Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and penicillin-streptomycin (100 units of penicillin per ml media, and 100 μg streptomycin per ml media; Invitrogen). Cells were cultured at 37°C in 5% CO2. At 24 h before the labeling experiments, fibroblasts were plated on 35-mm glass-bottom cell culture dishes (MatTek, Ashland, MA) coated with collagen I (1 μg/cm2; Cohesion Tech, Palo Alto, CA).
On the day of the experiments, the cell confluency had reached ∼60%. At room temperature, cells were then fixed with 3.7% formaldehyde in phosphate buffered saline (PBS, Mediatech) for 10 min, washed twice with PBS, and extracted with 0.1% Triton X-100 in PBS for 5 min. To reduce nonspecific background staining, fixed cells were then incubated in PBS containing 1% bovine serum albumin (Polysciences, Warrington, PA) for 20 min. For F-actin labeling, cells were then incubated with 165-nM AlexaFluor 532 phalloidin (Molecular Probes) for 20 min and washed three times with PBS.
RESULTS
Imaging fluorescent polystyrene spheres
A comparison of conventional TIRF PSF and 1D SW-TIRF PSF images is shown in Fig. 4. The sample contains fluorescent polystyrene microspheres in water (see Materials and Methods). The intensity profiles in the SW direction show that SW-TIRF PSF has narrowed by 239% over TIRF in terms of FWHM. However, SW-TIRF has two prominent side lobes with amplitudes slightly <30% of the main peak. This observation is consistent with theoretical prediction, Eq. 2, if the NA of the microscope objective is ∼1.1. Although the theoretical NA of the objective is supposed to be 1.45, the lower NA value is consistent with conventional TIRF measurement where PSF with FWHM of 260 nm is measured. This performance characteristic of the objective is consistent with in-house measurement by Olympus (Eiji Yokoi, Olympus America, Inc., personal communication, 2005) and will be further investigated in the Discussion. Nonetheless, since the relative height of side lobes compared to the main peak is <30%, linear deconvolution can quickly eliminate side lobes as shown in Fig. 4 c (18). The PSF FWHM of the main peak remains unchanged after linear deconvolution. No filtering to the raw images before image reconstruction was applied.
FIGURE 4.

Effective PSF measurement of (A) conventional TIRF, (B) 1D SW-TIRF, and (C) 1D SW-TIRF with linear deconvolution with a 0.04-μm fluorescent bead. The evanescent standing excitation is in the vertical direction. The corresponding vertical intensity profile is (a), (b), and (c). These vertical profiles show a narrower PSF FWHM for SW-TIRF in comparison with that of conventional TIRF. (Experimental condition is the same as that of numerical simulation: NA = 1.45, 
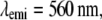 n = 1.52, and
n = 1.52, and 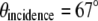 ± 0.5° except the contrast was typically measured ∼0.9.)
± 0.5° except the contrast was typically measured ∼0.9.)
Imaging semiconductor nanocrystals (quantum dots)
Quantum dots are gaining popularity for biological imaging. Their long-term photostability is far superior to organic fluorescent dye molecules. They also possess broad excitation and narrow emission spectrums, allowing simultaneous labeling of different structures (21). The imaging of single molecular quantum dots allows us to demonstrate the sensitivity of this technique.
Quantum dots are also ideal objects for PSF measurements due to their small size of 10–25 nm, which is far below the diffraction limit. The result is displayed in Fig. 5 with vertical intensity profiles of several regions of interests (ROI). The total input power into the objective was ∼4.6 mW. The results of 1D SW-TIRF imaging showed lateral resolution enhancement of more than 2.5 times in ROI I and II.
FIGURE 5.

The comparison of conventional TIRF, 1D SW-TIRF, and SW-TIRD images of semiconductor quantum dots. The evanescent SW direction is along the vertical direction, the exposure time is 1.0 s, and other experimental conditions are the same as in Fig. 4.
Interestingly, some spots did not show enhanced resolution, which is likely due to the blinking of the quantum dot. Although the mechanism for quantum dot blinking is not fully understood, several different ways of blinking suppression were documented (22). We have tried a number of these approaches without great success except for the use of betamercaptoethanol; however, betamercaptoethanol is toxic to biological specimen and hence has minimal long-term utility. Recently, we observed significantly reduced blinking behavior in quantum dot products from a different source (Evident Technology, Troy, NY). The manufacturing process of quantum dots seems to play an important role in their blinking behavior. Finally, we also found that quantum dots mounted on microscope slides tend to lose their signals and degrade in a few days. Unless there are reliable methods to suppress the blinking of quantum dots and to maintain their photostability in specimens, they may be less than ideal probes for SW-TIRF microscopy.
Imaging actin cytoskeleton: 1D SW-TIRF
Imaging of biological specimen is demonstrated in Fig. 6. Since the SW propagation is in the vertical direction, 1D SW-TIRF gives resolution enhancement only in the vertical direction. WF image A (a) has a higher background noise level, whereas conventional TIRF image B (b) shows significantly lower background noise compared to WF due to the shallow evanescent excitation of ∼73 nm. However, the lateral resolution remains about the same with WF and TIRF. Images in B (c) and B (d) show the SW-TIRF image and that with linear deconvolution, respectively. The presence of horizontal stripes in B (c) is due to the side lobes of the original 1D SW-TIRF image and is reasonably suppressed after linear deconvolution in B (d). The SW-TIRF images reveal finer detail of the cellular actin cytoskeleton, resulting in 235% enhancement in terms of lateral resolution compared to the WF image, which demonstrates the high resolution capability of this system in the application of imaging cellular actin cytoskeleton.
FIGURE 6.

F-actin cytoskeleton in mouse fibroblast cells (NIH 3T3) imaged with (a) WF, (b) conventional TIRF, (c) 1D SW-TIRF, and (d) 1D SW-TIRF with linear deconvolution (SW-TIRD). B (a–d) are enlarged insets of corresponding image from A (a–d). B (a1–d1) are the vertical profiles of dashed line in the corresponding image B (a–d). The evanescent standing excitation is in the vertical direction.
Imaging actin cytoskeleton: 2D SW-TIRF
The F-actin cytoskeleton structure of a single fibroblast cell was imaged by 2D SW-TIRF in Fig. 7 with the resolution enhancement in both directions. Here, the superposition of two perpendicular directional 1D SW-TIRF was used to generate one 2D SW-TIRF image. Fig. 7, A (a) and (b), show that TIRF decreases the out-of-focus noise to improve the signal/background ratio compared to conventional WF imaging at the cost of observing only near the surface region. In inset 1 of Fig. 7 B, SW-TIRF can reveal the 190 nm separated actin fibers, which are not resolvable with either WF or TIRF. In Fig. 7 C, second row, FWHM of actin fiber is narrowed from 286 nm to 127 nm by more than twofold. In the third row, SW-TIRF can distinguish actin fibers separated by 190 nm. It is recognized that not all the actin fibers in one image are in focus due to the slight changes of actin fiber height from the bottom surface. These results clearly demonstrate the superior high resolution capability of SW-TIRF over conventional WF or TIRF. A major difficulty of imaging biological specimen compared to the fluorescent beads is the photobleaching issue. The beads are bright and can last a relatively long period under continuous excitation, whereas the actin phalloidin needs extra care to minimize the total illumination time before fading away. Since the intermediate images were recorded sequentially at three phases and along two orthogonal directions, some photobleaching is unavoidable. To compensate for photobleaching, the two 1D SW-TIRF image set along two orthogonal directions are superposed after normalization based on the maximum intensity level of each image. However, the photobleaching of the three sequential images along one direction was not compensated since the degree of photobleaching was not significant. For samples that may be photobleaching faster, further compensation can be applied by assuming a simple linear photobleaching rate model (23).
FIGURE 7.

F-actin cytoskeleton in mouse fibroblast cells (NIH 3T3) imaged with (a) WF, (b) conventional TIRF, (c) 2D SW-TIRF, and (d) 2D SW-TIRF with linear deconvolution (SW-TIRD). Subfigure A compares the four different imaging modes and subfigures B and C are the enlarged insets 1 and 2 marked in subfigure A. In subfigure C, the second and third rows are the vertical profiles of (1) and (2) in subfigure C (a).
DISCUSSION
TIRF microscopy and a lateral version of SW microscopy were combined. This new technique has been demonstrated with fixed biological specimen. The comparison between WF, TIRF, and SW-TIRF shows that SW-TIRF can reveal the fine structural details of actin cytoskeleton with a high signal/background ratio close to the basal plasma membrane of cells. Other complementary imaging modalities such as atomic force microscopy could be combined with TIRF (24,25) to provide high resolution imaging on the apical surface. We have further implemented SW-TIRF in objective-launched geometry instead of using a prism-launched method to facilitate the imaging of cellular specimens. One drawback of objective-launched TIRF is the presence of stray illumination light arising from the scattering inside the objective contributing nonevanescent fluorescence background.
From the theory of diffraction-limited imaging provided by the Airy function, the ideal PSF FWHM is calculated to be 199 nm with an NA of 1.45 and an emission wavelength of 560 nm. In contrast, the measured PSF FWHM was 260 nm under the same condition with a 44-nm bead. Since in SW-TIRF the height of side lobes depends on the original PSF, a discussion regarding the origin of this nonideal PSF might be useful. First, the high NA microscope objective may not have an ideal modulation transfer function from manufacturing due to strong attenuation of marginal rays (19). Second, the emission from the fluorescent beads is not actually monochromatic at 560 nm but peaked at 560 nm with a long spectral tail into the longer wavelength region. Third, the effect of bead size adds some broadening to the final PSF FWHM. However, numerical simulation by convoluting the finite bead model with the Airy function gives the broadening of PSF FWHM of <5% with a 44-nm-size bead. Fourth, there exists cross talk between CCD pixels due to the coupling fibers which connect the microchannel plate in the image intensifier to the CCD chip. The pixelation error is increased by a factor of 1.5 due to the fiber coupling ratio, resulting in the effective pixel size of 22.6 μm instead of the physical pixel size of 15 μm (26). In fact, iCCD is not our first detector choice for two reasons but is used since it is available in our laboratory. Our image field of view is significantly limited by the relatively small CCD chip (512 × 512 pixels) used in this camera. Our imaging speed (∼11 fps) is limited by the slow readout rate of this device. A larger format, electron bombardment CCD, with a fast readout rate may be a better choice. Other potential reasons could be the misalignment of the optical setup and the aberration of relay optics. The broadening of PSF FWHM by 20% was also reported in other systems (13).
In principle, the SW-TIRF resolution is not inherently limited by the emission PSF. But it is mostly limited by how small structured excitation could be generated. If a nonlinear excitation is incorporated, even further resolution could be achieved (9,15). Without incorporating any nonlinear optical technique, the PSF FWHM could have reached down to ∼90 nm with the setting here. However, the Gaussian laser beam needs a small margin to generate a uniform SW overlapping field onto the field of view. A further resolution of 69 nm could be achieved if the currently available NA 1.65 objective with 457-nm excitation is used.
We are currently developing a real-time SW-TIRF microscope for live cell imaging with multicolor capability. Semiconductor quantum dots might be suitable chromophores for long-term live cell imaging because of their excellent photostability and low photobleaching (21). Most importantly, different colors of quantum dots can be efficiently excited at a single excitation wavelength, eliminating the instrumental and computational complications of imaging using different colors and periods of standing evanescence waves. However, quantum dots exhibit characteristic blinking behavior. Blinking could interfere with SW-TIRF microscopy where precise intensity information is needed for image processing. The problem might be overcome by biochemical suppression methods (22). This development could allow the study of dynamic processes of live cells such as cellular attachment or migration as well as endo/exocitosis, even at higher resolution and with single molecular sensitivity.
Further resolution improvement may require nonlinear modalities such as STED or two-photon microscopy with the additional advantage of side-lobe suppression. However, an even higher signal/noise ratio may be obtained by utilizing surface plasmon resonance-coupled emission (27). The resolution of any imaging system depends on the signal/noise ratio, which is ultimately limited by the number of collected photons. For fluorescence methods, this imposes an intrinsic limit to any approaches to improve resolution by decreasing the size of PSF due to the relatively increased noise in low light fluorescence imaging (26). On the other hand, the nonfluorescent or scattering version of SW-TIR with novel metal nanoparticles may open up broader applications (28,29). Since the photon scattering process is instantaneous instead of having a finite lifetime as in fluorescence processes, the signal photon production rate of metal nanoparticles in biological systems is only limited by heating processes and can be orders of magnitude higher than fluorescence. Further, there is no concern about photobleaching for metal nanoparticles. These noble metal nanoparticles with very high scattering power may serve as fluorescent analogs in biology for SW-TIRF imaging although the imaging process is coherent rather than incoherent, as in fluorescence imaging.
CONCLUSION
TIRF microscopy has provided a way to peek into the dynamics of subcellular structures in the near membrane region of cells, and a high lateral resolution version of TIRF has been described in this article. Utilizing the subdiffraction limit evanescent SW excitation, the lateral resolution has been improved to reveal finer details of biological specimen which otherwise would be hidden in a blur of out-of-focus fluorescence. The first, to our knowledge, two-dimensional superresolution SW-TIRF imaging with cellular actin cytoskeleton is demonstrated by achieving more than 230% lateral resolution improvement. A 1D SW-TIRF is proved to be sufficient for substantial improvement of lateral resolution in the SW direction by imaging beads, achieving a lateral resolution of 109 ± 10 nm beyond the classical diffraction limit. In addition, we have shown this system has enough sensitivity to image single quantum dot molecules with superresolution. We have demonstrated a WF high resolution imaging technique that has significantly faster imaging speed over the point-by-point scanning approaches. SW-TIRF microscopy may contribute to resolve the dynamic structural and biochemical changes of the biomolecules near the basal cell membrane.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank Ms. Maxine Jonas of Massachusetts Institute of Technology for assisting in the preparation of biological specimens and Wai Teng Tang of the National University of Singapore for technical assistance.
This research was funded by the National Science Foundation (research grant MCB-9604382) and the National Institutes of Health (P01HL64858).
Editor: Enrico Gratton.
References
- 1.Axelrod, D. 1981. Cell-substrate contacts illuminated by total internal-reflection fluorescence. J. Cell Biol. 89:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axelrod, D. 2001. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2:764–774. [DOI] [PubMed] [Google Scholar]
- 3.Webb, S. E. D., S. R. Needham, S. K. Roberts, and M. L. Martin-Fernandez. 2006. Multidimensional single-molecule imaging in live cells using total-internal-reflection fluorescence microscopy. Opt. Lett. 31:2157–2159. [DOI] [PubMed] [Google Scholar]
- 4.Tokunaga, M., K. Kitamura, K. Saito, A. H. Iwane, and T. Yanagida. 1997. Single molecule imaging of fluorophores and enzymatic reactions achieved by objective-type total internal reflection fluorescence microscopy. Biochem. Biophys. Res. Commun. 235:47–53. [DOI] [PubMed] [Google Scholar]
- 5.Goodman, J. W. 1996. Introduction to Fourier Optics. McGraw-Hill, New York.
- 6.Westphal, V., and S. W. Hell. 2005. Nanoscale resolution in the focal plane of an optical microscope. Phys. Rev. Lett. 94:143903. [DOI] [PubMed] [Google Scholar]
- 7.Westphal, V., L. Kastrup, and S. W. Hell. 2003. Lateral resolution of 28 nm (lambda/25) in far-field fluorescence microscopy. Appl. Phys. B-Lasers Opt. 77:377–380. [Google Scholar]
- 8.Hell, S. W., and J. Wichmann. 1994. Breaking the diffraction resolution limit by stimulated-emission- depletion fluorescence microscopy. Opt. Lett. 19:780–782. [DOI] [PubMed] [Google Scholar]
- 9.Gustafsson, M. G. L. 2005. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA. 102:13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansfield, S. M., and G. S. Kino. 1990. Solid immersion microscope. Appl. Phys. Lett. 57:2615–2616. [Google Scholar]
- 11.Gustafsson, M. G. L. 2000. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. (Oxford). 198:82–87. [DOI] [PubMed] [Google Scholar]
- 12.Fedosseev, R., Y. Belyaev, J. Frohn, and A. Stemmer. 2005. Structured light illumination for extended resolution in fluorescence microscopy. Opt. Lasers Eng. 43:403–414. [Google Scholar]
- 13.Frohn, J. T., H. F. Knapp, and A. Stemmer. 2000. True optical resolution beyond the Rayleigh limit achieved by standing wave illumination. Proc. Natl. Acad. Sci. USA. 97:7232–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gliko, O., G. D. Reddy, B. Anvari, W. E. Brownell, and P. Saggau. 2006. Standing wave total internal reflection fluorescence microscopy to measure the size of nanostructures in living cells. J. Biomed. Opt. 11:064013. [DOI] [PubMed] [Google Scholar]
- 15.So, P. T. C., H. S. Kwon, and C. Y. Dong. 2001. Resolution enhancement in standing-wave total internal reflection microscopy: a point-spread-function engineering approach. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 18:2833–2845. [DOI] [PubMed] [Google Scholar]
- 16.Cragg, G. E., and P. T. C. So. 2000. Lateral resolution enhancement with standing evanescent waves. Opt. Lett. 25:46–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung, E., D. K. Kim, and P. T. C. So. 2006. Extended resolution wide-field optical imaging: objective-launched standing-wave total internal reflection fluorescence microscopy. Opt. Lett. 31:945–947. [DOI] [PubMed] [Google Scholar]
- 18.Hanninen, P. E., S. W. Hell, J. Salo, E. Soini, and C. Cremer. 1995. 2-photon excitation 4pi confocal microscope—enhanced axial resolution microscope for biological-research. Appl. Phys. Lett. 66:1698–1700. [Google Scholar]
- 19.Neil, M. A. A., R. Juskaitis, and T. Wilson. 1997. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 22:1905–1907. [DOI] [PubMed] [Google Scholar]
- 20.Palm, W. J. 2000. Modeling, analysis, and control of dynamic systems. Wiley, New York.
- 21.Michalet, X., F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir, and S. Weiss. 2005. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 307:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hohng, S., and T. Ha. 2004. Near-complete suppression of quantum dot blinking in ambient conditions. J. Am. Chem. Soc. 126:1324–1325. [DOI] [PubMed] [Google Scholar]
- 23.Yu, W., P. T. C. So, T. French, and E. Gratton. 1996. Fluorescence generalized polarization of cell membranes: a two-photon scanning microscopy approach. Biophys. J. 70:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathur, A. B., G. A. Truskey, and W. M. Reichert. 2000. Atomic force and total internal reflection fluorescence microscopy for the study of force transmission in endothelial cells. Biophys. J. 78:1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar, A., R. B. Robertson, and J. M. Fernandez. 2004. Simultaneous atomic force microscope and fluorescence measurements of protein unfolding using a calibrated evanescent wave. Proc. Natl. Acad. Sci. USA. 101:12882–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stelzer, E. H. K. 1998. Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: fundamental limits to resolution in fluorescence light microscopy. J. Microsc. (Oxford). 189:15–24. [Google Scholar]
- 27.Kostov, Y., D. S. Smith, L. Tolosa, G. Rao, I. Gryczynski, Z. Gryczynski, J. Malicka, and J. R. Lakowicz. 2005. Directional surface plasmon-coupled emission from a 3 nm green fluorescent protein monolayer. Biotechnol. Prog. 21:1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yguerabide, J., and E. E. Yguerabide. 1998. Light-scattering submicroscopic particles as highly fluorescent analogs and their use as tracer labels in clinical and biological applications: I. Theory. Anal. Biochem. 262:137–156. [DOI] [PubMed] [Google Scholar]
- 29.Yguerabide, J., and E. E. Yguerabide. 1998. Light-scattering submicroscopic particles as highly fluorescent analogs and their use as tracer labels in clinical and biological applications: II. Experimental characterization. Anal. Biochem. 262:157–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.