

Abstract
As exciting light in a scanning confocal microscope encounters a cell and its subcellular components, it is refracted and scattered. A question arises as to what proportion of the exciting light is scattered by subcellular structures and whether cells in the vicinity of the imaged area, i.e., cells that are not directly illuminated by the laser beam, can be affected by either an exposure to scattered light and ensuing phototoxic reactions, or by the products of photoactivated reactions diffusing out of the directly illuminated area. We have designed a technique, which allows us to detect subtle cell photodamage and estimate the extent and range of phototoxic effects inflicted by interaction between scattered exciting light and fluorescent probes in the vicinity of the illuminated area. The technique is based on detecting an increased influx of acridine orange into photodamaged cells, which is manifested by a change of color. We demonstrate that phototoxic effects can be exerted not only on the illuminated cell, but also on fluorescently labeled neighboring cells. The damage inflicted on neighbors is due to exposure to light scattered by the imaged (i.e., directly illuminated) cell, but not phototoxic products diffusing out of the directly illuminated area. When light encounters a cell nucleus, scattering is so intense that photodamage can be inflicted even on fluorescently labeled cells located within a radius of ∼90 μm, i.e., several cell diameters away. This range of scattering is comparable with that caused by the glass bead resting on a coverslip (up to 120 μm). The intense scattering of exciting light imposes limits on FRAP, FLIP, and other techniques employing high intensity laser beams.
INTRODUCTION
Scattering of light by live cells
Tissues, individual cells in culture, and subcellular structures scatter visible light extensively (1–3). Light scatter provides contrast in standard and confocal dark field imaging of individual cells (4,5). Scattering has been exploited in automatic cell counters and subsequently in flow cytometry, where low and right angle scatter was found to convey information about cell size and granularity (6). Light scatter has also been investigated as a means of detecting pathological changes in skin and cancerous changes in tissues (7–10) and employed in quantitating changes of mitochondrial shape induced by calcium overload (11).
Light scatter in fluorescence microscopy
The absolute refractive index (RI) of culture medium, plasma membrane, and subcellular structures of cells grown in vitro can differ dramatically. Although the RI of water is 1.3, culture medium and saline 1.31, and cytoplasm 1.35 (12), the RI of cell membranes is as high as 1.46–1.60 (13); mitochondria is 1.4 (14,15) and nucleus may be 1.4 as well (15). Consequently, extensive scattering of exciting light as well as emitted fluorescence occurs at the surface and inside live cells. In wide-field fluorescence microscopy scattered exciting light is not detected in the image, but scattered fluorescence is seen as a bright background. This background causes deterioration of image contrast and, thus, degrades image resolution. This problem was largely eliminated in a scanning confocal microscope (M. Minsky, Massachusetts Institute of Technology, Cambridge, MA; 1961, U.S. patent No. 3013467) (16,17), where scattering of exciting light is minimized by using a focused beam, and scattered fluorescence is eliminated from the image by a confocal aperture (17,18). Although the scattering of exciting light is minimized in a focused beam scanning microscopy, it can still be extensive, as demonstrated in this report. Such scattering may constitute a particular problem in live cell imaging, fluorescence recovery after photobleaching (FRAP) experiments, and new high resolution imaging techniques employing laser beams of high intensity. Scattered exciting light may potentially complicate confocal imaging in three ways: by causing unexpected photobleaching in regions surrounding the directly illuminated volume, by causing phototoxic effects even at a large distance from the illuminated area, and by making it impossible to focus a laser beam into a diffraction-limited spot. An added complication arises from the fact that scattering depends on the size and shape of scattering particles and the wavelength of incident light, in a complicated way, which cannot be predicted by simple models (18,19).
This report focuses on estimating the range of scattering of exciting light on live cells in scanning confocal microscopy, describes an intense scattering on cell nucleus, and demonstrates that interaction between scattered light and fluorescent probes can cause photodamage to neighboring labeled cells. To quantitate the adverse effects of scattered light, we use a new method of detecting and quantitating subtle photodamage inflicted on cells.
MATERIALS AND METHODS
Reagents
Chromatographically pure acridine orange (AO) (Molecular Probes, Eugene, OR) was a generous gift of Prof. Z. Darzynkiewicz. AO stock was dissolved in water at 1 mg/ml and kept at 4°C. Propidium iodide (PI) and ethidium bromide (EB) (Sigma, Poznan, Poland) stocks (1 mg/ml) were kept frozen. Verapamil (Sigma) was prepared fresh as 50 mg/ml solution in water, and subsequently added to culture medium to a final concentration of 1 mM. Glass beads (Sigma, St. Louis, MO) had diameters in the range 90–150 μm.
Cell cultures
HeLa cells were cultured in DMEM (Sigma), supplemented with 10% fetal calf serum (Gibco, Paisley, UK), using standard procedures.
Confocal microscopy
Images of luminescence of AO and PI were recorded using a Bio-Rad MRC1024 confocal system, interfaced with a Nikon Diaphot (Nikon, Amsterdam, The Netherlands) inverted microscope equipped with a 100-mW Ar ion laser (ILT, Salt Lake City, UT), 25 mW KrAr laser (ALC, Salt Lake City, UT), three detection channels, and a Nikon PlanApo 60× oil immersion lens (NA 1.4). The fluorescence detection conditions were: AO – exc. 458 nm, primary dichroic 510DCLP (VHS filter block), secondary dichroic 565DRLP (A2 filter block), green emission filter 540DF30, red emission filter RG630 (these settings ensured that the cross talk was negligible (21)); PI, excitation 568 nm, primary triple (488/568/647) dichroic (T1 filter block), secondary dichroic 560DRLP (T2A); 8-bit, 512 × 512 images were collected at a rate of 1/s or 0.3/s. Laser light power was measured through a PlanApo 4× NA 0.2 lens, using LaserCheck light meter (Coherent, Santa Clara, CA) and was 0.5 mW for the 458 line and the VHS filter block. When inflicting photodamage on cells, the confocal plane was placed within 1 μm of the glass surface. Images of acridine orange were recorded 15–20 min after inflicting photodamage.
Image analysis
Images were analyzed with LaserSharp v.3.2 (Carl Zeiss, Jena, Germany), ImageJ (http://rsb.info.nih.gov/ij/), or AutoDeblur (Bitplane AG, Zurich, Switzerland), using standard procedures.
Definition of terms
The key terms used in this text are defined in the following way: “sublethal cell damage” and “subtle damage” cell damage, which is not resulting in an immediate loss of plasma membrane integrity, with no reference to a capacity to complete the next mitosis; “lethal damage” cell damage, which results in an immediate loss of plasma membrane integrity, detectable by dye exclusion assays, generally leading to inability to complete the next mitosis; “phototoxic effects” and “photodamage”, any adverse effects resulting from interaction of light with intrinsic or exogenous fluorescent compounds present in or around cells, regardless of the molecular mechanism, involvement of oxygen, etc.
RESULTS
Quantitating sublethal cell damage
To detect the phototoxic effects exerted by interaction between the scattered light and fluorescent labels in the vicinity of the illuminated area we required a microscopic method of rapidly quantitating sublethal cell damage. The common endpoints in assays designed to detect cell damage are plasma membrane integrity, clonogenic capacity, rate of cell divisions, metabolic activity, or DNA synthesis (21). None of these assays can be used to rapidly detect (under a microscope) and quantitate sublethal damage inflicted on cells as a result of interaction between exciting light and a fluorescent probe. We designed a new method to perform this task; the principle is demonstrated in Fig. 1 I. This approach is based on detecting impairment of drug efflux in damaged cells. In this assay the endpoint is the increase of intracellular concentration of a fluorescent drug, acridine orange, over a detectable threshold. Under standard conditions healthy cells pump out the drug efficiently so the intracellular concentration of AO is maintained below the extracellular level. In damaged cells, however, inhibition or impairment of the system responsible for pumping out the drug occurs. Consequently, the balance between a gradient-driven influx and an enzyme-driven efflux is shifted toward drug entry. This shift is dependent on the extent of damage and is manifested by a change of color, according to the following mechanism.
FIGURE 1.

(I) Efficiency of drug efflux can be estimated by the color of cytoplasmic AO. In all panels extracellular concentration of AO was 8 μg/ml, i.e., a concentration when ds and ss nucleic acids are stained differentially, but the access of AO to cell interior was different, as described: (A) image of AO luminescence in a cell fixed with formaldehyde. Because internal and plasma membranes are fully permeable, the intracellular and extracellular concentrations of AO are equal. Single stranded RNA in cytoplasm and nucleoli stain red, double stranded DNA in nucleus stains green. Bar, 20 μm. (B) Image of AO fluorescence in the cytoplasm of live, intact cells. Cells pump out AO, thus, intracellular concentration of AO is lower than extracellular and AO stacks are not formed. The cytoplasm as well as nucleus (N) and nucleoli (NC) emit green fluorescence. Acidic endosomes (ES) accumulate AO and the high intraendosomal concentration of AO promotes formation of stacks, resulting in red luminescence. (C) Image of AO luminescence following mechanical damage to plasma membranes. The cell monolayer was scratched with a needle resulting in mechanical damage to some cells (arrowheads) located at the edge of the wound (arrow). This damage caused a rapid entry of AO into cells and an increase of intracellular AO concentration to the level of the surrounding medium. As a result, the cytoplasm emits red luminescence in damaged cells (arrowheads). Intact cells (at a distance from the wound) maintain the AO gradient and still emit only green fluorescence. (D) Image of AO in cells treated with verapamil, a drug, which inhibits drug efflux. The intracellular concentration of AO increases following exposure to verapamil and cytoplasm assumes orange color, derived from the presence of both, AO stacks (red) and AO monomers (green). The presence of stacks indicates that the intracellular concentration of AO has increased above the level of ∼5 μg/ml. (E) Image of AO in cells deprived of energy by maintaining in culture without medium replenishment for 4 days. Drug efflux is less efficient and intracellular concentration of AO rises. The increasing intracellular concentration of AO leads to formation of stacks and a relative increase of red luminescence. (F) Image of AO in a cell (arrow) after exposure to a high intensity exciting light (458 nm, 50 μW beam, pixel 30 nm, 0.3 scan/s, 10 scans). Interaction of light with AO (and presumably subsequent reactions with oxygen) leads to various phototoxic effects. These effects are manifested by the inhibition of drug efflux and increased permeability of plasma membranes. As a consequence, the intracellular concentration of AO increases and stacks are formed on RNA. AO in cytoplasm emits not only green fluorescence, as in a control, but red luminescence as well. Endosomes are damaged and lose the accumulated AO. Neighboring cells also suffer subtle damage, as demonstrated by red emission of cytoplasm; this damage is less severe as shown by the presence of endosomes that are still capable of maintaining accumulated AO. (II) Subtle photodamage is manifested by less efficient drug efflux and can be assessed on the basis of the relative increase of the intracellular concentration of AO; PI exclusion can only detect heavy photodamage. The images describe photodamage in fluorescently labeled cells illuminated by exciting light of various intensities. Top rows, AO luminescence; bottom rows, fluorescence of propidium. (A) Control, unilluminated cells with efficient drug efflux. AO in cytoplasm and nucleus emits green fluorescence (except for acidic endosomes); red luminescence of AO bound to RNA is undetectable. Propidium is excluded from cells. A slight bleed-through of the intense red luminescence of AO from endosomes is detected in the red channel dedicated to propidium. Bar, 20 μm. (B and C) AO in cells exposed to excitation light of a low intensity (B) All cells in the field of view were exposed to low light levels and suffered subtle photodamage; (C) the damaged cell is marked with an arrow. Green AO luminescence and a detectable red component indicates that drug efflux is less efficient than in cells not subjected to illumination. The lack of intracellular fluorescence of PI indicates that the integrity of plasma membrane is not compromised. (D–F) AO and PI luminescence in cells exposed to high doses of exciting light (arrow). Cell damage is manifested by red luminescence. Integrity of plasma membrane is lost and the cells are unable to exclude propidium. (Bottom) A graph showing the parameter γ as a function of the doses of exciting light used in B–F above. Higher light doses cause more pronounced damage to drug efflux mechanisms (and presumably the structure of plasma membrane) and the damage is reflected in a higher concentration of cytoplasmic AO. In the most heavily damaged cells plasma membrane integrity is lost and cells no longer exclude propidium (the gray area in the graph). The test with AO can detect subtle damage, whereas PI exclusion detects only heavy, lethal damage manifested by a complete loss of plasma membrane integrity. (III) Sublethal damage can be inflicted on neighbors of the illuminated cell due to interaction between the scattered light and fluorescent labels residing in these cells. The damage is manifested by impairment of drug efflux. (Left column) Schematics of the illuminated sample, (center left) a graph showing values of the parameter γ as a function of the distance from the exciting beam (blue bars, control; pink bars, cells affected by scattered light); (center right and far right) images before and after exposure to exciting light (458 nm, 50 μW laser beam, 10 scans, 0.3 scan/s). The illuminated area (17 × 17 μm) is marked with an arrow. (A) Exciting light is incident on a glass outside of cells; neighboring cells are intact. Bar, 20 μm. (B) Exciting light is incident on cytoplasm of two adjacent cells in an area with few acidic endosomes. Only a small area of the cytoplasm shows signs of damage; neighbors remain intact. (C) Exciting light is incident on nucleus; neighbors within 70–90 μm are damaged. (D) Exciting light is incident on a glass bead (diameter 100 μm) resting on a coverslip. The point of contact of the bead with the coverslip is marked with an arrow. Neighbors within a radius of 120 μm (beyond the field of view shown) are damaged.
AO is a metachromatic dye, which exhibits two modes of binding to nucleic acids. At concentrations below 5 μg/ml, AO intercalates into single stranded (ss) and double stranded (ds) nucleic acids and yields green fluorescence (22). At higher concentrations (10 μg/ml and above) AO readily forms stacks on ss and ds nucleic acids. At a range of 5–10 μg/ml AO differentially stains ds and ss nucleic acids; stacks predominate on ss, whereas intercalated monomers predominate on ds nucleic acids. This difference in predominant binding modes leads to a perceived red luminescence of RNA in cytoplasm (and nucleoli) and green fluorescence of nuclear DNA in fully permeable cells (Fig. 1 I A). The staining pattern of live cells is different, however. When live HeLa cells with intact plasma membranes are submerged in culture medium supplemented with acridine orange at 8 μg/ml, the dye still enters cells, stains nucleic acids, and accumulates in acidic endosomes (Fig. 1 I B) (22). In contrast to permeabilized cells, however, the cytoplasmic concentration of AO is maintained below the extracellular concentration, through the action of an efficient drug efflux mechanism. Intracellular concentration of AO is maintained sufficiently low so as the stacks are not formed on RNA and, as a result, cytoplasm, nuclei, and nucleoli of live cells with functional membranes acquire only green fluorescence (Fig. 1 I B, only acidic endosomes accumulate AO and fluoresce red (22)). If the integrity of plasma membrane is compromised by mechanical damage, however, AO can enter the damaged cell freely and the intracellular (cytoplasmic and nuclear) concentrations of dissolved AO equilibrate with the extracellular environment, allowing for formation of stacks, as shown in Fig. 1 I C. Under these conditions AO stacked on RNA in cytoplasm and nucleoli emits red luminescence. A similar effect can be observed when drug efflux is impaired by verapamil—an inhibitor of P-glycoprotein, an enzyme involved in drug efflux (Fig. 1 I D), or as a result of depriving cells of energy (Fig. 1 I E). In both cases inhibition of drug efflux results in an increased entry of AO, formation of stacks on the cytoplasmic RNA, and a relative increase of red luminescence. The effect is dose dependent, can be quantitated, and is used to assess photodamage, as demonstrated below.
Subtle photodamage is manifested by drug influx
Fig. 1 I F demonstrates that cell damage resulting from interaction between exciting light and AO leads to the effects similar to those observed after starvation or a treatment with an inhibitor of drug efflux. In a cell exposed to exciting light, the cytoplasmic concentration of AO also increases above the level, which promotes formation of stacks. A gradual loss of a gradient between intra- and extracellular concentration of the dye may be due to impairment of drug efflux. The postulated subtle photodamage does not affect the integrity of plasma membrane; this is demonstrated by the inability of stably charged ions of propidium to enter cells (data not shown). As a result, this type of damage remains undetected by standard tests of plasma membrane integrity, as shown in Fig. 1 II, B and C. When fluorescently labeled cells are illuminated with a higher dose of exciting light, heavy damage is inflicted on plasma membranes and not only AO, but also cations like propidium can enter cell interior (Fig. 1 II, D–F). Thus, the growing relative intensity of red luminescence in cytoplasm (in areas outside endosomes), conveys information about the extent of photodamage (Fig. 1 II, graph).
It is conceivable to suspect that the red emission in cytoplasm of damaged cells that reside in medium supplemented with AO might arise from spillage of AO from damaged acidic endosomes. We have demonstrated in control experiments that when AO is removed from culture medium and AO-loaded endosomes are damaged as a result of photodynamic effect, the amount of AO released is not sufficient to cause any detectable increase of red luminescence of the cytoplasm (data not shown). Thus, the red component of AO luminescence, which occurs in cells insulted with exciting light, verapamil, or starvation, is derived from AO, which entered cells when drug efflux became impaired.
As described above, the red luminescence of AO bound to RNA carries information about the severity of cell damage. However, calibration of the method has to take into account the fact, that due to different amounts of RNA, intensity of detected fluorescence varies between cells and areas of the image, and cell preparations. It is also affected by photobleaching (23). To quantitate photodamage and minimize the influence of cell population heterogeneity, instrumental variables, photobleaching, and inhomogeneous subcellular distribution of AO on the assessment of cell damage, we defined the following parameter, similar to the parameter used in (24):
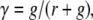 |
where g, r are the integrated intensity of green (g) and red (r) luminescence in a selected area of cytoplasm (excluding acidic endosomes). This parameter decreases when a cytoplasmic concentration of AO becomes elevated as a result of cell damage. Fig. 1 II (graph) describes the values of γ in cells exposed to increasing intensities and doses of excitation light, and shows the range of doses that cause various degrees of damage, ranging from subtle to a complete loss of plasma membrane integrity.
It is important to note that there is a range of light doses (below ∼30 kJ/cm2) within which the increased AO influx can be detected, but plasma membrane is still impermeable to propidium (Fig. 1 II, graph). It is apparent that the test based on drug efflux can detect subtle cell damage, which remains undetected by the propidium exclusion assay. Thus, the AO efflux assay is a much more sensitive method of detecting and quantitating photodamage than a standard PI exclusion assay.
We have observed that the color of cytoplasm and the parameter γ were sensitive to the ratio between the number of cells and the amount of AO added to a culture dish, rather than the absolute concentration of AO. This is expected because the conditions permitting formation of stacks occur after saturating all intercalation binding sites with AO. The concentration of AO and cell density needs to be carefully controlled to obtain reproducible readings of γ. Moreover, the comparison between the values of parameter γ in various experiments is only valid if the optical filters, detection settings, i.e., the gain in both detection channels and the confocal apertures are maintained constant.
We have reported previously that the red luminescence of AO bleaches faster than the green emission (23). This phenomenon could potentially result in underestimating the degree of sublethal cell photodamage. However, when AO is used as a reporter only, the light exposure required to record AO images is not sufficient to distort the assessment of cell damage (Supplementary Material, section 1). The measurement itself requires only one to two scans to record the image and calculate the parameter γ, whereas the difference between the relative strength of the red and green emission of AO bound to nucleic acids becomes measurable after scanning a field of view as many as ∼50 times ((23) (Figs. 4, 6, and 8 therein)).
Underestimating the damage may become detectable when AO is used as photosensitizer as well as reporter, and the intensity of light incident on AO is high. This situation is represented by a slight increase of γ when the dose is increased from 200 to 680 kJ/cm2 (Fig. 1 II). These light doses are so high, that bleaching, heavy cell damage, and loss of plasma membrane integrity occur. In summary, differential bleaching of AO emissions does not influence the assessment of sublethal cell damage but may result in a slight underestimate of a degree of heavy, lethal damage.
The increased influx of acridine orange into photodamaged cells becomes manifested by red luminescence within a few minutes of inflicting damage. We noticed that the intensity of red emission of AO in sublethally damaged cells stabilized within 10–15 min (Supplementary Material, section 2). However, in some heavily damaged cells, red luminescence was gradually increasing within the next hour, presumably due to a progressing loss of cell functions. Thus, we chose the time of 15–20 min after inflicting damage as the optimal time window to assess and compare damage.
The incident light inflicts damage on labeled neighbors of the illuminated cell
We used the method described in the previous sections to demonstrate phototoxic effects exerted on neighbors of the illuminated cell by interaction between scattered exciting light and fluorescent label residing in these cells. Cells were stained with AO and a small area of the specimen was illuminated with exciting light (for details see Fig. 1 III). Subsequently, exclusion of propidium and the parameter γ were measured in a directly illuminated cell as well as in other cells in the vicinity. When light was incident on coverglass, between cells, no damage was detected in neighboring cells (Fig. 1 III A). When light was incident on cytoplasm, in the area with no acidic endosomes, the damaged region was confined to the illuminated cell and usually exceeded the illuminated region only slightly (see also Supplementary Material, section 2). The cells in the vicinity remained intact (Figs. 1 III B and 2, only at severalfold higher light intensities was the damage detectable throughout the whole illuminated cell and in neighboring cells; data not shown). When the exciting light encountered the nucleus, the damage was inflicted on the illuminated cell and, interestingly, sublethal damage was also found to occur in labeled neighboring cells located as far as 70–90 μm away (Fig. 1 III C). When a glass bead was placed on a coverslip in a small area devoid of cells, the exciting light scattered by the bead caused damage to AO-stained cells located even 120 μm away (Fig. 1 III D; data not shown).
FIGURE 2.

Photodamage does not spread throughout the AO stained cell (see also Supplementary Material, section 2). Low intensity exciting light was incident on a selected small area (5 × 5 μm) of cytoplasm (arrow points at the region that was illuminated and acquired red luminescence). After 2 min still only a small area of the cytoplasm shows signs of damage. Red staining expanded by no more than 2 μm beyond the originally illuminated region. This may be a result of light scatter and limited range of diffusion of toxic products of reactions activated by exciting light and AO. Bar, 10 μm.
The damage described above is not a consequence of diffusion of any phototoxic products, signaling molecules, etc., originating in the illuminated cell. It is entirely a result of interaction of the light scattered on a directly illuminated cell and its subcellular structures with a fluorescent label in neighboring cells (Fig. 2; see Discussion and Supplementary Material, section 2).
The light beam incident on a cell has a shape of a cone. This diverging shape of the light beam might be suspected to contribute to the extensive size of the damaged region. When a nucleus is illuminated (at the bottom, 1 μm above the glass) the size of the illuminated square is 17 × 17 μm in a confocal plane, ∼22 × 22 μm at the glass surface, and 41 × 41 μm at a distance of 5 μm above the confocal plane. The total light dose, and hence the photodamage, is similar in 17 × 17 μm squares at various distances directly above the confocal plane (17,23), but falls sharply toward the edges of the 41 × 41 μm square located 5 μm above the confocal plane. The region where cells show the increased drug influx has a diameter of ∼140–180 μm. Thus, the “hour glass” shape of the laser beam is not the cause of the observed large size of the area where cells are damaged.
The adverse effects of interaction between the scattered light and a fluorescent label residing in cells vary between different fluorescent probes. In the experiments described above AO served as a photosensitizer as well as a reporter of cell damage. AO is highly phototoxic, therefore even low intensity light can lead to detectable photodamage. In a similar experiment we have also tested eGFP bound to core histones in the nucleus (Fig. 3) and ethidium bound to nucleic acids (Supplementary Material, section 3). eGFP excited by blue light caused no measurable cell damage (Fig. 3). This observation indicates that the phototoxic effects of eGFP excited by scattered blue light may be significantly less pronounced than the damage we observed in the case AO excited by the 458-nm light. The significantly lower phototoxicity of eGFP than AO or EB may arise from the fact that the chromophore of eGFP is shielded by the polypeptide barrel, which prevents it from direct interaction with molecular oxygen (25,26). Low molecular weight dyes, like propidium or ethidium, are more likely to interact with oxygen directly and generate singlet oxygen.
FIGURE 3.

Phototoxic effects of eGFP bound to histone H2B in cell nucleus are not detectable. Exciting blue light (488 nm, 40 μW laser beam reaching the specimen, 10 scans, area 10 × 10 μm, 512 × 512 pixels) was incident on a rectangular area within the cell nucleus (the bleached region). Following illumination, the cells were submerged in medium containing AO to detect cell damage. No increased drug influx has been detected, demonstrating that eGFP did not cause detectable impairment of drug efflux or breach of plasma membrane integrity. Bar, 20 μm.
Estimates of the proportion of light scattered by cytoplasm and nucleus
Within a certain range of light intensities, the cell damage, which causes a loss of balance between AO efflux and influx, is reflected by a growing concentration of cytoplasmic AO and a decreasing value of the parameter γ. Thus, in our experiments, the intensity of the light incident on a cell can be roughly estimated from the value of γ. We used this approach to estimate the proportion of exciting light, which is scattered by cytoplasm, cell nucleus, or a glass bead. These calculations demonstrate that an immediate neighbor of the illuminated cell may be exposed to a light of the intensity not exceeding 1% of the beam incident on cytoplasm, but as much as 10% of exciting light incident on the nucleus.
DISCUSSION
In a typical confocal microscope a focused laser beam scans the selected area of a specimen. In book diagrams the scanning beam is often drawn passing through the imaged cell as if light did not diffract or scatter. In reality, however, scattering on cells may be extensive. When the beam encounters the cell surface and subcellular structures, diffraction and scattering occur at every interface dividing the regions differing in refractive index. The scattering structures include cell surface, nucleus, mitochondria, membranes of endoplasmic reticulum, Golgi cisternae, lipid droplets, peroxisomes, etc. As a result of light scattering in the imaged region, neighboring cells may be exposed to a significant dose of light, even though they are not exposed to a scanning beam directly. We demonstrate that under typical imaging conditions the amount of scattered light is sufficiently high not only to reach neighboring cells, but also to cause adverse, phototoxic effects in fluorescently labeled cells located even 70–90 μm away from the focused beam.
Quantitating photodamage: drug efflux
We used two endpoints to detect cell damage caused by exciting laser light: integrity of plasma membrane and capacity of cellular transport systems to efficiently pump out drugs. The latter is driven by a number of proteins involved in multidrug resistance, including glycoprotein P (27). The loss of plasma membrane integrity was readily detected by an established method based on entry of propidium into cells and subsequent staining of nucleic acids. A loss of a capacity to efficiently pump out drugs from cells with intact plasma membrane was assessed by a new technique, based on detecting the formation of aggregates (stacks) of AO when it binds to RNA in cytoplasm. The aggregates are formed only when the intracellular concentration of AO rises above a certain threshold.
A loss of plasma membrane integrity is a sign of extensive, and usually lethal, damage. However, interaction between exciting light and fluorescent labels may lead to numerous other adverse effects in imaged cells (28). These effects are not immediately manifested, and thus, not readily detectable. Inhibition of drug efflux mechanisms, which leads to increased intracellular concentration of the drug, appears to be one of these less conspicuous effects occurring after insulting a fluorescently labeled cell with relatively low doses of light. Thus, an increase of drug influx into a cell with intact plasma membrane constitutes a sensitive test of cell damage. Here, we used this test to detect photodamage exerted by scattered light and estimate the range of scattering.
Photodamage to labeled neighbors is due to light scatter
The data presented in this report demonstrate that, in scanning confocal microscopy, sublethal photodamage can occur in the vicinity of the area, which was illuminated by the exciting beam. The exciting light may generate toxic products in the illuminated region when it interacts with the fluorescent label. Thus, the effects seen in the vicinity could be ascribed to the action of these toxic substances diffusing out of the illuminated area. However, we postulate that the phototoxic effects observed in cells that had not been exposed to a laser beam directly are caused only by the interaction between the scattered light and fluorescent labels, and not by the diffusing toxic products of photoactivated reactions. This conclusion is derived from the following observations. First, when light interacts with AO monomers dissolved in culture medium, excitation of the dye does occur and toxic photoproducts are generated, but scattering of excitation light is negligible and, under these conditions, no photodamage is observed in neighboring cells (Fig. 1 III A). Second, when light is incident on a small area in the cytoplasm, the damage is detected exclusively in this region, and does not spread throughout the entire illuminated cell (Fig. 1 III B, and Supplementary Material, Section 2). On the other hand, it is known that the most likely toxic photoproduct to occur is singlet oxygen (29,30) with the expected diffusion distance in a cell of only 10–20 nm (29). It is also known that even a large molecule like eGFP equilibrates across a single cell in a few seconds. Thus, any long-lived toxic photoproduct, of either a small or large molecular size, could be expected to diffuse throughout the illuminated cell within seconds. Because spreading of photodamage detected within several minutes following illumination of a small area of cytoplasm does not exceed 2 μm, we conclude that it can be a result of light scatter and photoproducts diffusing a short distance only. Summarizing the data reported here support the notion that the damage to neighboring cells, as demonstrated in this experimental system, is due entirely to the interaction between the scattered light and a fluorescent label in a neighboring cell, but not to photoproducts diffusing out of the directly illuminated area. As would be expected, scattering of light in a culture medium is negligible, but scattering on a nucleus is extensive. Thus the experimenter who studies cells by confocal microscopy can reasonably expect that, in the case of cells labeled with a fluorescent dye like acridine orange, the damage zone may extend beyond the illuminated cell to a distance as large as several cell diameters. Moreover, the shape of the damage zone may be irregular because the scatter on subcellular structures may vary as a function of direction.
Light scattered by a nucleus
Scattering of exciting light in scanning confocal microscopy is no surprise to anyone who ever watched the scanning beam move over the coverglass with a sparse cell culture. However, the extent of scatter on cell nucleus is greater than could be predicted by an intuitive guess. Apparently, a nucleus can scatter nearly as efficiently as a glass bead placed in water. If we assume that the rough endoplasmic reticulum (ER) immediately adjacent to a nucleus has RI of 1.4–1.6, whereas the intranuclear volume near the envelope may have RI of 1.31, the relative RI between nucleus and ER is as large as between water and glass (1.3 vs. 1.5). In fact, an even greater scattering can be expected at the surface of nucleoli, as their RI is clearly significantly different from the surrounding nuclear matter—after all this is the reason why we can see nucleoli in a standard optical microscope so easily. The extensive scattering of the nucleus is consistent with the observations described by others (31,32). Pawley demonstrated that an image of a flat fluorescent plane located behind a nucleus assumes a concave shape.
Light scatter: consequences for FRAP and FLIP
The observations described above may have important consequences for FRAP and FLIP studies of live cells. FRAP is based on the assumption that the bleached and unbleached populations are spatially separated at the beginning of the experiment and that no photodamage is exerted on cells during the initial bleach insult (33). The data reported here demonstrate that it may not be justified to assume that the part of the cell, which is not illuminated, remains in the dark. We demonstrate that exciting light can be scattered extensively and interact with fluorescent labels so as to evoke phototoxic effects in the wide region surrounding the directly illuminated area (Fig. 1 III C; see also Supplementary Material, section 4). It has been suggested that the absence of phototoxic effects can be proven by demonstrating that the cell under study can complete the next mitosis. However, very few reports provide this information or any alternative means of demonstrating the absence of phototoxic effects. Light intensities used in FRAP and FLIP are very high (several mW, for a fraction of a second) in comparison with the intensities used in our experiments (several μW, 1–30 s) or in live cell imaging in general (150 nW (34) in a number of short intervals over minutes and hours). One can reasonably expect that even if only a small percentage of exciting light is scattered into the unilluminated part of the cell, the intensity of the scattered light may exceed the levels that cause no adverse effects. Thus, not only will the bleaching beam cause cell damage in a directly illuminated region, but also the scattered light may cause adverse physiological effects in the adjacent labeled “unilluminated”, nonbleached area of the cell as well. Moreover, the data presented in this report indicate that serious disturbances to cell physiology can occur not only in areas not subject to illumination but even in cell neighbors, as a result of interaction between fluorescent labels and light scattering by the cell nucleus. This cell damage will remain undetected by crude “viability” assays like exclusion of propidium. It is not known how this subtle cell damage influences protein dynamics because there is no independent method to measure the dynamics of proteins in cells that were not exposed to high doses of exciting light. A somewhat soothing conclusion is derived from the fact that fluorescent proteins are generally much less phototoxic than low molecular dyes (Fig. 3).
Can light be diffraction focused inside a cell?
An interesting problem is raised regarding new microscopy methods that employ high intensity laser pulses. Based on the data presented above, one may conclude that whereas a laser beam can in principle be diffraction focused in water or clear solution, achieving the same small size of a focal spot in a living cell, especially in the nucleus, may not be possible. Fluorescence, which builds the image, is subject to diffraction and scattering as well. The difficulty in controlling the focal volume and the lightpath of the emitted fluorescence in optically inhomogeneous cell interior may result in resolution worse than theoretically predicted.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Prof. Z. Darzynkiewicz, Brander Cancer Research Institute, New York, and Dr. G. McConnell, Biophotonics Center, Glasgow, for stimulating discussions. Excellent technical assistance of B. Czuba-Pelech is gratefully acknowledged.
This work was supported by research grants from The Wellcome Trust, London, Polish Ministry of Science and Higher Education, and Foundation for Polish-German Cooperation in Warsaw.
Editor: Alberto Diaspro.
References
- 1.Cheong, W.-F., S. A. Prahl, and A. J. Welch. 1990. A review of the optical properties of biological tissues. IEEE J. Quantum Electron. 26:2166–2185. [Google Scholar]
- 2.Perelman, L. T., J. Wu, I. Itzkan, and M. S. Field. 1994. Photon migration in turbid media using path integrals. Phys. Rev. Lett. 72:1341–1344. [DOI] [PubMed] [Google Scholar]
- 3.Yodh, A. G., and B. Chance. 1995. Spectroscopy and imaging with diffusing light. Phys. Today. 48:34–40. [Google Scholar]
- 4.Oldfield, R. 1994. Light Microscopy. An Illustrated Guide. Wolfe Publishing, London.
- 5.Torok, P., Z. Laczik, and J. N. Skepper. 1996. Simple modification of a commercial scanning laser microscope to incorporate dark-field imaging. J. Microsc. 181:260–268. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro, H. 2003. Practical flow cytometry, Chapter 7. In Parameters and Probes, 4th ed. Wiley, New York. 273–410.
- 7.Marchesini, R., M. Brambilla, C. Clemente, M. Maniezzo, A. E. Sichirollo, A. Testori, D. R. Venturoli, and N. Cascinelli. 1991. In vivo spectrophotometric evaluation of neoplastic and non-neoplastic skin pigmented lesions–I. Reflectance measurements. Photochem. Photobiol. 53:77–84. [DOI] [PubMed] [Google Scholar]
- 8.Mourant, J. R., I. J. Bigio, J. Boyer, R. L. Conn, T. Johnson, and T. Shimada. 1995. Spectroscopic diagnosis of bladder cancer with elastic light scattering. Lasers Surg. Med. 17:350–357. [DOI] [PubMed] [Google Scholar]
- 9.Mourant, J., I. Bigio, J. Boyer, T. Johnson, and J. Lacey. 1996. Elastic scattering spectroscopy as a diagnostic for differentiating pathologies in the gastrointestinal tract: preliminary testing. J. Biomed. Opt. 1:192–199. [DOI] [PubMed] [Google Scholar]
- 10.Mourant, J. R., A. H. Hielscher, A. A. Eick, T. M. Johnson, and J. P. Freyer. 1998. Evidence of intrinsic differences in the light scattering properties of tumorigenic and nontumorigenic cells. Cancer. 84:366–374. [PubMed] [Google Scholar]
- 11.Boustany, N. N., R. Drezek, and N. V. Thakor. 2002. Calcium-induced alterations in mitochondrial morphology quantified in situ with optical scatter imaging. Biophys. J. 83:1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charney, E., and F. S. Brackett. 1961. The spectral dependence of scattering from a spherical alga cell and its implication for the state of organization of the light accepting pigments. Arch. Biochem. Biophys. 92:1–12. [DOI] [PubMed] [Google Scholar]
- 13.Meyer, R. A. 1979. Light scattering from biological cells: dependence of backscatter radiation on membrane thickness and refractive index. Appl. Opt. 18:585–588. [DOI] [PubMed] [Google Scholar]
- 14.Quinby-Hunt, M. S., and A. J. Hunt. 1988. Effects of structure on scattering from marine organisms: Rayleigh-Debye and Mie predictions. SPIE Ocean Optics. IX925:288–295. [Google Scholar]
- 15.Beuthan, J., O. Minet, J. Helfmann, M. Herrig, and G. Mueller. 1996. The spatial variation of the refractive index in biological cells. Phys. Med. Biol. 41:369–382. [DOI] [PubMed] [Google Scholar]
- 16.Petran, M., M. Hadravsky, J. Benes, R. Kucera, and A. Boyde. A. 1985. The tandem scanning reflected light microscope. Part 1. The principle and its design. Proceedings of the Royal Microscopical Society 20:125–129. [Google Scholar]
- 17.Pawley, J. B. 2006. Handbook of Biological Confocal Microscopy, 3rd ed. Plenum Press, New York.
- 18.Hulst. van de H.C. 1981. Light Scattering by Small Particles. Dover Publications, New York.
- 19.Bohren, C. F., and D. R. Huffman. 1988. Absorption and Scattering of Light by Small Particles. Wiley, New York.
- 20.Dobrucki, J., and Z. Darzynkiewicz. 2001. Chromatin condensation and sensitivity of DNA in situ to denaturation during cell cycle and apoptosis: a confocal microscopy study. Micron. 32:645–652. [DOI] [PubMed] [Google Scholar]
- 21.Freshney, R. I. 2000. Culture of Animal Cells. A Manual of Basic Technique, 4th ed. Wiley, New York.
- 22.Darzynkiewicz, Z., and J. Kapuscinski. 1990. Acridine orange: a versatile probe of nucleic acids and other cell constituents. In Flow Cytometry and Sorting, 2nd ed. Wiley-Liss, New York. 291–314.
- 23.Bernas, T., E. K. Asem, J. P. Robinson, P. R. Cook, and J. W. Dobrucki. 2005. Confocal fluorescence imaging of photosensitized DNA denaturation in cell nuclei. Photochem. Photobiol. 81:960–969. [DOI] [PubMed] [Google Scholar]
- 24.Darzynkiewicz, Z. 1994. Acid-induced denaturation of DNA in situ as a probe for chromatin structure. Methods Cell Biol. 41:527–541. [DOI] [PubMed] [Google Scholar]
- 25.Greenbaum, L., C. Rothmann, R. Lavie, and Z. Malik. 2000. Green fluorescent protein photobleaching: a model for protein damage by endogenous and exogenous singlet oxygen. Biol. Chem. 381:1251–1258. [DOI] [PubMed] [Google Scholar]
- 26.Bernas, T., M. Zarebski, P. R. Cook, and J. W. Dobrucki. 2004. Minimizing photobleaching during confocal microscopy of fluorescent probes bound to chromatin: role of anoxia and photon flux. J. Microsc. 215:281–296. [DOI] [PubMed] [Google Scholar]
- 27.Choi, C. H. 2005. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 4:5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight, M. M., S. R. Roberts, D. A. Lee, and D. L. Bader. 2003. Live cell imaging using confocal microscopy induces intracellular calcium transients and cell death. Am. J. Physiol. Cell Physiol. 284:C1083–C1089. [DOI] [PubMed] [Google Scholar]
- 29.Moan, J., and K. Berg. 1991. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 53:549–553. [DOI] [PubMed] [Google Scholar]
-
30.Dobrucki, J. W. 2001. Interaction of oxygen-sensitive luminescent probes
 and
and  with animal and plant cells in vitro. Mechanism of phototoxicity and conditions for non-invasive oxygen measurements. J. Photochem. Photobiol. B. 65:136–144. [DOI] [PubMed] [Google Scholar]
with animal and plant cells in vitro. Mechanism of phototoxicity and conditions for non-invasive oxygen measurements. J. Photochem. Photobiol. B. 65:136–144. [DOI] [PubMed] [Google Scholar] - 31.Mourant, J. R., M. Canpolat, C. Brocker, O. Esponda-Ramos, T. M. Johnson, A. Matanock, K. Stetter, and J. P. Freyer. 2000. Light scattering from cells: the contribution of the nucleus and the effects of proliferative status. J. Biomed. Opt. 5:131–137. [DOI] [PubMed] [Google Scholar]
- 32.Pawley, J. B. 2002. Limitations on optical sectioning in live-cell confocal microscopy. Scanning. 24:241–246. [DOI] [PubMed] [Google Scholar]
- 33.Carrero, G., D. McDonald, E. Crawford, G. de Vries, and M. J. Hendzel. 2003. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods. 29:14–28. [DOI] [PubMed] [Google Scholar]
- 34.Manders, E. M., A. E. Visser, A. Koppen, W. C. de Leeuw, R. van Liere, G. J. Brakenhoff, and R. van Driel. 2003. Four-dimensional imaging of chromatin dynamics during the assembly of the interphase nucleus. Chromosome Res. 11:537–547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.