

Abstract
Control of organelle abundance is a fundamental unsolved problem in cell biology. Mechanisms for number control have been proposed in which organelle assembly is actively increased or decreased to compensate for deviations from a set-point, but such phenomena have not been experimentally verified. In this report we examine the control of centriole copy number. We develop a simple scheme to represent organelle inheritance as a first-order Markov process and describe two figures of merit based on entropy and convergence times that can be used to evaluate performance of organelle number control systems. Using this approach we show that segregation of centrioles by the mitotic spindle can shape the specificity of the steady-state centriole number distribution but is neither necessary nor sufficient for stable restoration of centriole number following perturbations. We then present experimental evidence that living cells can restore correct centriole copy number following transient perturbation, revealing a homeostatic control system. We present evidence that correction occurs at the level of single cell divisions, does not require association of centrioles with the mitotic spindle, and involves modulation of centriole assembly as a function of centriole number during S-phase. Combining our experimental and modeling results, we identify two processes required for error correction, de novo assembly and number-limiting, and show that both processes contribute to robust and stable homeostatic control of centriole number, yielding a system capable of suppressing biological noise at the level of organelle abundance.
INTRODUCTION
A fundamental question in cell biology is how cells regulate organelle abundance, that is, how cells measure and control the quantity of a given organelle. This question represents one instance of the more general problem of biological homeostasis, which has emerged as a key problem for systems biology (1–3). For a given cell type, different organelles will tend to be present at characteristic levels of abundance, suggesting the set-point of the organelle abundance control system is under genetic control. Organelle number control raises interesting questions about mechanism. Do cells “know” how many of a given organelle they have? Can cells count? Can organelle abundance be regulated following perturbations? Does regulation take place at the level of organelle formation, segregation, degradation, or some combination of these? What role do different methods of organelle partitioning or segregation play in number control?
Previous studies of organelle abundance have focused on membrane-bound organelles such as chloroplasts, mitochondria, endosomes, golgi, and peroxisomes. Several quantitative studies measured the degree to which these organelles are equally partitioned between daughter cells during mitosis (4–6). The results of these studies generally suggest that partitioning is either entirely random, governed by the binomial distribution, or else somewhat less random, such that the variability in numbers inherited by each daughter is less than that predicted from the binomial distribution (5). In either case, the fact that partitioning is either entirely or partially random suggests a need for an active mechanism to control numbers, otherwise the number of organelles in different lineages would gradually diverge. Birky has discussed several possible mechanisms by which number variability could be reined in (7). For instance, based on studies of chloroplast number distributions, it has been proposed that cells with too many chloroplasts might block chloroplast replication for one division to bring the number down to half its original value (5). Similarly, cells with too few chloroplasts might perform multiple additional rounds of chloroplast replication to bring numbers back up to the desired set-point. These elegant mechanisms would be exceedingly interesting because they would require that cells have a way to count the number of organelles they contain. Unfortunately, to our knowledge these models have never been tested experimentally. This is for two reasons. First, in general, organelle abundance depends on size, shape, and number, which tends to obscure what we mean by “abundance”. Second, the large number of organelles found in a given cell makes counting difficult.
In contrast to chloroplasts or other membrane-bound organelles, centrioles present a uniquely tractable situation for studying number control. First, centrioles in a given cell type always have the same size and shape. Abundance can thus be described entirely by a single integer value, namely the number of centrioles per cell, such that centrioles represent a discretized version of the more general organelle abundance control problem. Second, the normal number of centrioles is exactly two per cell (8), making any deviation from this number extremely easy to detect. As a result, genetic screens have already succeeded in identifying mutants with defective centriole number control. Abnormal centriole number is a feature of tumor cells and may contribute to genomic instability (9), further highlighting the interest in understanding how number is controlled.
New centrioles form adjacent to, and at right angles with, preexisting centrioles in a remarkable process termed duplication. During S-phase of the cell cycle, each centriole duplicates to produce exactly one new centriole, which is termed the “daughter”. During mitosis, a mother-daughter centriole pair joined together by connecting fibers moves to each of the two spindle poles, so that when the cell divides, each daughter cell receives a pair of centrioles (schematized in Fig. 1 A). From the viewpoint of number control, all these complex molecular events boil down to this: centriole duplication results in a net doubling of centriole number, whereas centriole segregation during mitosis results in a net halving of centriole number; thus the combined duplication/segregation system can propagate centriole copy number from one generation to the next.
FIGURE 1.

Modeling centriole number control. (A) Centriole inheritance. A cell starts out with two centrioles in the G1 stage of the cell cycle. Each of these gives rise to a “daughter” centriole before division, and then during mitosis two centrioles associate with each pole of the mitotic spindle. This spindle-mediated segregation allows each daughter cell to inherit exactly two centrioles. (B) Markov process model for studying centriole copy-number control. At generation k, number distribution represented by vector whose element vj represent the fraction of cells in the containing j centrioles. Changes in number from one generation to the next are modeling using transition probability matrix whose elements amn specify the probability that a cell with n centrioles will produce a daughter cell with m centrioles. (C) Hypothetical model of perfect duplication and segregation. Whatever number of centrioles a cell has, is propagated exactly to both of its daughters. This system does not have a unique steady-state solution. (D) Hypothetical model including pairwise segregation of mother and daughter centrioles. After duplication, centrioles associate in pairs as shown in the cartoon. (E) Addition of de novo assembly to the pairwise segregation model. The changes to the first column reflect the ability of a cell with zero centrioles to produce a daughter having one centriole, as illustrated in cartoon. (F) Model including number-limiting, de novo assembly, and pairwise segregation. Number limiting prevents cells with three or more centrioles from undergoing duplication. (G) Model in which centriole pairs segregate randomly to daughter cells due to loss of spindle-mediated partitioning. Cartoon illustrates one possible outcome in which a cell with three centrioles segregates all three to one daughter, producing one daughter cell lacking centrioles entirely.
What if something goes wrong? Regulatory mechanisms exist to prevent multiple rounds of centriole duplication (10–12), but whereas this reduplication block helps prevent errors in centriole duplication, it cannot correct errors once they have occurred. If a cell should somehow acquire too many centrioles, or too few, and each centriole faithfully duplicates exactly once per division, then the abnormal centriole number would be continually propagated unless a compensatory error should occur to revert the number back to two. The spontaneous error rate is unknown, but number errors occur naturally in situations where centrioles form de novo, such as during early development in some species, because de novo assembly always leads to a random number of centrioles per cell, as has been documented extensively in many different cell types (13–16). We therefore anticipate that even if the spontaneous error rate is low, organisms may have evolved mechanisms to restore number following such errors,
In this report we explore the homeostatic control system by which cells actively correct errors in centriole copy number so as to regulate centriole abundance.
MATERIALS AND METHODS
Experiments
Strains and reagents
All strains were obtained from the Chlamydomonas Genetic Center, Duke University, except for the vfl2 ts100021 double-mutant strain, which we constructed using standard genetic methods. The vfl2ts line used in our experiments was the temperature sensitive vfl2 intragenic revertant allele R15 described by Taillon (17). Cells were grown in TAP media (18) under continuous light. Fla10 antibodies were a gift from Doug Cole (University of Idaho, Moscow, ID). Centrin antibodies were a gift from Jeff Salisbury (Mayo Clinic, Rochester, MN). IFT52 antibodies were a gift from Joel Rosenbaum (Yale University, New Haven, CT). Acetylated tubulin antibodies were purchased from Sigma (St. Louis, MO). All secondary antibodies were goat anti-Mouse or goat anti-Rabbit and were obtained from Jackson Immunoresearch (West Grove, PA).
Cell growth and imaging
Immunofluorescence was performed using standard procedures for Chlamydomonas (19). Temperature downshift experiments (Fig. 2, B and C) were performed by inoculating 2mL TAP cultures at low density, growing at 34°C for 4–5 days in a roller drum in an illuminated incubator (Precision model 818), and then transferring to 21°C, maintaining continuous illumination. Cultures were maintained in continuous log phase growth by daily checks of cell density followed by dilution when necessary, to make sure that cell division continued throughout the course of the experiment. Single cell pedigree analysis (Fig. 3 C) was performed by embedding cells in soft agarose pads and imaging by phase contrast or differential interference contrast microscopy, as previously described (14).
FIGURE 2.

Experimental testing of centriole copy number homeostasis. (A) The vfl2ts mutant allows reversible generation of centriole number errors. Graph shows distribution of centriole copy numbers in vfl2ts grown at 21°C (blue) and 34°C (red). Copy-number distribution in constitutive vfl2 mutant is included for comparison (gold). (Inset) Images to show recovery of centrin fibers (34°C) on left lacks recognizable fibers when cells are stained with anticentrin antibodies, but after one day of growth following downshift to permissive temperature (21°C) assembly of centrin fibers has been recovered. (B) Dynamic recovery of copy number in vfl2ts following downshift. Copy number determined by counting flagella and computing rms error relative to the nominal wild-type copy number of two per cell. Blue diamonds indicate measured data points. Red dotted line indicates rms error in cells grown continuously at permissive temperature. Graph indicates that copy number error returns to the resting level within six generations following restoration of VFL2 gene function after temperature downshift. Error bars represent 95% confidence intervals. An average of 472 cells were scored per time point. (C) Copy-number restoration confirmed by immunofluorescence. Cells were fixed and stained with rabbit anti-FLA10 kinesin and monoclonal antiacetylated tubulin antibodies, both of which recognize centrioles. Centrioles were detected as foci that stained with both antibodies. Plots indicate fraction of cells containing either too few centrioles (less than two per cell, indicated by blue diamonds) or too many centrioles (more than two per cell, indicated by red circles). Gray shaded box represents the time interval during which cells lack the centrin-based connecting fibers responsible for proper centriole segregation as judged by centrin immunofluorescence. Plot shows that centriole number begins returning to the wild-type distribution as soon as centriole segregation is restored. An average of 212 cells were scored per time-point. (D) Copy-number restoration is not due to selective cell death. We examined individual cell divisions and determined the frequency with which vfl2 cells produce inviable daughter cells. Results from a total of 401 live cell divisions are reported.
FIGURE 3.

Error correction still occurs when centrioles are dissociated from spindle. (A) Modeling restoration without segregation. A matrix describing inheritance with de novo assembly and number limiting but random segregation (Fig. 1 G) was used to simulate recovery in cells lacking centriole association with mitotic spindle. Initial distribution was set either to a uniform distribution (black line), all cells containing zero centrioles (red line) or all cells containing four centrioles (green line). Successive rounds of matrix multiplication were used to simulate changes in distribution per generation of cell division. At each generation, the distribution was compared to the measured distribution in vfl2 mutants and the difference in calculated and measured distributions was computed using the variational distance measure. (B) Restoration of steady-state vfl2 centriole distribution following perturbation. Mutant vfl2 cells have a variable number of centrioles per cell, with numbers found in a characteristic distribution (see Fig. 2 A). Individual vfl2 cells, having zero, one, or two centrioles, were seeded into wells of 96-well microtiter plate and allowed to undergo multiple rounds of division. At regular time points, cells were observed and the motility of all cells in a well scored to determine the number of centrioles present. For each well, at each generation observed, the distribution of numbers in the well was compared with the normal population-level distribution seen in vfl2 and the difference characterized by the χ-squared statistic. Results for each generation were averaged and plotted. Results were obtained from a total of 176 individual cells (116 with zero centrioles, 27 with one, and 33 with two) tracked from 0 to 4 days. (C) Error correction detected during division of living cells. Individual vfl2 cells were embedded in agarose and were observed before and after division. Graph shows average number of new centrioles (represented on the vertical axis by ΔNc, the change in the number of centrioles) made in a single division plotted versus centriole copy number (Nc) of the parent cell. Only successful cell divisions resulting in viable progeny (as judged by cell morphology and ability to continue dividing at least once more) were included in the plot. Gray line shows prediction for ideal duplication in which each centriole produces exactly one new centriole (hence a line with a slope of 1). Values falling above the line for low Nc and below the line for Nc indicate modulation of duplication in response to centriole copy number. Error bars represent standard error of the mean. Data based on observation of 254 successful cell divisions. Inset illustrates how ΔNc is calculated, by indicating all observed outcomes for division of a cell with two flagella and therefore two centrioles, and showing the calculated value for ΔNc in each case, found by summing the total number of centrioles (as judged by flagella) in both daughters and subtracting the number of centrioles in the mother.
Computer simulations
As described in the text, steady-state solutions can be calculated directly from the eigenvectors of the transition matrix, but to analyze short-term dynamics of the system we used numerical simulations. Simulations of number homeostasis were performed by representing the centriole number distribution as a vector of real numbers representing the frequency of each number class from zero to four. For each iteration of the simulation a column vector representing the distribution was left-multiplied by the transition matrix being simulated, and the resulting vector normalized so that its elements sum to 1. This then produces a new vector corresponding to the next generation. All simulations were implemented in MATLAB.
To simulate error correction during the temperature downshift experiment (see Fig. 5 B) the distribution was initialized to equal the measured distribution in vfl2 mutants, and the transition matrix was initialized to a predicted wild-type matrix with de novo assembly and assuming perfect number limiting, as follows:
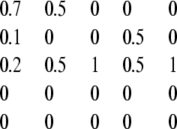 |
FIGURE 5.

Role of de novo assembly and number limiting in homeostatic control. (A) Model correctly predicts steady-state vfl2 copy-number distribution. Graph plots prediction of centriole number distribution in vfl2 mutants based on eigenvalue analysis of a transition matrix (inset) derived from experimental outcome probabilities measured in the live-cell pedigree analysis of Fig. 3 C. (Gold) Measured centriole copy number distribution in vfl2 cells. (Gray) Predicted centriole copy-number distribution obtained from the eigenvector corresponding to the predicted steady-state solution. (B) Model correctly predicts recovery kinetics in vfl2ts after downshift. (Blue solid line) Results of simulation as described in Materials and Methods, in which the centriole number distribution is initialized to the actual experimentally observed vfl2 distribution (A, gold bars) and then simulated through multiple rounds of cell division by multiplication with a transition matrix that includes templated duplication, de novo assembly, and number limiting. (Black dotted line with squares) Experimental data taken from Fig. 2 B. (Red, magenta, and green lines) Simulation results for models with reduced probabilities Pdenovo (Pd) and Plimiting (Pl) of activation of de novo assembly and number-limiting, respectively, in cells for which these processes would normally be active. (Red) Pd = Pl = 0.5, (magenta) Pd = Pl = 0.25, (green) Pd = Pl = 0. (C) Contributions of de novo assembly and number limiting to “restoration”. Graph of the restoration figure of merit FR as a function of the probabilities Pdenovo (Pd) and Plimiting (Pl). Graph is color coded with separation of 0.02 between contour lines. Darker green indicates slower predicted restoration of the steady state following a perturbation. (D) Role of de novo assembly and number limiting in noise suppression. Graph plots change of mean-squared centriole copy number error during computer simulation of two rounds of cell division following transient random perturbation of the initial distribution. Axes correspond to Pdenovo and Plimiting as in panel C. Graph is color coded according to the ratio of mean-squared error after cell division to that before cell division. Contour lines give values of the ratio and define distinct regions of parameter space. Regions with a ratio greater than one indicate noise amplification (red and dark red), whereas regions with a ratio less than one indicate noise suppression.
This matrix was constructed by taking the probability of de novo assembly producing zero, one, or two centrioles (0.7, 0.1, and 0.2, respectively) as equal to the probabilities observed experimentally for division of centriole-less vfl2 cells (14). This yields the first column, which corresponds to all possible outcomes for a cell starting with zero centrioles. The remaining columns were set equal to the matrix in Fig. 1 F to represent number limiting and spindle-mediated segregation. The trajectory of the centriole number distribution over time was found by iterative matrix multiplication operations, each corresponding to one generation of cell division. After each iteration, the root mean-square (rms) error relative to the wild-type copy number distribution of two per cell was calculated and plotted.
To explore the role of de novo assembly and number limiting in homeostasis, we define a matrix as a function of the efficacies of de novo and number limiting as follows. Assuming that this pathway is normally operating at full efficiency, we would then predict that when activated the pathway would produce de novo centrioles in the same frequency as observed in pedigree analysis of vfl2 mutants (14). This rate therefore defines the expected de novo assembly behavior if the de novo efficacy were 100%. The actual rate of de novo assembly is therefore this maximal rate multiplied by the de novo efficacy Pd. To model number limiting at variable efficacy, we assume that if a cell does not execute the number-limiting process, it would duplicate all of its centrioles at the full wild-type rate and then segregate the mother-centriole pairs to daughter cells such that a cell with eight centrioles distributes them 4:4, and a cell with six distributes them 2:4. The relative proportion of outcomes is determined by the efficacy of number-limiting Pl. With these assumptions, we can compute the transition probability matrix for any given value of Pl and Pd as follows:
 |
We then used this matrix to study the response of the system, iterating both Pd and Pl through the range 0–1 in intervals of 0.01 and calculating the figures of merit FR and FS at each set of values to generate the plot in Fig. 5 C. To augment the analysis of this model, we have also calculated the eigenvalues of this matrix in closed form as a function of the parameters Pd and Pl. Two of the eigenvalues are constants independent of Pl or Pd and are equal to 1 and 0, respectively. The other three eigenvalues each depend either on Pl, or on Pd, but never on both: λL = 1 − Pl, which only depends on the efficacy of number limiting, and 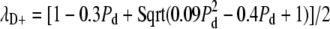 and
and 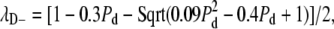 both of which only depend on the efficacy of de novo assembly. We will be concerned primarily with the magnitude of the second largest eigenvalue, and it is therefore important to note that the magnitude of λD+ is always greater than that of λD−, so that in calculating the second largest magnitude eigenvalue we need only consider λL and λD+. We note that all three eigenvalues vary monotonically as the probability (Pd or Pl) upon which they depend varies from 0 to 1.
both of which only depend on the efficacy of de novo assembly. We will be concerned primarily with the magnitude of the second largest eigenvalue, and it is therefore important to note that the magnitude of λD+ is always greater than that of λD−, so that in calculating the second largest magnitude eigenvalue we need only consider λL and λD+. We note that all three eigenvalues vary monotonically as the probability (Pd or Pl) upon which they depend varies from 0 to 1.
To simulate noise suppression or amplification as a function of de novo and number limiting (Fig. 5 D), the system was repeatedly initialized to the wild-type distribution [0 0 1 0 0]T and then subjected to perturbation by adding a zero-mean Gaussian random variable to each element of the distribution vector, applying a floor/ceiling operation to constrain all values between zero and 1, and then normalizing the vector elements by scalar multiplication to have a total summed probability of 1. A perturbation term was used that had a standard deviation of 0.1, but we obtained qualitatively comparable results with a range of other values. To perform the simulation, the noise-perturbed initial state was then multiplied by a transition matrix for the specified number of generations (two in the case of Fig. 5 D), and the whole process repeated 3000 times with different random perturbations. The mean-squared error relative to a distribution of two centrioles per cell was computed before and after each simulation run, and then the average ratio of mean-squared error after division to mean-squared error before division was determined and plotted in Fig. 5 D. This process was repeated for each value of Pd and Pl.
RESULTS
Representing organelle inheritance during number regulation
We begin by representing organelle abundance exclusively in terms of the number of individual organelles, without keeping track of their individual sizes. This representation is specifically motivated by our goal of using the model to study the abundance of centrioles, whose size is invariant.
We denote the distribution of centriole numbers in a population of dividing cells as a vector v such that element vj of the vector v represents the fraction of cells in the population that contain j centrioles. We assume that changes in centriole number over time can be modeled as a first-order Markov process and represent the action of centriole duplication and inheritance as a transition probability matrix A whose elements amn specify the probability that a cell with n centrioles will produce a daughter cell with m centrioles (Fig. 1 B). This matrix can be empirically determined by measuring centriole numbers before and after cell division (see experiments below), or it can be derived theoretically to represent any hypothetical model for centriole number control. The matrix A indicates how the distribution of centriole copy numbers v will change in a population of cells after one round of cell division. The distribution v(k) of centrioles at generation k will give rise to a new distribution of centrioles v(k + 1) at the next generation, according to the simple relation v(k + 1) = Av(k).
The steady-state number distributions are given by the eigenvectors of the matrix A for which the corresponding eigenvalue is 1. For stochastic matrices the theorems of Perron and Frobenius (20) guarantee all eigenvalues have magnitude of 1 or less, and that at least one eigenvalue is equal to 1. In principle, however, the matrix A could have multiple eigenvalues of 1, corresponding to multiple steady-state solutions. In this case, any linear combination of steady-state solutions will also be a steady-state solution, hence the system will have a continuum of steady states and will not be able to restore a unique state following a perturbation.
We note for completeness that because the elements of vectors in our model represent probability distributions, their elements v0–v4 should sum to 1, hence they must lie on a hyperplane v0 + v1 + v2 + v3 + v4 = 1. However, when computing the eigenvectors of these matrices, the values of the elements of the vector need not sum to 1, and therefore they do not directly represent a probability distribution. However, because a scalar multiple of an eigenvector is also an eigenvector, it is always possible to normalize the eigenvectors so that their elements sum to 1. All eigenvectors reported in this article will be in this normalized form.
Experiments have demonstrated that centrioles, once formed, are stable and do not turn over (21). Hence, an implicit assumption throughout all our modeling work is that the only process by which centriole number can decrease is the partitioning of centrioles into daughter cells during cell division. For some organelles, number control may involve active degradation of extraneous copies when the number becomes too high, by processes such as autophagy (22–24). Such changes can easily be incorporated into the model if necessary.
We note that an alternative scheme has recently been employed to model centriole inheritance in the specific case of Drosophila spermatogenesis (25) following a modeling procedure used to model plasmid copy number control (26). Because plasmids cannot form de novo, the specific model of Seneta and Tavaré is not directly applicable to centriole inheritance, requiring the authors of the previous centriole inheritance study to assume, incorrectly, that de novo assembly never occurs. Our model can easily incorporate de novo formation of random numbers of centrioles, such as have been clearly shown to occur in many cell types including mammalian cells (14–16), by appropriately populating the first column of A.
Centriole segregation and duplication are not sufficient for number control
Using the conceptual framework outlined above, we consider how centriole number may be controlled. When we say that organelle number is controlled, we mean that the cell is able to maintain the number of organelles within some narrow distribution, without drifting over time, and such that the mean can be restored following a perturbation. The concept of number homeostasis thus involves two separate aspects, which we term “specificity” and “restoration”. By “specificity” we mean the narrowness of the distribution around a single unique number, and by “restoration” we mean how rapidly, if at all, the system is able to return to the correct organelle number following a perturbation. We want to ask, within the context of the simple model described above, what features of organelle inheritance, as expressed in the form of the inheritance transition probability matrix A, can contribute to specificity and restoration.
This task requires us to define figures of merit for specificity and stability. Specificity refers to the degree to which one, or possibly more than one, copy numbers are preferentially emphasized in the steady-state probability distribution. We therefore seek as a figure of merit for specificity some measure of how far away the steady-state distribution is from a uniform distribution in which all numbers are equally likely. Using the Entropy Normalized Kullback-Leibler Divergence (26) to measure distance from a uniform distribution, we obtain a figure of merit for specificity
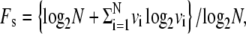 |
(1) |
where N is the dimension of matrix A and vi is the probability of having i centrioles in the steady-state distribution. The second term in the numerator will be recognized as being proportional to the Shannon Entropy. The specificity can thus be interpreted as a measure of how much information is needed to explicitly distinguish the actual number distribution from a uniformly distributed random distribution. The expression is normalized so that a system in which all centriole numbers are equally likely (i.e., completely lacking specificity) would have Fs = 0, whereas a system that forces all cells to have the same number of centrioles (i.e., perfect specificity) would have Fs = 1. In the case of multiple eigenvalues of 1, we take an average of the corresponding steady state distributions and use this averaged distribution to compute Fs. We find (data not shown) that qualitatively similar results are obtained using an alternative figure of merit based on the Bhattacharyya distance (27).
To develop a figure of merit for “restoration”, we start by identifying the second eigenvalue (taken in decreasing order of magnitude) of the transition probability matrix A, denoted λ2, which is a standard indicator of the convergence time of a Markov process (see, for example, (28)). The smaller the magnitude of λ2, the more rapid the exponential decay of the distribution component defined by its corresponding eigenvector, hence the more rapidly the state of the system will converge to the steady-state distribution. Eigenvalues smaller than λ2 will lead to even faster decay, hence the convergence rate is limited by λ2. From this, we define a figure of merit for restoration as
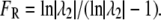 |
(2) |
The fact that A is a stochastic matrix guarantees that |λ2| is in the range (0,1). From the way we define FR we can see that if the matrix has more than one eigenvalue of 1, in which case the system cannot return to a unique steady-state value as discussed above, we obtain FR = 0, indicating a zero rate of restoration. At the other extreme, as λ2 approaches 0, FR approaches 1, which is reasonable since λ2 = 0 implies there is a single eigenvalue of 1 and all others are zero, in which case the steady state will be achieved in a single division, thus representing “perfect” restoration. We note that both FR and FS are generic in the sense that they describe system performance independent of the actual set point. The fact that the normal number of centrioles per cell happens to be 2 is not used in their evaluation, hence they can be applied in principle to evaluate performance of any organelle number control system.
With these figures of merit in hand we can evaluate the performance of hypothetical organelle number control systems. First we consider a hypothetical situation (Fig. 1 C) in which the only mechanisms at work are perfect duplication (such that each centriole duplicates once per cell cycle) and perfect segregation (such that centrioles are passed on to daughter cells in a maximally equal way). This would presumably require the mitotic spindle to equalize the numbers associated with each pole, a mechanism that may function for other organelles besides centrioles (29). In the following discussion we restrict our model to a range of 0–4 centrioles per cell but the approach can be easily extended to any maximum number. With these considerations in mind, the corresponding transition matrix would just be the identity matrix I, since a cell with any particular number of centrioles would exactly double the number, and then distribute exactly half to the two daughter cells, resulting in no net change in number. Such a system obviously cannot reach a unique steady-state number. Applying the definitions above we obtain figures of merit FS = 0 and FR = 0, showing the system completely lacks specificity or restoration.
In fact, centriole segregation cannot be entirely equal between daughter cells. Newly formed mother-daughter centriole pairs remain associated with each other, and are segregated in pairs to the daughter cells. This pairwise cosegregation leads to the modified inheritance matrix shown in Fig. 1 D. The first column indicates that cells lacking centrioles cannot make new ones. The second column, which describes the outcomes for a cell with one centriole, indicates that this one centriole will duplicate to form a single mother-daughter pair, which will then be inherited by one daughter cell, the other daughter inheriting none. The third column describes the outcome for a cell with two centrioles, and indicates that each will duplicate to produce a mother-daughter pair, and then each daughter cell will inherit a mother-daughter centriole pair, such that each daughter cell will contain two centrioles. The fourth column describes the outcome for a cell with three centrioles, and indicates that after all three duplicate, one daughter cell will inherit two mother-daughter centriole pairs, to have a copy number of four, whereas the other will inherit just one pair and have a copy number of 2. The final column indicates that in a cell with four centrioles, all four will duplicate, and the four mother-daughter pairs then segregate 2:2 to the two daughter cells, giving each daughter cell four centrioles. The values of 1 on the diagonal indicate that a cells with 0, 2, or 4 centrioles will produce only daughters with 0, 2, or 4 centrioles. The eigenvalues of this matrix are, in order of magnitude, (1,1,1,0,0) and the eigenvectors corresponding to the three eigenvalues of 1, which denote steady-state solutions, are [1 0 0 0 0]T, [0 0 1 0 0]T, and [0 0 0 0 1]T, that is to say, distributions consisting entirely of zero centrioles, two centrioles, or four centrioles. Any linear combination of these distributions will also be a steady state. This simple inheritance system is therefore unable to attain a unique steady state. In terms of our previously defined figures of merit, the pairwise cosegregation of mother-daughter centriole pairs yields FS = 0.32 and FR = 0. Mother-daughter association increases specificity but does not improve restoration.
This idealized model neglects the well-established fact that centrioles can form de novo (13–16,30). If we now introduce de novo assembly, for instance, by supposing that a cell with no centrioles will produce a single new centriole per generation and then pass it to one of its two daughters, we obtain the revised matrix shown in Fig. 1 E, which now has eigenvalues (1, 1, 0.81, −0.31, 0). De novo assembly eliminates the eigenvalue of 1 that corresponded to a distribution of zero centrioles per cell. However the system still has multiple steady states corresponding to all linear combinations of [0 0 1 0 0]T and [0 0 0 0 1]T. Under these assumptions, FS = 0.6 and FR = 0, showing an increase in specificity but a continued lack of restoration.
Prior studies of chloroplast number control have lead to a proposal that when the organelle number exceeds a threshold, production of new organelles is downregulated (5). We will refer to a block of organelle duplication when number exceeds a fixed threshold as “number limiting”. If we add number limiting to the previous matrix, so that cells with more than two centrioles do not make any new centrioles but simply distribute their preexisting centrioles to their daughters, we get the further revised matrix shown in Fig. 1 F, which has eigenvalues (1, 0.81, 0.31, 0, 0). The single eigenvalue of 1 corresponds to an eigenvector representing two centrioles per cell, just as is seen in actual cells. For this system, FS = 1 and FR = 0.17. Therefore, addition of de novo assembly and number limiting convert the system into a globally stable system in which any initial distribution will eventually evolve into a distribution in which all cells have the correct copy number. Note that even in this highly idealized case, FR < 1, indicating that multiple generations will be required for restoration.
Nonrandom segregation is dispensable for number control
The foregoing argument suggests that pairwise segregation mediated by the mitotic spindle is not sufficient for number control. We next ask if segregation is even necessary. By assuming de novo and number limiting as above, but random segregation of mother-daughter pairs, we obtain the matrix shown in Fig. 1 G. This matrix has eigenvalues (1, 0.5, −0.05 ± 0.23i, −0.21) with a predicted steady-state distribution of [0.377, 0.22, 0.3, 0.023, 0.08]T. The figures of merit for this system are FS = 0.16 and FR = 0.60. Compared to the hypothetical situation described above of perfect mother-daughter pairwise segregation in the absence of de novo or number limiting, this random-segregation system has a worse selectivity figure, but actually shows a somewhat better restoration figure. This suggests that segregation is not necessary for restoration of number following perturbation, but may be necessary for precise specificity of the steady-state number distribution. The steady-state distribution calculated from the first eigenvector of this matrix predicts that in a mutant in which centrioles lose their association with the mitotic spindle, centrioles with zero, one, or two centrioles should be observed with frequencies roughly 40%, 20%, 30%, with three or four centrioles being much less frequently observed. This type of number distribution is in fact observed in mutants that lose centriole-spindle associations (14).
To summarize these simple models, we find that number control requires de novo assembly and number limiting that actively correct errors. Such process may seldom be observed in normal cells since the process of centriole duplication and segregation normally operates with very high fidelity. However, we predict that if centriole number could be experimentally perturbed in living cells, they would be able to restore the proper number distribution after several generations. The model also predicts that although mitotic spindle association-mediated segregation is important for specificity of the number distribution, it is not necessary for restoration following perturbation, so if perturbations could be applied to mutant cells with centrioles dissociated from the mitotic spindle, restoration of some steady-state distribution should still be observed even if the final distribution shows less specificity than in wild-type cells. We next test these predictions experimentally.
Centriole number can be corrected following perturbation
To test for homeostatic restoration of centriole number, it is necessary to generate cells with copy-number errors, and ask whether the normal number distribution can be restored. For this purpose, we employed mutants of the unicellular green algae Chlamydomonas reinhardtii, which is a well-established genetic system for the study of centrioles (31). We generate cells with copy number errors by using a conditional allele of the VFL2 gene, which encodes the conserved EF-hand protein centrin (17,32,33). Centrin forms fibers that link centrioles to the spindle poles in green algae during mitosis (34), and constitutive vfl2 mutants show errors in centriole segregation, presumably due to this loss of spindle association, such that cells in a population of vfl2 mutants show a variable number of centrioles, between zero and six per cell (35). Conditional vfl2ts mutants (17), when grown at the permissive temperature (21°C), have normal centrin fibers joining the centrioles to the spindle poles, and a normal centriole copy number (two per cell). The normal copy number demonstrates that centriole duplication functions properly in these cells when they are growing at the permissive temperature. However, when grown at the nonpermissive temperature (34°C), the centrin fibers linking centrioles to the spindle are lost, and the cells display a random number of centrioles per cell comparable to a constitutive allele of vfl2 (Fig. 2 A). We therefore asked whether this randomization of copy number seen at 34°C can be corrected once the wild-type VFL2 gene function is restored by shifting the cells back to the permissive temperature.
We grew vfl2ts cells at the nonpermissive temperature until they developed the variable centriole number phenotype seen in Fig. 2 A. We then shifted the cells back to the permissive temperature and asked whether the correct copy number (two per cell) could be restored. As shown by centrin immunofluorescence in Fig. 2 A (inset), we first verified that within one generation after the shift back to permissive temperature, centrin function was restored as judged by restored assembly of centrin into fibers connecting the centrioles with the nucleus. Having thus verified that gene function has been restored, we asked whether the perturbation in number can be corrected. To track the distribution of centriole copy number over multiple generations, we took advantage of the fact that in vfl2 mutants during G1, all centrioles are active as basal bodies to produce flagella, hence one can measure the number of centrioles in a vfl2 cell simply by counting flagella (14). Using this method, we found that within several generations following downshift the centriole number error was corrected (Fig. 2 B). Similar kinetics of recovery were measured, without relying on flagella as a marker, by using immunofluorescence imaging of centrioles in cells fixed at time points following the downshift (Fig. 2 C). We conclude from these data that a population of cells containing variable centriole numbers per cell is able to restore the correct copy number.
This result raises the possibility that Chlamydomonas cells may contain an error-correction mechanism for centriole copy number. It is formally possible that the results of Fig. 2, B and C, which were obtained on populations of cells, could be explained by selective death of cells with incorrect copy number, rather than by active modulation of centriole assembly during cell division. It is unclear a priori whether centriole number would have much impact on viability. The complete removal of centrioles from cells appears to have no effect on their ability to divide or progress through the cell cycle (36), and the presence of multiple centrioles does not generally produce multipolar spindles due to a clustering mechanism (37). In Chlamydomonas, mutants in which centrioles are either highly reduced (38) or missing altogether (39) are fully viable. In fact, we did not find any marked change in the number of viable progeny as a function of initial centriole number (Fig. 2 D), suggesting that selective cell death cannot account for the observed kinetics of recovery. We will revisit this question below when we present experimental evidence that modulation of centriole assembly as a function of preexisting centriole number occurs during individual cell divisions. First, however, we will consider how the known aspects of centriole segregation and duplication may contribute to number restoration.
Centriole number control still occurs in a mutant with random segregation
The theoretical analysis presented in the first part of this report suggested that mitotic spindle-based segregation would not be necessary for restoration. To further refine this prediction, we estimate the time course of restoration expected in such a mutant by iterative matrix multiplication with a transition matrix described above (Fig. 1 G) that represents random segregation but normal de novo and number limiting. Results of this simulation, plotted in Fig. 3 A, lead to a quantitative prediction that not only should number restoration occur in mutants lacking spindle-associated segregation, it should take place on the timescale of roughly four generations.
To test these predictions we employ a constitutive vfl2 mutant, in which centriole segregation is random due to lack of attachment of centrioles to the mitotic spindle poles (40). The ability of vfl2 cells to restore their steady-state distribution has previously been suggested by experiment of Jarvik and co-workers (35), who showed that when a vfl2 culture is streaked out to single colonies on plates, and then the number distribution within each colony is measured, all colonies attain the same steady-state number distribution despite the fact that they arose from single cells that presumably had different numbers of centrioles. However those prior results did not establish the kinetics with which the number was restored, nor did they directly measure the number of centrioles in the founding cells for each colony.
We therefore measured the ability of vfl2 cells to restore their steady-state centriole number distribution, by distributing a liquid culture of cells at high dilution to wells of 96-well microtiter plates. We observed each well to locate wells that contained single cells, and then analyzed the swimming motion of the cells to distinguish those that were nonmotile (and therefore lacked flagella), those that spun in place (and therefore had a single flagellum), and those that swam normally (and therefore had two flagella). Since, as discussed above, vfl2 mutant centrioles are all competent to make flagella, we can infer that the cells with zero, one, or two flagella contained zero, one, or two centrioles, respectively. We could not, with this analysis, distinguish cells with more than two flagella from those that had two, however such cells are exceedingly rare in the population; although it is formally possible that we misclassified a cell with, for example, three flagella, as having only two, this would have only a slight statistical effect on the measured results.
Once individual cells in the microwells were classified based on swimming type, we then allowed them to grow for 4 days, periodically observing each well and counting the cells in each swimming class. We then calculated, as a function of the number of generations elapsed, the deviation in the individual distributions, measured as a χ-squared statistic using the vfl2 steady-state distribution for comparison. The results, plotted in Fig. 3 B, show that the steady-state vfl2 number distribution is restored over a period of roughly five to eight generations, a rate comparable to the recovery kinetics seen in the experiments of Fig. 2, B and C, when segregation was normal. This experimentally measured recovery is, strictly speaking, of a similar order of magnitude as the predicted rate for a segregation defective mutant (Fig. 3 A), but it does take one to three generations longer than the prediction. This discrepancy is likely a result of the fact that the hypothetical random segregation model used to generate Fig. 3 A assumed for expository convenience that 100% of centriole-less cells form a new centriole by de novo formation, and that duplication is completely blocked when the number of centrioles exceeds two, whereas as we shall see below, real de novo assembly is less efficient. Thus the comparison between Fig. 3 A and Fig. 3 B should be limited to general features and not numerical specifics.
At any rate, the recovery of number distribution in mutants with centrioles dissociated from the mitotic spindle confirms the theoretical prediction that segregation of centrioles by the mitotic spindle poles is not required for homeostatic restoration of a steady-state number distribution even though, as with the theoretical prediction, the actual distribution seen at steady state lacks the specificity of wild-type cells.
Experimental test of number-limiting mechanism
Having found that centriole number is under homeostatic control even in the absence of spindle-mediated segregation, we next asked whether this homeostatic process demonstrates de novo assembly and number limiting as predicted by the model. To this end, we monitored division of individual vfl2 mutant cells embedded in agarose pads and measured the number of centrioles present before and after division as previously described (14,35). By comparing the number of centrioles present in the two daughter cells following cell division to the number of centrioles present in the parent cell, we calculated the total number of new centrioles made per division (Fig. 3 C). We found that cells with one or two centrioles make one or two new centrioles on average. Cells lacking centrioles make roughly 0.5 new centrioles per cell by de novo synthesis, as previously reported (14–16), satisfying the requirement for de novo assembly predicted above in our simple model. Strikingly, cells with three or four centrioles make very few new centrioles, implying that cells have a centriole copy-number proofreading mechanism that can detect the presence of supernumerary centrioles and shut off duplication. This confirms the prediction of the simple model presented above, in which a “number-limiting” mechanism was predicted as a way to eliminate an undesired steady-state solution at n = 4. The key features of number control, namely de novo production and number limiting, are therefore confirmed by this single-cell imaging data. Because these data show that error correction occurs at the level of single cell division as judged by live-cell imaging of cells that divided successfully to produce two viable progeny, we argue that the error correction seen in Figs. 2 B and 3 B is not trivially explained by selective death of cells with incorrect copy number (see also Fig. 2 D), but actually involves an active modulation of centriole assembly as a function of centriole number. These data thus reveal a control system that can restore centriole number in individual cells following transient perturbations.
Centriole number correction acts by modulating duplication during S-phase
We next asked when this control system acts relative to the timing of normal centriole duplication. Centrioles duplicate during S-phase in Chlamydomonas, just as in other species (10,14). Thus we can use double mutants of the vfl2 mutation and the temperature-sensitive S-phase arrest cell cycle block mutant ts100021 (41) to generate a population of S-phase arrested cells with varying centriole copy number. We then wish to count the number of newly formed centrioles versus the number of preexisting centrioles by staining cells with antibodies that can distinguish the preexisting centrioles from the new ones that have formed during S-phase to calculate the duplication efficiency as a function of preexisting number. As a marker for preexisting centrioles, we employ the intraflagellar transport protein IFT52, which localizes to the transitional fibers of mature centrioles (42). These transition fibers are not present on centrioles when they form during S-phase, but instead these fibers only assemble when centrioles mature during mitosis (43). The same is true of the corresponding fibers (the distal appendages) in mammalian centrioles (44), and indeed proteins localized to these distal appendages in centrioles are well known to act as specific markers for mother centrioles (45,46). Electron microscopy and immunofluorescence studies showed that these distal structures remain stably attached to the mother centriole once they form, and are still attached at the time of centriole duplication in the following S-phase (43,44).
Since IFT52 localizes to a structure that is only assembled on new centrioles after entry into mitosis, and which is missing when centrioles first form during S phase, we reasoned that IFT52 would be a marker for mature versus immature centrioles that would allow us to distinguish the centrioles initially present upon entry into S-phase (which had therefore formed in the previous cell cycle) from those that might accumulate during successive rounds of centriole duplication during prolonged S-phase arrest. We verified this prediction experimentally, by growing the temperature sensitive S-phase arrest mutant (41) ts100021 at 34°C for 24 h. This mutant has the normal wild-type centriole copy number of two per cell, but during S-phase arrest, it has been shown that multiple centrioles accumulate in these cells (14). As illustrated in Fig. 4 A, staining with the FLA10 antibody, which recognizes both newly formed and preexisting centrioles, shows that almost half of the cells contained at least five centrioles (in some cases, as many as 12 centrioles) by 24 h of arrest. In contrast, when the same cells were stained with IFT52 antibodies, they all showed exactly two centrioles per cell. We infer that these were the two parental centrioles that the cell first contained when it entered S-phase. This result confirms the prediction, based on the localization of IFT52 to transition fibers that only form during mitosis, that IFT52 immunofluorescence localization is specific for preexisting centrioles during S-phase arrest, and does not localize to the newly formed daughters.
FIGURE 4.

Centriole number correction occurs during S-phase. (A) IFT52 antibody only recognizes preexisting centrioles during S-phase. ts100021 mutants were grown at 34°C for 24 h, during which time most cells accumulated at least three, and in some cases many more, new centrioles as detected by FLA10 immunofluorescence, as indicated by the colored regions of the first bar in the graph. In contrast, all cells only showed two foci of transition-fiber specific IFT52 staining, confirming that this antibody only recognizes the two centrioles that a cell had when it entered S-phase, and not the newly formed ones, as predicted from the fact that transition fibers, the locus of IFT52 recruitment, do not assemble onto new centrioles until mitosis. (B) Centriole number is not corrected before S-phase. (Blue) Centriole copy-number distribution in vfl2 cells during G1. (Red) Copy-number distribution of preexisting centrioles during S-phase arrest in vfl2 ts100021 as judged by localization of IFT52p. Inset shows typical images in which additional centrioles detectable by centrin immunofluorescence accumulate during S-phase arrest but do not stain with antibodies to IFT52. Graph indicates that the distribution of number at onset of S (as judged by preexisting centrioles) is the same as that seen during G1, indicating that error correction did not occur before the G1-S transition. (C) Duplication efficiency during S-phase is modulated by centriole copy number. Plot shows duplication efficiency, as determined by the number of new centrioles made during a 24-h S-phase arrest, per preexisting centriole, normalized to 1 for cells with two centrioles, plotted on log scale versus initial centriole number. Plot indicates increased duplication for cells with too few centrioles relative to correct copy number, and decreased duplication for cells with too many centrioles. Error bars are standard deviation.
We next used this system to ask about the timing of number control. One possible model one might imagine would be for cells to adjust number before centriole duplication to avoid duplicating errors. To test this model, we asked whether number correction occurs before S-phase, the normal time of centriole duplication. Using the IFT52 antibody specific for mature, preexisting centrioles, we counted the number of preexisting centrioles present in vfl2 ts100021 cells that had undergone S-phase arrest for 24 h (Fig. 4 B). The vfl2 mutation was used to introduce number errors to allow for testing of restoration. Although new centrioles still formed as judged by the more general FLA10 staining, the number of preexisting centrioles in vfl2 ts100021 S-phase arrested cells, as judged by IFT52 staining, matched the number in vfl2 cells during G1, showing that error correction had not taken place before S-phase entry.
A second possibility is that number control might act at the level of centriole assembly, with new assembly modulated as a function of preexisting centriole number. To address this possibility, we compared the total number of centrioles following arrest (by using an antibody that recognizes all centrioles including both preexisting and newly formed centrioles during S-phase arrest) to the number of preexisting centrioles (as judged by IFT52 antibody staining) to determine the total number of new versus old centrioles. From this, we calculated the efficiency with which preexisting centrioles give rise to daughters during S-phase arrest. The results of this analysis, plotted in Fig. 4 C, show that centriole duplication efficiency appears to be altered as a function of copy number, such that duplication efficiency is reduced as the number of preexisting centrioles increases. These results roughly mirror the results seen during live cell divisions as plotted in Fig. 3 C, suggesting that modulation of centriole duplication during S-phase may be a primary mechanism for achieving centriole copy number control.
Direct comparison of model predictions with experimental results
We next wanted to use our simple model to explore variations on the number control system. First, however, we tested whether the simple first-order Markov model as we have described is sufficient to account for observed behaviors. In the best-studied instance of biological number control, namely plasmid copy-number regulation in bacteria, it has been found that a more complex branching process model is needed to represent observed behaviors (26). The same branching process model used for plasmids has recently been applied to centrioles (25) although, as discussed above, that study neglected de novo assembly. Nevertheless the fact that the only published mathematical model of centriole segregation used a much more complex framework than our simple Markov model, raised the serious possibility that our model may somehow not be adequate to account for actual behavior. Indeed, our model makes many simplifications, including the assumption that the duplication and segregation of centrioles at any given generation depends only on the number of centrioles initially present at that generation, and not on prior numbers, thus restricting us to a first-order Markov process. From a practical standpoint, the tremendous simplifications in computation and intuition that result from the use of such a model clearly justify its application, but only if the simplification is not bought at the expense of failure to model real behavior.
First we asked whether our simple model could predict the centriole number distribution in the vfl2 mutant. As described above, a purely theoretical model of a segregation-defective mutant gave a good match to the centriole number distribution seen in vfl2 mutants. But as a further test, we wanted to check whether the model, when given experimentally measured data as an input, would produce a consistent prediction. We took the single-cell pedigree data from vfl2 cells embedded in agarose pads obtained in the experiment of Fig. 3 C, arranged the experimentally measured outcome probabilities into a matrix (Fig. 5 A, inset), and computed the steady-state solution. The eigenvalues of this matrix were 1, 0.48, 0.08, and 0.009 ± 0.027i, with a predicted steady-state distribution plotted in Fig. 5 A. This matrix gave figures of merit FS = 0.35 and FR = 0.42. As indicated by the experimental data plotted alongside the predicted distribution in Fig. 5 A, the prediction correctly matches the steady-state copy number distribution that is actually seen in vfl2 mutants.
We next tested whether the model can predict the kinetics of number restoration seen in our temperature downshift experiments, by using numerical simulations (see Materials and Methods), and again found a remarkable agreement with the experimental data (Fig. 5 B). For comparison, we also simulated recovery using transition matrices with reduced efficacy of de novo assembly and number limiting. We define the quantities Pdenovo (abbreviated Pd) and Plimiting (abbreviated Pl), to describe the probability of activating the de novo assembly and number-limiting processes, as follows. We interpret Pd as the probability that a cell that has the opportunity to form a centriole de novo (because it lacks centrioles) actually activates the de novo assembly pathway. Similarly, we interpret Pl as the probability that a cell potentially eligible for number limiting, because it has too many centrioles, actually limits duplication in response to the number cue.
As illustrated by the red, magenta, and green lines in Fig. 5 B, we found that decreasing efficacy of these two control processes led to slower rates of recovery, with a complete loss of recovery when Pd = Pl =0. The model assuming full efficacy of de novo and number limiting (Pd = Pl =1) gave a numerically close fit to the experimental data as judged by a mean-squared residual fitting error of 0.012. The alternative models with de novo and number-limiting efficacies of 0.5, 0.25, and 0, gave substantially higher mean-squared residual fitting errors of 0.15, 0.36, and 0.76, respectively.
We note that the inheritance matrix used for the simulation of recovery in Fig. 5 B did not correspond to the matrix measured for mutant cells, but rather to an idealized wild-type matrix as discussed in Materials and Methods. This was because recovery took place after the conditional mutation was rescued by shift to growth at the permissive temperature, where our own studies have shown centrin assembly is visibly restored. Therefore, number control is not simply a byproduct of the reduced centrin levels caused by the vfl2 mutation but occurs in cells that are effectively wild-type with regard to their centrin content.
These results suggest the first-order Markov model presented here adequately encapsulates the phenomenological behavior of the homeostatic control system for centriole abundance.
Stability and robustness of centriole copy-number homoeostasis
Next we used this model to analyze the importance of de novo assembly and number limiting for centriole homeostasis. To ascertain the relative contributions of de novo assembly and number limiting, we computationally generated a series of hypothetical transition matrices characterizing a range of efficiency of these two processes as a function of Pd and Pl (see Materials and Methods for details of the simulation). For this analysis it was assumed that spindle-mediated segregation was fully active. We then calculated the performance of the system in terms of the restoration figure of merit FR. Note that only FR was plotted because FS does not change unless Pd or Pl becomes zero. This is because unless Pd or Pl is zero, the matrix always has just one eigenvalue equal to 1 and this always corresponds to a steady-state distribution of two centrioles per cell giving FS = 1. Unless Pd or Pl becomes zero, this will be the only eigenvalue of 1, hence for any nonzero values of Pd and Pl, the steady-state solution will always be [0 0 1 0 0]T, i.e., two centrioles per cell.
The outcome of this analysis is that FR varies from 0 to 0.21, taking its maximum value when Pd = Pl = 1 (Fig. 5 C). We note that FR decreases smoothly as Pd and Pl are reduced, and does not show a catastrophic breakdown in restoration until either de novo or limiting is completely eliminated. If both parameters are zero, the matrix becomes equivalent to the nonrestoring matrix of Fig. 1 D.
The overall features of this graph can be readily interpreted in terms of the eigenvalues of the transition matrix. As discussed in Materials and Methods, the second eigenvalue of the transition matrix, which determines FR, will always be either λL = 1 − Pl, which only depends on the efficacy of number limiting, or 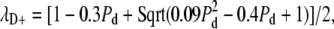 which only depends on the efficacy of de novo assembly. Whichever is larger in magnitude will determine the second eigenvalue and hence determine FR according to Eq. 2. The fact that FR depends on only one of the two eigenvalues for any particular value of Pd and Pl explains the straight edges seen in the contour lines for the following reason. For any value of Pd, there is a corresponding value for Pl given by the expression
which only depends on the efficacy of de novo assembly. Whichever is larger in magnitude will determine the second eigenvalue and hence determine FR according to Eq. 2. The fact that FR depends on only one of the two eigenvalues for any particular value of Pd and Pl explains the straight edges seen in the contour lines for the following reason. For any value of Pd, there is a corresponding value for Pl given by the expression 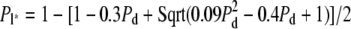 at which λL and λD+ are equal. Increasing Pl beyond this point reduces λL below λD+, so that the latter now determines FR. Further increasing Pl beyond this equivalence point has no effect on FR since λD+ is independent of Pl, giving rise to a straight horizontal line in the contour plot. Similarly, increasing Pd beyond the point at which the two eigenvalues are equal decreases λD+ so that now FR is determined only by λL, hence the contour follows a vertical line. This accounts for the rectangular shape of the contour lines.
at which λL and λD+ are equal. Increasing Pl beyond this point reduces λL below λD+, so that the latter now determines FR. Further increasing Pl beyond this equivalence point has no effect on FR since λD+ is independent of Pl, giving rise to a straight horizontal line in the contour plot. Similarly, increasing Pd beyond the point at which the two eigenvalues are equal decreases λD+ so that now FR is determined only by λL, hence the contour follows a vertical line. This accounts for the rectangular shape of the contour lines.
These considerations also explain why FR attains a maximum of ∼0.21. Both λL and λD+ attain their minimal values when Pd and Pl equal 1. For these values of the parameters, λL becomes zero while λD+ drops only to 0.765. This value is thus the smallest value possible for the second eigenvalue, hence it determines the upper bound on FR. Substituting 0.765 into Eq. 2 gives 0.211 as the value for this upper bound. Since the upper bound on FR is set by a function of Pd rather than Pl, it means that de novo assembly is the limiting factor constraining the rate of restoration. If cells could form centrioles de novo more efficiently, they could restore the number distribution more rapidly. We also note that in contrast to recovery by de novo assembly, which is limited to a finite rate of correction, λL can become zero when Pl becomes 1, indicating that number limiting can eliminate supernumerary centrioles within a single generation. This presumably accounts for the fact that mutants lacking spindle-mediated segregation (which can be interpreted as causing an intrinsically high error rate in number) are much more likely to have too few centrioles versus too many (see for example Fig. 5 A).
A similar argument accounts for the fact that FR depends more strongly on Pl than on Pd. Differentiating the expressions for λL and λD+ given above, we find that |dλL/dPl| = 1, whereas |dλD+/dPd| ≤ 0.25, and since FR is always determined by one or the other of these eigenvalues, we conclude that the variation in FR as a function of Pl to the left of the curve λL = λD+ should be steeper than the variation in FR as a function of Pd to the right of the curve.
Small variations in centriole number distributions can be interpreted as a type of noise, and restoration as the suppression of this noise. To explore this effect numerically, we simulated a population of cells with a resting distribution of two centrioles per cell, subjected the distribution to a random perturbation (noise), and then compared the mean-squared error in centriole number, relative to the nominal value of two per cell, before and after two generations of cell division as simulated by repeated multiplication by the transition matrix. For each set of parameters, Pd and Pl, we asked whether the noise, measured as mean-squared error in copy number, is increased or decreased after division.
As illustrated in Fig. 5 D, we find that both processes make a clearly measurable contribution to noise suppression. When de novo assembly and number-limiting efficiencies are reduced below a threshold, the resulting defective error correction system actually makes the noise worse, as indicated by the red zones on the plot. As the efficacy of the two processes improves, a point is reached at which the noise decreases after division, and progressive reduction in noise is seen as the efficiency of the two processes increases.
We can now consider the related questions of stability and robustness. We define stability in the following sense: a number control system will be called stable if the steady-state number distribution can be restored in the limit of infinite time following a perturbation to some other distribution. Stability in this sense is therefore equivalent to saying that the steady-state copy number distribution is attractive. This definition of stability (which is one of several standard definitions used to describe stability of dynamical systems) makes no assertions regarding the transient behavior of the system and is therefore to be distinguished from Liapunov stability. In the context of centriole homeostasis, stability describes the ability of the system to return to the correct copy number following any transient perturbation in the distribution. The rate of this return is measured by the “restoration” figure of merit FR. The simulation of Fig. 5 B shows that our current model is sufficient to produce a stable steady-state solution to which the system will return following a perturbation, as predicted a priori by eigenvalue analysis of the inheritance matrix.
In contrast to stability, which describes the response to a perturbation in the centriole number distribution, robustness describes the qualitative behavior of the system following small changes in the parameters of the system itself. In this sense robustness can be interpreted as structural stability. To be more specific, we will define robustness in the following way: a number control system is robust if it remains stable (in the above sense) and retains a single unique steady-state solution, when parameters of the system are subjected to small perturbations. As discussed above, for any nonzero values of Pl and Pd, the matrix will have a single eigenvalue of 1, and this will always correspond to a single steady-state solution, namely, two centrioles per cell. Thus, any variation of Pd or Pl, as long as they remain greater than zero, will preserve the existence of a unique steady state. Similarly, Fig. 5 C shows that FR > 0 (indicating stability) for all nonzero values of Pd and Pl. Hence we conclude the system is robust. We note that these definitions of robustness and stability make no guarantees concerning transient behavior of the system. It is possible for the number distribution to become farther away from the desired set point, at least for a few generations, although ultimately it will always return to two per cell. Fig. 5 D provides a view of the transient response to a perturbation, and indicates that when the operating point reaches the point at which noise suppression becomes noise amplification (indicated by the contour line labeled 1.0), the system may transiently increase the error relative to its ultimate steady state. Overall, however, these results demonstrate that the homeostatic control of centriole number is a robust process that can tolerate quantitative changes in internal parameters and still stably restore itself to a unique steady state.
DISCUSSION
Biological significance of centriole homeostasis
The importance of homeostasis as an error-correction system depends on the rate of spontaneous error. Therefore we expect homeostasis to have its biggest biological effect in cases where centriole number is likely to become randomized. The two situations where this may occur are tumor progression and early development.
Tumor cells often show abnormal numbers of centrioles, and there has been considerable debate about whether the centriole abnormality might play a causal role in genomic instability. The fact that some tumor cells having chromosome loss or other genomic aberrations can be found in which the centriole number appears normal appears to cast doubt on centriole abnormalities playing a universal causal role. However, our results suggest that this argument needs to be reexamined. Because of centriole homeostasis, it is possible for a centriole abnormality to occur at some point during tumor progression, persist long enough to cause genomic instability due to multipolar spindles, and then become corrected. Indeed, one would imagine that for a tumor cell to grow robustly and propagate, it would need a way to compensate for the deleterious effects of abnormal centriole number. Although a compensation mechanism involving centriole clustering has been proposed (37,47), the centriole homeostasis system identified in this report might provide an alternative mechanism.
Centriole errors are also anticipated during development in species that rely on de novo centriole formation during their normal life cycle (30). Unlike normal centriole duplication, which has a high intrinsic fidelity, it is well established that de novo centriole assembly results in production of a random number of centrioles (13–16). The high variance in the number of centrioles that form during a round of de novo assembly is reminiscent of the high variance in bacteriophage burst size during single-step growth experiments, and is probably a fundamental characteristic of a self-assembly process. This variability becomes a potential problem whenever de novo centriole assembly occurs in development. For example, in the mouse, it has been reported that centrioles are absent from the early cleavage divisions (48,49), implying that the centriole present during later development must form de novo in individual blastomeres. Similarly, de novo centriole formation is a general feature of parthenogenetic development in many animal species from invertebrates to mammals (50,51). Development of embryos that start out with a variable number of centrioles in different blastomeres due to de novo assembly will require a mechanism to restore the correct copy number of two centrioles per cell following the initial burst of de novo assembly. In the mouse, normal centriole pairs are consistently detectable by the 32–64 cell stage (48). If de novo assembly began at the four-cell stage, this would mean that the embryo has three to four generations to restore the correct copy number. This is roughly consistent with the rates of restoration observed in our experiments and simulations (Fig. 5 B).
Implications for other organelles
It is interesting to consider whether the general scheme for number control presented here could serve as a paradigm for controlling abundance of other organelles. Most other organelles have variable sizes and shapes, and it is unclear whether one might be able to ignore these variations and simply count the number of separate individuals present in the cell, in which case the same type of model could be applied. Although centrioles are probably unique in their highly precise duplication process, some organelles do appear to arise from preexisting ones by some sort of discrete duplication (52). Moreover, simple fission of an organelle is equivalent to duplication if one simply considers organelle numbers and ignores their sizes. If such a model were to prove adequate for other organelles, our results with centrioles predict that robust copy number control for other organelles should require: a), a de novo assembly pathway that is activated when the organelle is missing (for example, by construction of a new organelle precursor out of endoplasmic reticulum-derived vesicles) and b), downregulation of organelle fission when the copy number becomes too high. Such a model has previously been proposed for number control of chloroplasts (5), however to our knowledge this model was never tested experimentally but instead was inferred based on the statistical distribution of copy numbers in cells. It will be of great interest to test these predictions for membrane-bound intracellular organelles.
Acknowledgments
This work was initiated when the author was a postdoctorate in the laboratory of Joel Rosenbaum, and I gratefully acknowledge his support and insights. I thank Doug Cole, Dennis Diener, and Jeff Salisbury for antibodies and advice. I also thank members of my lab for careful reading of the manuscript, and Clifford Marshall and Francois Nedelec for advice about matrix computations.
This work was supported by National Institutes of Health grant R01 GM077004, a Hellman Family Award for Early Career Faculty, and the Searle Scholars program.
Editor: Gaudenz Danuser.
References
- 1.Stelling, J., U. Sauer, Z. Szallasi, F. J. Doyle, and J. Doyle. 2004. Robustness of cellular functions. Cell. 118:675–685. [DOI] [PubMed] [Google Scholar]
- 2.El-Samad, H., J. P. Goff, and M. Khammash. 2002. Calcium homeostasis and parturient hypocalcaemia: an integral feedback perspective. J. Theor. Biol. 214:17–29. [DOI] [PubMed] [Google Scholar]
- 3.Tyson, J. J., K. C. Chen, and B. Novak. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15:221–231. [DOI] [PubMed] [Google Scholar]
- 4.Wilson, E. B. 1916. The distribution of the chondriosomes to the spermatozoa in scorpions. Proc. Natl. Acad. Sci. USA. 2:321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennis, A. S., and W. Birky. 1984. Stochastic partitioning of chloroplasts at cell division in the alga Olisthodiscus, and compensating control of chloroplast replication. J. Cell Sci. 70:1–15. [DOI] [PubMed] [Google Scholar]
- 6.Bergeland, T., J. Widerberg, O. Bakke, and T. W. Nordeng. 2001. Mitotic partitioning of endosomes and lysosomes. Curr. Biol. 11:644–651. [DOI] [PubMed] [Google Scholar]
- 7.Birky, C. W. 1982. The partitioning of cytoplasmic organelles at cell division. Int. Rev. Cytol. Suppl. 15:49–89. [DOI] [PubMed] [Google Scholar]
- 8.Mazia, D. 1978. Origin of twoness in cell reproduction. In Cell Reproduction. E. R. Dirksen, D. M. Prescott, and C. F. Fox, editors. Academic Press, New York. 1–14.
- 9.Doxsey, S. 2002. Duplicating dangerously: linking centrosome duplication and aneuploidy. Mol. Cell. 10:439–440. [DOI] [PubMed] [Google Scholar]
- 10.Sluder, G., and E. H. Hinchcliffe. 1999. Control of centrosome reproduction: the right number at the right time. Biol. Cell. 91:413–427. [PubMed] [Google Scholar]
- 11.Wong, C., and T. Stearns. 2003. Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat. Cell Biol. 5:539–544. [DOI] [PubMed] [Google Scholar]
- 12.Tsou, M. F., and T. Stearns. 2006. Mechanism limiting centrosome duplication to once per cell cycle. Nature. 442:947–951. [DOI] [PubMed] [Google Scholar]
- 13.Dingle, A. D. 1970. Control of flagellum number in Naegleria. J. Cell Sci. 7:463–482. [DOI] [PubMed] [Google Scholar]
- 14.Marshall, W. F., Y. Vucica, and J. L. Rosenbaum. 2001. Kinetics and regulation of de novo centriole assembly. Implications for the mechanism of centriole duplication. Curr. Biol. 11:308–317. [DOI] [PubMed] [Google Scholar]
- 15.Khodjakov, A., C. L. Rieder, G. Sluder, G. Cassels, O. Sibon, and C. L. Wang. 2002. De novo formation of centrosomes in vertebrate cells arrested during S phase. J. Cell Biol. 158:1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaTerra, S., C. N. English, P. Hergert, B. F. McEwen, G. Sluder, and A. Khodjakov. 2005. The de novo centriole assembly pathway in HeLa cells: cell cycle progression and centriole assembly/maturation. J. Cell Biol. 168:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taillon, B. E. 1993. Proteins Associated with the basal body apparatus in Chlamydomonas reinhardtii: a molecular genetic and cell biological analysis. PhD thesis. Carnegie Mellon, Pittsburgh, PA.
- 18.Harris, E. H. 1989. The Chlamydomonas Sourcebook. Academic Press, San Diego, CA. [DOI] [PubMed]
- 19.Cole, D. G., D. R. Diener, A. L. Himelblau, P. L. Beech, J. C. Fuster, and J. L. Rosenbaum. 1998. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory cilia. J. Cell Biol. 141:993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gantmacher, F. R. 1959. Matrix Theory, Vol. II. Chelsea Publishing, New York.
- 21.Kochanski, R. S., and G. G. Borisy. 1990. Mode of centriole duplication and distribution. J. Cell Biol. 130:1599–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan, M., N. Rayapuram, and S. Subramani. 2005. The control of peroxisome number and size during division and proliferation. Curr. Opin. Cell Biol. 17:376–383. [DOI] [PubMed] [Google Scholar]
- 23.Monastryska, I., and D. J. Klionsky. 2006. Autophagy in organelle homeostasis: peroxisome turnover. Mol. Aspects Med. 27:483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernales, S., K. L. McDonald, and P. Walter. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettencourt-Diaz, M., A. Rodrigues-Martins, L. Carpenter, M. Riparbelli, L. Lehmann, M. K. Gatti, N. Carmo, F. Balloux, G. Callaini, and D. M. Glover. 2005. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15:2199–2207. [DOI] [PubMed] [Google Scholar]
- 26.Seneta, E., and S. Tavare. 1983. Some stochastic models for plasmid copy number. Theor. Pop. Biol. 23:241–256. [DOI] [PubMed] [Google Scholar]
- 26.Khayam, S. A., and H. Radha. 2003. Markov-based modeling of wireless local area networks. 6thAnnual ACM International Workshop on Modeling, Analysis, and Simulation of Wireless and Mobile Systems Proceedings. 100–107.
- 27.Kailath, T. 1967. The divergence and Bhattacharyya distance measures in signal selection. IEEE Trans. Commun. 15:52–60. [Google Scholar]
- 28.Pritchard, G., and D. J. Scott. 2001. Empirical convergence rates for continuous-time Markov chains. J. Appl. Probab. 38:262–269. [Google Scholar]
- 29.Warren, G. 1993. Membrane partitioning during cell division. Annu. Rev. Biochem. 62:323–348. [DOI] [PubMed] [Google Scholar]
- 30.Mizukami, I., and J. Gall. 1966. Centriole replication. II. Sperm formation in the fern, Marsilea, and the cycad. Zamia. J. Cell Biol. 29:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutcher, S. K. 2003. Elucidation of basal body and centriole functions in Chlamydomonas reinhardtii. Traffic. 4:443–451. [DOI] [PubMed] [Google Scholar]
- 32.Salisbury, J. L., A. T. Baron, and M. A. Sanders. 1988. The centrin-based cytoskeleton of Chlamydomonas reinhardtii: distribution in interphase and mitotic cells. J. Cell Biol. 107:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taillon, B. E., S. A. Adler, J. P. Suhan, and J. W. Jarvik. 1992. Mutational analysis of centrin: an EF-hand protein associated with three distinct contractile fibers in the basal body apparatus of Chlamydomonas. J. Cell Biol. 119:1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright, R. L., J. Salisbury, and J. W. Jarvik. 1985. A nucleus-basal body connector in Chlamydomonas reinhardtii that may function in basal body localization or segregation. J. Cell Biol. 101:1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuchka, M. R., and J. W. Jarvik. 1982. Analysis of flagellar size control using a mutant of Chlamydomonas reinhardtii with a variable number of flagella. J. Cell Biol. 92:170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uetake, Y., J. Loncarek, J. J. Nordberg, C. N. English, S. LaTerra, A. Khodjakov, and G. Sluder. 2007. Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J. Cell Biol. 176:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quintyne, N. J., J. E. Reing, D. R. Hoffelder, S. M. Gollin, and W. S. Saunders. 2005. Spindle multipolarity is prevented by centrosomal clustering. Science. 307:127–129. [DOI] [PubMed] [Google Scholar]
- 38.Dutcher, S. K., N. S. Morrissette, A. M. Preble, C. Rackley, and J. Stanga. 2002. Epsilon-tubulin is an essential component of the centriole. Mol. Biol. Cell. 13:3859–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuura, K., P. A. Lefebvre, R. Kamiya, and M. Hirono. 2004. Bld10p, a novel protein essential for basal body assembly in Chlamydomonas: localization to the cartwheel, the first ninefold symmetrical structure appearing during assembly. J. Cell Biol. 165:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright, R. L., S. A. Adler, J. G. Spanier, and J. W. Jarvik. 1989. Nuclear-basal body connector in Chlamydomonas: evidence for a role in basal body segregation and against essential roles in mitosis or in determining cell polarity. Cell Motil. Cytoskeleton. 14:516–526. [DOI] [PubMed] [Google Scholar]
- 41.Howell, S. H., and J. A. Naliboff. 1973. Conditional mutants in Chlamydomonas reinhardtii blocked in the vegetative cell cycle. J. Cell Biol. 57:760–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deane, J. A., D. G. Cole, E. S. Seeley, D. R. Diener, and J. L. Rosenbaum. 2001. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 11:1586–1590. [DOI] [PubMed] [Google Scholar]
- 43.Gaffal, K. P. 1988. The basal body-root complex of Chlamydomonas reinhardtii during mitosis. Protoplasma. 143:118–129. [Google Scholar]
- 44.Vorobjev, I. A., and Y. S. Chentsov. 1982. Centrioles in the cell cycle. I. Epithelial cells. J. Cell Biol. 93:938–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishikawa, H., A. Kubo, S. Tsukita, and S. Tsukita. 2005. Odf2-deficient mother centrioles lack distal-subdistal appendages and the ability to generate primary cilia. Nat. Cell Biol. 7:517–524. [DOI] [PubMed] [Google Scholar]
- 46.Delgehyr, N., J. Sillibourne, and M. Bornens. 2005. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J. Cell Sci. 118:1565–1575. [DOI] [PubMed] [Google Scholar]
- 47.Brinkley, B. R. 2001. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 11:18–21. [DOI] [PubMed] [Google Scholar]
- 48.Szollosi, D., P. Calarco, and R. P. Donahue. 1972. Absence of centrioles in the first and second meiotic spindles of mouse oocytes. J. Cell Sci. 11:521–541. [DOI] [PubMed] [Google Scholar]
- 49.Calarco-Gillam, P. D., M. C. Siebert, R. Hubble, T. Mitchison, and M. Kirschner. 1983. Centrosome development in early mouse embryos as defined by an autoantibody against pericentriolar material. Cell. 35:621–629. [DOI] [PubMed] [Google Scholar]
- 50.Miki-Noumura, T. 1977. Studies on the de novo formation of centrioles: aster formation in the activated eggs of sea urchin. J. Cell Sci. 24:203–216. [DOI] [PubMed] [Google Scholar]
- 51.Szollosi, D., and J. P. Ozil. 1991. De novo formation of centrioles in parthenogenetically activated, diploidized rabbit embryos. Biol. Cell. 72:61–66. [DOI] [PubMed] [Google Scholar]
- 52.He, C. Y., M. Pypaert, and G. Warren. 2005. Golgi duplication in Trypanosoma brucei requires Centrin2. Science. 310:1196–1198. [DOI] [PubMed] [Google Scholar]