

Abstract
The physiological importance of cholesterol in the cell plasma membrane has attracted increased attention in recent years. Consequently, the use of methods of controlled manipulation of membrane cholesterol content has also increased sharply, especially as a method of studying putative cholesterol-enriched cell membrane domains (rafts). The most common means of modifying the cholesterol content of cell membranes is the incubation of cells or model membranes with cyclodextrins, a family of compounds, which, due to the presence of relatively hydrophobic cavity, can be used to extract cholesterol from cell membranes. However, the mechanism of this activity of cyclodextrins is not completely established. Moreover, under conditions commonly used for cholesterol extraction, cyclodextrins may remove cholesterol from both raft and non-raft domains of the membrane as well as alter the distribution of cholesterol between plasma and intracellular membranes. In addition, other hydrophobic molecules such as phospholipids may also be extracted from the membranes by cyclodextrins. We review the evidence for the specific and non-specific effects of cyclodextrins and what is known about the mechanisms for cyclodextrin-induced cholesterol and phospholipid extraction. Finally, we discuss useful control strategies that may help to verify that the observed effects are due specifically to cyclodextrin-induced changes in cellular cholesterol.
Keywords: Membrane cholesterol, membrane rafts, cyclodextrin
1. INTRODUCTION
Numerous studies have shown that a variety of cellular functions are affected when cells are exposed to β-cyclodextrins (βCDs), a class of pharmacological agents commonly used to remove membrane cholesterol. The goals of this review are to summarize the evidence for cholesterol-specific and non-specific effects of βCDs, to discuss whether cholesterol-specific effects can be attributed to the raft-disruption or to general membrane cholesterol depletion, to describe what is known about the biophysical and biochemical mechanisms of βCD-induced cholesterol depletion, and finally to discuss strategies for verifying the specificity of these effects for cholesterol depletion.
1.1. What are β-Cyclodextrins?
Cyclodextrins (CDs) are cyclic oligosaccharides consisting of α-(1–4)-linked D-glycopyranose units, which are primary degradation products of starch (reviewed by Davis and Brewster (2004) [1] and Uekama (2004) [2]). These compounds have been long recognized as potent carriers for hydrophobic drugs because, although they are water soluble, they contain a hydrophobic cavity which may encapsulate various hydrophobic molecules. CDs typically exist as hexamers (αCDs), heptamers (βCDs) or octomers (γCDs). The degree of polymerization defines the size of the hydrophobic cavity and, consequently, the affinity of the carrier to specific classes of compounds [1, 2]. β-cyclodextrins have the highest affinity for inclusion of cholesterol and are the most efficient in extracting cholesterol from erythrocyte and model membranes [3, 4, 5]. αCDs, on the other hand, are the most efficient in extracting phospholipids [4]. These differences have been attributed to the size and hydrophobicity of the CD inner cavities. Specifically, the cavity of αCDs appear to be too small to accommodate cholesterol molecule and the cavity of γCDs is not as hydrophobic as that of βCDs [4, 5]. It is also important to note that the cavity of single βCD molecule (~8 Ǻ) is too small to screen a cholesterol molecule (~18 Ǻ) from water and two stacked βCD molecules seem to be required [6]. Another difference between α–, β–, and γCDs is their water solubility, which at ambient conditions is 2% weight by weight for βCDs vs. 13% and 26% for αCDs and γCDs respectively [1]. Water solubility of βCDs, however, can be significantly improved by using hydrophilic modifications, such as methylated, 2-hydroxypropylated, sulfobutylether and branched CD derivatives [2]. Methyl-β-cyclodextrins (MβCD) and 2-hydroxyl-β-CD (2OHpβCD) are most widely used in cell biology to deplete cells of cholesterol.
2. β-CYCLODEXTRINS AS CHOLESTEROL DONOR-ACCEPTOR SYSTEM
2.1. β-Cyclodextrins are efficient in removing cholesterol from cellular membranes
Numerous studies have shown that exposing cells to βCDs results in removal of cellular cholesterol. The degree of cholesterol depletion is a function of the βCD derivative used, its concentration, incubation time, temperature and type of cells. MβCD was shown to be the most efficient as acceptor of cellular cholesterol, when compared to 2-hydroxypropyl (2OHPβCD), carboxymethyl, and tetradecasulfated β-cyclodextrins [7–9] and it is most commonly used. When cells are exposed to high concentration of MβCD (5–10 mM) for a prolong period of time (>2 hours) 80–90% of total cellular cholesterol can be removed (e.g. [7, 10]). Under these conditions, cells typically lose their morphology, round up and in extreme cases become nonviable. Indeed, hemolysis was one of the earlier noted effects of cyclodextrins [11]. Decreasing the concentration of the CD and/or using shorter incubation times results in milder depletion effects. Typical examples of MβCD effects on the level of cellular cholesterol are shown in Table 1. It is important to note that the degree of cholesterol depletion may differ significantly between cell types even when comparable CD concentrations and exposure times are applied [7, 8, 10, 12–19]. Most surprisingly, Fulop et al (2001) [19] showed that at low concentrations (0.5 mM for 30 min) MβCD induced an increase rather than decrease in cellular cholesterol of T lymphocytes isolated from young subjects and that the effect was reversed by prolonging the exposure to 60 min. An MβCD-induced increase in cellular cholesterol was observed only in cells isolated from young but not from elderly subjects. Fulop et al (2001) attributed the MβCD-induced cholesterol increase to disruption of membrane rafts. These observations underscore the importance of verifying the effect of cyclodextrins on the specific cell type and experimental condition.
Table 1.
Examples of MβCD-induced depletion of cellular free cholesterol.
| MβCD/time | cell type | Depletion | Reference |
|---|---|---|---|
| 20 mM/1.5 h | COS-7 | ~50% | [12] |
| 10 mM/8 h | mouse L-cell fibroblasts | ~100% | [7] |
| 10 mM/2 h | rod disk membranes | ~65% | [13] |
| 10 mM/1 h | mast cells | ~60% | [14] |
| 10 mM/1 h | arterial rings | ~20% | [15] |
| 10 mM/0.5 h | BHK | ~60% | [16]* |
| 10 mM/1 h | MDCK | ~70% | [16]* |
| 5 mM/8 h | mouse L-cell fibroblasts | ~80% | [7] |
| 5 mM/2 h | CHO | ~60% | [17] |
| 5 mM/0.5–1 h | A431 | ~30%/~40% | [18] |
| 5 mM/2 h | aortic endothelium | ~90% | [10] |
| 2.5 mM/6 h | rat hepatoma | ~70% | [8] |
| 0.5 mM/0.5 h | T lymphocytes, young | 2 fold up | [19] |
| 0.5 mM/1 h | T lymphocytes, young | ~30% | [19] |
| 0.5 mM/1 h | T lymphocytes, elderly | ~40% | [19] |
The levels of free (unesterified) cholesterol were measured either by gas-liquid chromotography [7, 8, 10, 17] or using colorimetric or fluorometric cholesterol oxidase assays [14–16, 19]. We have compared the two methods directly and obtained identical results (not shown). It is important to note that the variability between the different studies may be attributed not only to the type of cell and concentration and duration of the CD exposure but also to differences in cell density, passage, temperature, and other experimental conditions that may not be specified in the original papers.
In this study, cells were first pretreated with lovastatin/mevalonate to inhibit cholesterol synthesis
2.2. β-Cyclodextrins may alter cholesterol distribution between different cellular membranes
Another important issue to consider is the impact of cyclodextrins on cholesterol distribution between different cellular membranes. Several studies have shown that plasma membranes have significantly higher cholesterol levels than intracellular membranes [20–23]. Specifically, ~90% of free cholesterol was estimated to reside in the plasma membrane in several cell types [22–24]. However, van Meer (1989) [25] estimated that plasma membrane contains only 25–40% of total cellular cholesterol. Thus, exposing cells to cyclodextrins may also affect cholesterol levels in the intracellular membranes, which were suggested to constitute the slow kinetic pool of cholesterol as estimated by cyclodextrin-induced cholesterol efflux [26], (see discussion below). In fact, it has been shown that cholesterol level in the intracellular membranes is regulated by cholesterol level in the plasma membrane [27, 28]. Furthermore, small changes in plasma membrane cholesterol may induce more dramatic changes in the cholesterol level in the intracellular membranes. For example, Lange et al (1999) [27] showed that exposing fibroblasts to 2% hydroxypropyl-β-cyclodextrin resulted in a ~25% depletion of total membrane cholesterol, at the same time causing an 80% depletion of the cholesterol level in the endoplasmic reticulum. In contrast, treatment of epithelial cell line BHK with 10 mM MβCD resulted in a strong decrease in cholesterol-specific fluorescence in the plasma membrane, as estimated by filipin binding, whereas the intracellular staining was affected to a lesser degree [16]. Thus, it is essential to take into account that exposing cells to βCDs may alter the relative distribution of cholesterol between different cellular compartments with corresponding physiological effects.
2.3. Cholesterol depletion from different membrane domains
Much of the evidence reported using various methods and cell or model systems suggests that cholesterol distribution in the membrane is heterogeneous and that it is concentrated in cholesterol-rich and sphingomyelin-rich membrane domains (membrane rafts). The exact nature and even presence of membrane rafts in cellular rafts is a hotly debated issue, and until very recently no accepted definition of these structures existed. The recent Keystone Symposium on Lipid Rafts and Cell Function (March 23–28, 2006 in Steamboat Springs, CO) advanced the following consensus definition: “Membrane rafts are small (10–200 nm), heterogeneous, highly dynamic, sterol- and sphingolipid-enriched domains that compartmentalize cellular processes” [29]. Morphology, size, density and molecular composition of rafts in cellular membranes are still controversial, as summarized recently in several excellent reviews [33, 35–47] and a detailed discussion of these topics is beyond the scope of the current review. The goal of this section of our review is to analyze the evidence suggesting that MβCD extracts cholesterol from the raft membranes.
Typically, membrane rafts are identified as low-density membrane fractions separated by sucrose gradient (with or without detergent extraction). These fractions were estimated to contain ~30%–50% of total [61] [62] [63] or plasma membrane [64, 65] cholesterol in several cell types, including MDCK cells, macrophages and monocytes (since it is difficult to get a pure plasma membrane fraction, it is likely that some contamination of internal membranes is present in plasma membrane preparations). We have observed a slightly different cholesterol distribution with ~80% in low-density membrane fractions in Chinese Hamster Ovary cells (Tikku et al. Ms. submitted) and only the low density fractions contained measurable amounts of cholesterol in smooth muscle cells [67]. In contrast, only 4% of membrane cholesterol was found in the caveolae fraction isolated from plasma membranes of human fibroblasts [66]. A possible explanation for these discrepancies is the differences in the isolation protocols. Specifically, Gaus et al (2005) [65] showed that while at low detergent concentration (0.2% Triton), detergent and non-detergent protocols yield similar results, increasing the Triton concentration to 1% resulted in a dramatic redistribution of cholesterol from low- to high-density fractions. Similarly, we observed that in CHO cells, Triton-insoluble fractions contained only ~25% of the total membrane cholesterol, whereas low-density membrane fractions isolated with the non-detergent method contained ~80% of total membrane cholesterol (Figure 1). In addition, using different detergents also yields different cholesterol distributions [50, 65, 68]. However, detergent-free extractions may also be dependent on experimental conditions, and be susceptable to artifacts [51]. Thus, while the precise distribution of cholesterol between different membrane domains is controversial, it appears that cholesterol does not reside exclusively in the rafts but that significant amount of it is found in the non-raft fractions.
Figure 1.
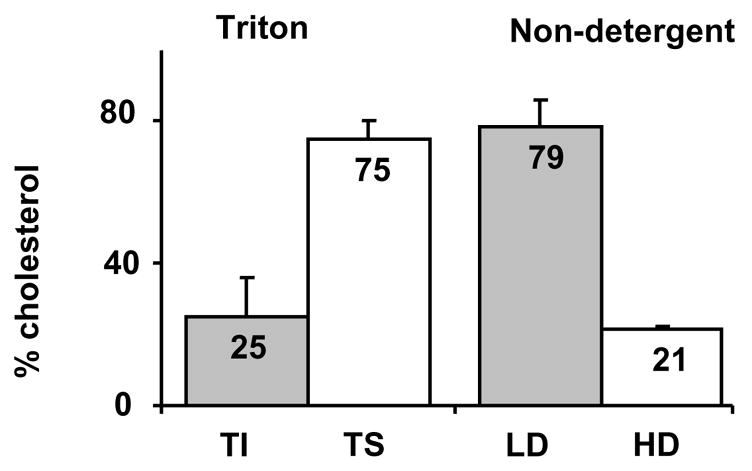
Raft and non-raft cholesterol contents in Triton-Insoluble (TI) and Triton-Soluble (TS) membrane fractions isolated using 1% cold Triton (A) or Low Density (LD) and High Density (HD) membrane fractions isolated using non-detergent sonication protocol (B). Briefly, for both procedures, cells were scraped into buffer A (in mM): 150 NaCl, 20 HEPES, 5 EDTA, pH 7.4, 1x Protease Inhibitor Cocktail (PIC) (Roche, Indianapolis, IN), 1 μg/ml pepstatin, and homogenized in a Dounce tissue grinder (40 strokes), and centrifuged for 10 min at 1,000 g. The pellet was resuspended, dounced, and re-centrifuged for 10 min at 1,000 g. Combined supernatant was centrifuged for 1 h at 200,000 g to obtain the “high speed pellet” (SW40Ti rotor, Beckman Coulter, Fullerton, CA). For preparing Triton-soluble and Triton-insoluble fractions, the high-speed pellet was resuspended in 1 ml of Buffer A, sonicated 3×10 s, and supplemented with a small volume of concentrated solution of Triton X-100 to a final concentration of 1%. After 15 min incubation on ice, the suspension was centrifuged for 1 h at 200,000 g. Then, the pellet (Triton insoluble fraction) was resuspended in Laemmli buffer. For isolation of membrane fractions using non-detergent method, the total membrane pellet was resuspended in 1 ml of 45% sucrose solution, sonicated and layered on sucrose gradient (35%-5%). The sucrose gradient was then centrifuged for 18 hr at 100,000 g. After centrifugation, 11 fractions were collected, protein was precipitated by TCA and measured using BCA Protein Assay Kit (BioRad, Hercules, CA). The samples were then resolved on 12% SDS PAGE at reducing conditions followed by transfer to PVDF membranes.
It is also important to put these findings in context of the fraction of the cell surface suggested to be occupied by membrane rafts. Here also a wide range of values (13–80%) was reported, depending on the specific method and cell type used (reviewed by [42]). Briefly, Schutz et al (2000) [69] reported that the regions of confined slow lipid diffusion, that they associated with membrane rafts, occupy 13% of the cell surface of smooth muscle cells. A similar estimate (10–15%) was reported by Gaus et al (2003), [59] in living macrophages using two-photon microscopy of environmentally sensitive probe Laurdan whose fluorescence properties depend on the physical properties of the lipid membrane bilayer. However, Gidwani et al (2001), [58], using steady-state fluorescence anisotropy of two lipid probes estimated that roughly 40% of the plasma membrane surface of RBL-2H3 mast cells is occupied by ordered domains. A similar estimate was given by Simons and Toomre (2000) [36] simply on the basis of relative abundance of sphingolipids in the inner and outer leaflets. Using electrone spin resonance, Swamy et al (2006) [70] found that most of the living cell surface is occupied by the Lo phase (rafts), which is continuous, and contains smaller regions of Ld phase, consistent with the model suggested previously by Munro (2003) [40]. A model where the bulk of the membrane is occupied by “rafts” is also consistent with experiments in model systems [71].
The crucial question is whether βCDs may be used to selectively remove cholesterol from rafts or from non-rafts domains. Multiple studies have shown that cholesterol depletion results in disassociation of a variety of proteins from detergent resistant/low-density membrane fractions (e.g. [14, 52–55]). Furthermore, cholesterol depletion induces significant changes in the physical properties of these fractions, as revealed by measuring diffusion of raft-associated proteins [57], by fluorescent anisotropy measurements of the raft and non-raft domains [58], and by laurdan generalized polarization [59]. Cholesterol depletion also results in disappearance of caveolae [15, 60], and inhibition of membrane ruffling [18]. These observations, however, do not mean that cholesterol is extracted exclusively from rafts. In fact, several studies have shown that βCDs are capable of removing cholesterol from both low and high density membrane fractions (Table 2) [62–65, 72], suggesting that cholesterol is removed from both raft and non-raft fractions, or more precisely from both Lo and Ld phases. Nevertheless the efficiency of cholesterol removal may vary among various membrane domains, as discussed below.
Table 2.
βCD-induced cholesterol depletion from low-density and high-density membrane fractions.
| %depletion | ||||
|---|---|---|---|---|
| CD/time | Cell type | Rafts | Non-rafts | Reference |
| MβCD | ||||
| 0.5 mM/60 min | Lymphocytes | ~55% | ~55% | [63]a |
| 5 mM/30 min | neuronal cells | ~30% | ~50% | [72]b |
| 10 mM/30 min | neuronal cells | ~90% | ~60% | [72] |
| 10 mM/60 min | THP-1 macrophages | ~85% | ~45% | [65]c |
| 10 mM/2 min | Jurkat cells | ~100% | ~none | [62]d |
| 10 mM/10 min | Jurkat cells | ~100% | ~50% | [62] |
| HPβCD (hydroxypropyl β-cyclodextrin) | ||||
| ~1 mM/60 min | THP-1 macrophages | significant | little or none | [64]e |
| TMCD (trimethyl cyclodextrin) | ||||
| ~3 mM/60 min | THP-1 macrophages | significant | little or none | [64] |
In all of the studies listed above, membrane fractions either of total or of plasma membrane were separated by discontinuous sucrose gradients with or without detergents. Cholesterol levels were quantified either using HPLC or cholesterol oxidase assay.
Total membranes, sucrose gradient (5, 35, 42.5% w/v) in the presence of 0.5% Triton X-100. Fractions 1–3 were GM1 positive and defined as lipid rafts.
Total membranes, sucrose gradient (5, 35, 42.5% w/v) in the presence of 1% Triton X-100, Fraction 5 was found to be cholesterol rich and defined as “low-density”, fractions 9–11 were defined as “high density”.
Plasma membranes, sucrose gradient (5, 20, 25, 35, 45% w/w sucrose) either using a non-detergent method (sonication) or in the presence of low concentration of Triton X-100 (0.2%). Rafts were defined as fractions 2–4 and non-rafts were defined as fractions 8–10. The two methods yielded similar results [65].
Total membranes, sucrose gradient (0.2–0.9 M with 0.1 M step) in the presence of 0.5% Triton X-100. Fractions 2–4 were cholesterol rich and defined as rafts [62].
Plasma membranes separated by a sucrose gradient (5–45% w/w sucrose) in the presence of 0.2 %Triton X-100 or 1% Lubrol WX, Rafts were defined as fractions 2–4 and non-rafts as fractions 8–10 [64].
Several studies have shown that the efficiency with which βCDs remove cholesterol from the low-density fractions appears to be higher than cholesterol depletion from the high density fractions, both in the presence of detergents and in detergent-free isolations [62, 64, 65]. In other studies, however, the effects were comparable [63, 72] and depended on the specific exposure conditions [72]. It seems likely, therefore, that at least in some cell types the use of short exposures [62] or very low CD concentrations [65] allowed selective depletion of cholesterol from lipid rafts without having a significant effect on non-raft cholesterol. Taken together, these studies suggest that raft cholesterol may be removed faster than the non-raft cholesterol. It is important to note that similar conclusions were reached on model membranes, with characterized Lo and Ld phases (see section 2.5). It is thus possible under specific conditions to preferentially remove cholesterol from membrane rafts. Interestingly, Rouquette-Jazdanian et al (2006) [62] showed that depletion of cholesterol from the low-density membrane fractions has little effect on the distribution of lipid raft markers whereas further cholesterol depletion that removes it from the non-raft domains has the so-called “raft disrupting” effect. Clearly, more studies are needed to investigate the relationship between raft cholesterol and the integrity of raft protein complexes. In summary, the preponderance of evidence suggests that relatively high (≥10 mM) MβCD concentrations and relatively long (≥ 30 min) exposures will lead to cholesterol depletion from all membrane fractions. Conversely, short (2–10 min) exposures and/or low (≤ 1 mM) concentrations may preferentially remove cholesterol from the low-density membrane fractions.
2.4. β-Cyclodextrins are also efficient in enriching and/or replenishing cells with cholesterol
The high affinity of βCDs for cholesterol can be used not only to remove cholesterol from the biological membranes but also to generate cholesterol inclusion complexes that donate cholesterol to the membrane and increase membrane cholesterol level. βCD-cholesterol inclusion complexes are typically generated by mixing cholesterol suspension with a cyclodextrin solution, as described earlier [8, 10, 73]. The ratio between the amounts of cholesterol and cyclodextrin in the complex determines whether it will act as cholesterol acceptor or as cholesterol donor [8, 13, 74]. The efficiency of cholesterol transfer from βCD inclusion complex to biological membranes depends on βCD:cholesterol molar ratio, βCD-cholesterol concentration, and duration of the exposure [8, 10, 13, 17, 73, 74]. Thus, it is important to note that exposing cells to βCD-cholesterol complexes that contain saturating amounts of cholesterol typically results not just in replenishing cholesterol to control levels but in significant cholesterol enrichment. Cholesterol enrichment is observed even if the cells were first depleted of cholesterol and then exposed to βCD-cholesterol complexes.
Repletion of cholesterol-depleted cells
Klein et al (1995) [73] showed that exposing MβCD-depleted membrane vesicles isolated from guinea pig myometrium to increasing concentrations of MβCD-cholesterol labeled with [3H]cholesterol resulted in a maximum [3H]cholesterol incorporation upon incubation with 0.5 mM MβCD-cholesterol. The molar ratio of cholesterol to phospholipid in such membranes (1.0) was 54% higher than in untreated membranes (0.65). Exposing the membranes to lower concentrations of MβCD-cholesterol for the same period of time (30 min) resulted in a significantly smaller amount of cholesterol incorporation [73]. In a parallel study, Gimpl et al (1997) [75] showed that when the Sf9 insect cells which naturally have very low cholesterol level (C/P=0.04) are exposed to MβCD-cholesterol, their cholesterol levels increase up to C/P=0.65. Consistent with these studies, Sheets et al (1999) [14] have shown that the cholesterol content of mast cells after the depletion/repletion procedure was 3.0–3.5 fold higher than the cholesterol content of untreated control cells. In summary, the repletion procedure has to be optimized for every experimental system in order to avoid an inadvertent increase of cholesterol to the level far above the control. Importantly, it is not currently known whether cholesterol added to membranes in repletion experiments is distributed between rafts and non-rafts in a manner similar to the original distribution.
Cholesterol enrichment
As expected, saturated βCD:cholesterol complexes are most efficient as cholesterol donors [8, 17]. The amount of cholesterol needed to form the saturated βCD:cholesterol complex depends on the type of βCD used (MβCD requires a significantly lower amount of cholesterol than 2OHPβCD) and on the βCD concentration [8]. For example, MβCD becomes saturated with cholesterol at βCD:cholesterol molar ratios of ~20:1 for 1.5 mM whereas 2OHPβCD becomes saturated at βCD:cholesterol molar ratios of 70:1 and 40:1 for 10 mM and 25 mM solutions. The basis for the difference between the two βCDs in their ability to complex cholesterol is not well understood. As described above for cholesterol depletion, most cholesterol enrichment studies use the MβCD derivative, that was shown to be effective at significantly lower concentrations than 2OHPβCD [8, 76]. Typically, MβCD-cholesterol is used in the concentration range of 1–10 mM for MβCD, similar to concentrations of “empty” MβCD used for cholesterol depletion and the degree of cholesterol enrichment varies between ~30% to ~3-fold depending on the cell type (Table 3) [8, 10, 13, 18, 73, 77–79]. Typical exposures range between 30 min to several hours, but only limited amount of information is available about the time needed to achieve equilibrium. For example, Christian et al (1997) [8] have shown that exposing CHO cells to 5 mM MβCD-cholesterol (8:1 molar ratio) or 25 mM 2OHPβCD-cholesterol (40:1 molar ratio) resulted in a gradual increase of cellular cholesterol over a period of several hours (>6 hr). In other cell types, however, equilibrium was achieved after shorter exposures (1–2 hr) [13, 18]. Analysis of the kinetic parameters of cholesterol depletion has been suggested to provide insights into the mechanisms underlying βCD-mediated cholesterol flux, as discussed in the next section.
Table 3.
Examples of cholesterol enrichment induced by MβCD-cholesterol
| MβCD/time | Cell type | Enrichment | Reference |
|---|---|---|---|
| 10 mM/2 h | Rod Disk Membranes | ~ 3 fold | [13] |
| 10 mM/1.5 h | Oppossum Kidney cells | ~30% | [77] |
| 5 mM/7 h | CHO cells/rat hepatoma cells | ~ 3 fold | [8] |
| 5 mM/2 h | endothelial cells | ~50% | [10] |
| 5 mM/0.5–1h | A431 | ~50% | [18] |
| 2.5 mM/1 h | endothelial cells | ~50% | [78] |
| 1 mM/0.5 h | neurons | ~30% | [79] |
| 0.5 mM/0.5 h | plasma membrane vesicles | ~50% | [73] |
Cholesterol levels were measured as described in Table 1.
2.5. Mechanism of MβCD cholesterol removal
Much of the evidence for the elucidation of the mechanism of MβCD-induced cholesterol removal comes from the kinetic analysis of cholesterol efflux demonstrating that cellular free cholesterol exists in two kinetic pools. Specifically, Yancey et al (1996) [76] observed biexponential kinetics of cellular cholesterol efflux induced by 2OHPβCD for mouse L-cells, human skin fibroblasts, and Fu5AH hepatoma cells: a fast pool (half-time (t1/2) ~19–23 s) and a slow pool with t1/2 of 15–30 min [76]. Whereas the fast pool of cholesterol was interpreted as corresponding to the outer leaflet of the plasma membrane, the nature of the slow pool was less obvious. Yancey et al (1996) [76] considered three possibilities: 1) intracellular cholesterol, 2) cholesterol present in the cytoplasmic monolayer of the plasma membrane bilayer, and 3) a separate lateral lipid domain in the plasma membrane. Based on previous studies demonstrating that over 90% of cellular cholesterol is located in the plasma membrane [23, 80] and on their own finding that the sizes of this pool ranged from 50 to 80% of total cellular cholesterol, Yancey et al (1996) [76] concluded that the slow pool is also located in the plasma membranes. To support this argument, Yancey et al (1996) [76] found that two distinct kinetic pools of cholesterol were also observed with model membranes (large unilamellar cholesterol-containing vesicles). However, it was not clear whether the slow pool originated from the inner leaflet, the slow time in that case corresponding to the flip-flop rate of cholesterol, or from cholesterol-enriched laterally separated domains in the outer leaflet. Since Yancey et al (1996) [76] found that the exchange rate between the fast and slow pools was similar to the efflux rate from the slow pool, they could not exclude the possibility that cholesterol from the slow pool has to be transferred to the fast pool before it can be extracted by cyclodextrin. Even a simple binary system of cholesterol/POPC was shown to exhibit two kinetic pools of cholesterol exchange between the vesicles in an earlier study [81] with the results being interpreted as reflecting complex phase structure of the vesicles.
Based on their results, Yancey et al (1996) [76] proposed a model of cyclodextrin-induced cholesterol efflux whereby a cyclodextrin molecule diffuses into the immediate proximity of the plasma membrane so that cholesterol molecules can diffuse directly into the hydrophobic pocket of the cyclodextrin molecule, without the necessity of completely desorbing through the aqueous phase, which is substantially more efficient than phospholipid acceptors. Accordingly, the activation energy of cholesterol transfer to cyclodextrin (7 kcal/mol) is much lower than the transfer to a phospholipid absorbing particle (20 kcal/mol). Taking into account that the activation energy is proportional to the degree to which the diffusing cholesterol molecule is exposed to water, Yancey et al (1996) [76] estimated that about one third of the cholesterol molecule is exposed to water during the transfer to cyclodextrin.
Two cholesterol pools, with efflux half times of 15±5 s and 21±6 min were also found by Haynes et al (2000) [82] in CHO-K1 cells. Similarly to the earlier paper by Yancey et al (1996) [76], Haynes et al (2000) [82] argued that both pools are located at the plasma membrane. More recently, a similar finding was also reported for T lymphocytes [62]. The time constants of cholesterol efflux for the two kinetic pools were remarkably similar to those in previous studies (17 s and 15 min for the fast and slow pools respectively). In this study, the fast pool was associated with the raft domains whereas the slow pool was associated with non-raft membrane fractions [62]. Interestingly, two kinetic pools of plasma cholesterol with the fast pool associated with the raft domains were also reported for THP-1 macrophages exposed to Apoliprotein A-1 [64].
A significantly different interpretation was advanced by Hao et al (2002) [26], who also observed two pools (MβCD extracts) in TRVb-1 (modified CHO) cells of fluorescent cholesterol analog, with half-lives similar to those reported previously [76,82]. However, Hao et al (2002) [26], also showed that slower efflux was absent from energy-depleted cells. Accordingly, Hao et al (2002) [26] suggested that the slow cholesterol pool is due to a large amount of cholesterol (35±12% of the total) in the endocytic recycling compartment (ECR); the slow half-life corresponds to the energy-dependent efflux of ERC cholesterol into the plasma membrane. It is not clear how to reconcile these results with those of Yancey et al (1996) [76], especially in the light of the preponderance of evidence showing that ~90% of total cellular cholesterol is residing in plasma membranes.
However, only one kinetic pool was observed by Steck et al (2002) [83] for the exit of cholesterol from erythrocyte membranes (using MβCD), with a half-time of 1 sec, a single first order process, and an activation energy of 27–28 Kcal/mol. Thus, at least in erythrocyte membranes, there is only one, fast, cholesterol pool. Moreover, Steck et al (2002) [83] observed that over 90% of total cell cholesterol was transferred to MβCD with this short half-time, showing that cholesterol flip-flop from the inner to the outer leaflet must occur at even shorter times. Steck et al (2002) [83] considered three possible models for the transfer of lipids between membrane compartments: 1. simple collision mechanism that was suggested by some studies in late 80’s and early 90s primarily to describe the phospholipid transfer process [84, 85], although the cholesterol transfer by this mechanism was also reported [86]. 2. The most commonly accepted aqueous diffusion pathway, where desorbed from the donor lipid molecules diffuse into the aqueous medium prior to capture with an acceptor by collision [87] and 3. activation-collision model, where lipid molecules are activated via partial protrusion from the bilayer and then captured by an acceptor via collision or returned to the ground state [88]. Only the last, activation-collision mechanism, agrees with the data presented in Steck et al (2002) [83]. To address the discrepancy between their results and earlier stuides, Steck et al (2002) [83] argued against aqueous diffusion coupled with a physical barrier (e.g., unstirred water layer, glycocalyx) model [89] by stating that poor access to the surfaces of donor cells, or unstirred water layer, can slow the diffusion of the desorbed cholesterol molecule only by a matter of seconds. Furthermore, Steck et al (2002) [83] mention that small uncharged lipid vesicles have negligible unstirred water layers and lack protein coats but nevertheless show slow cholesterol transfer kinetics [89, 90]. Finally, the reported rapid transfer to acceptors of numerous polar membrane lipids and sterol derivatives (e.g. [88, 89, 91–93] argues against rate-determining diffusion barriers. To explain the slow exit kinetics from whole cells, Steck et al (2002) [83] suggested that in activation-collision process there is a competition between the bulky acceptor and cell surface (recapture) for the partially projected monomer. This possibility, however, does not take into account that Yancey et al (1996) [76] observed a slow kinetic pool with model membranes.
There is also an unresolved disagreement between Steck et al (2002) [83] and Yancey et al (1996) [76] regarding the activation energy of transfer. Steck et al (2002) [83] notice that in their system, the high observed activation energy of transfer (27–28 Kcal/mol) agrees with the typical values of cholesterol transfer which range from 10 to over 20 Kcal/mol [94–99], and similar values for synthetic phospholipids with 16–20 effective methylene units [87, 100, 101]. Yancey et al (1996) [76], on the other hand, reported that the activation energy is 7 Kcal/mol; and the cause of discrepancy between the results of Yancey et al (1996) [76] and Steck et al (2002) [83] is not clear. Overall, the critical question of the mechanism of cholesterol efflux from cells, including the existence and origin of two cholesterol pools in cellular and model membranes is currently unresolved. More studies are needed to decide among the suggested models.
3. IMPACT OF βCDS ON NON-CHOLESTEROL MEMBRANE COMPONENTS
3.1. β-Cyclodextrins may interact with membrane phospholipids
3.1.1. Model membranes
Several studies have demonstrated that βCDs may interact with membrane phospholipids. Specifically, Puglisi et al (1996) [102] showed that cyclodextrins affect thermodynamic parameters of dipalmitoylphosphatidylcholine (DPPC) main phase transition as estimated by differential scanning calorimetry (DSC), implying cyclodextrin-DPPC interaction. Furthermore, Leventis and Silvius (2001) [103] showed that MβCD greatly accelerated the rate of transfer of DPPC between LUVs (250-fold at 1 mM), which was a stronger effect than the acceleration of cholesterol transfer. Only about half of DPPC was available for the transfer, from which Leventis and Silvius (2001) [103] concluded that only the outer leaflet DPPC was available, and that MβCD does not affect the flip-flop rate of DPPC. These results open a new dimension in considering possible artifacts in biological applications of MβCD: MβCD-induced transfer and redistribution of phospholipids among membrane domains with normally distinct phospholipid compositions and slow exchange rates. Such an effect would be most prominent at high MβCD concentrations (10 mM or higher), the same concentration range that is typically used to test the effects of cholesterol depletion on multiple cellular functions (see Tables 1 and 2).
Consistent with these studies, Giocondi et al (2004) [104] showed that incubation with MβCD results in formation of holes in dioleoylphosphaditylcholine (DOPC)/sphingomyelin (SM) bilayers as observed by atomic force microscopy, leading to the interpretation that MβCD removes not only cholesterol, but also phospholipids [104]. Moreover, Giocondi et al (2004) [104] observed a triphasic response upon incubation of PC/SM bilayers with MβCD:cholesterol, with corresponding multiple phase transitions. Giocondi et al (2004) [104] suggested that these transitions are due to a complex exchange of SM and cholesterol between the membrane and the CD complex, which finally results in formation of a uniform bilayer in the liquid ordered phase. On the other hand, other studies reported that βCDs have a much stronger effect on membrane cholesterol than on DPPC or SM. Specifically, Irie et al (1992) [3] showed that HPβCD preferentially solubilized cholesterol (by about a factor of 10) as compared to DPPC or SM, whereas HPαCD has a strong preference for DPPC and SM. HPγCD solubilized cholesterol, DPPC, and SM similarly [3]. Furthermore, Ohvo and Slotte (1996) [5] reported that in mixed cholesterol/DPPC or cholesterol/SM monolayers, 1.4 mM βCD did not extract DPPC at all, and the rate of desorption of SM was only 5% that of cholesterol. They also noted, however, that di-10-PC was desorbed at a rate of 35% relative to cholesterol. In summary, while the extent of βCD interactions with different membrane lipids vary significantly between the different studies, it is important to take into account that MβCD -cholesterol complex may not only donate cholesterol to cellular membranes but at the same time, also remove other membrane components.
3.1.2. Cells
There is also no consensus on the percentage of phospholipids extracted from cells by CDs, probably at least partially due to different types of cells and other conditions (e.g., the type, concentration, and duration of exposure to CDs) employed. A number of studies reported only minimal release of phospholipids from cell membranes under exposure to CDs: less than 5% of the initial cell-associated [14]choline-labeled phospholipids was released after 1 hr exposure with 1 mM MβCD in various CHO cells [105] and at most 2% of cellular phospholipids were released after a 5 hr incubation with 5 mM MβCD from L-cells [7]. Several other observations are consistent with these studies: (a) removal of cholesterol from microvilli without affecting membrane phospholipids and glycosphingolipids by 2% (w/v) MβCD [106] (b) a limited efflux of choline-containing lipids from foam cell macrophages [107]: after 24 hr incubation with 0.7 mM TMCD, 8.76% of phospholipids were extracted, as compared with 9.34% for the media alone; and (c) no effect of βCD on phospholipids in rat small intestine, whereas αCD had a significant effect [108].
In contrast to these reports, however, Ottico et al (2003) [72] reported that rat cerebellar granule cells released 50.2%, 14.5%, and 17.1% of the total cell complement of cholesterol, SM, and glycosphingolipids, respectively, after 30 min treatment with 5 mM MβCD. All of the sphingolipid species studied, except for ceramide, but including gangliosides, neutral glycosphingolipids, and SM were extracted by MβCD to the same extent [72]. The results of Ottico et al (2003) [72] are consistent with earlier studies that demonstrated interaction of CDs with sphingolipids [109–112]. The glycerophospholipids were released from cells to a much lesser extent (about 2% of total cell component after 30 min treatment with 5 mM MβCD [72]. A significant extraction of phospholipids by βCD from brain endothelial cells was demonstrated by Monnaert et al (2004) [113]: βCD at 5 mM for 2 hr removed 19% PC and 63% SM. Ohtani et al (1989) [4] reported that CDs extracted phospholipids from erythrocytes with the potency of α>β≫γ, with about 25% of the phospholipids removed by βCD. Rawyler and Siegenthaler (1996) [114] showed that CDs (including MβCD) removed acid lipids from thylakoid membranes. Finally, Niu et al (2002) [13] noted that the amount of lipids extracted from outer disk membranes became significant at >15 mM MβCD. In summary, there is not enough consistent information to predict the impact of βCD on the non-cholesterol lipid components of the membrane and additional control strategies should be employed to assess the specificity of βCD -induced effects. Some of the possible control strategies are discussed at the end of this review.
3.1.3. Putative mechanism for βCD-phospholipid interaction
Anderson et al (2004) [115] noted that neither the solution-phase complexation model, nor the partition model are suitable for MβCD-POPC interaction (the latter because MβCD does not penetrate the membranes). Furthermore, Anderson et al (2004) [115] considered the relative dimensions of the MβCD cavity (0.8 nm), and POPC acyl chains (about 2 nm in length each), which would suggested that each acyl chain needs two MβCD molecules to form the inclusion complex. Taking the radius of the MβCD cavity of 0.3 nm, the total hydrophobic surface of 4 MβCD molecules will be 6 nm2 [115]. Accordingly, Anderson et al (2004) [115] developed a new model of MβCD -POPC interactions that incorporates features of two previous models. This mechanism involves the formation of an initial inclusion complex between bilayer-phase POPC and a single MβCD molecule, followed by incremental attachment of three more MβCD molecules. The first step involves the removal of a phospholipid molecule from the bilayer environment into the aqueous phase prior to, or simultaneous with, binding to a cyclodextrin; whereas the following three steps only involve binding of additional cyclodextrins to the nascent complex.
3.2. Cyclodextrins may interact with membrane proteins
Because of the hydrophobic character of its pocket, MβCD is expected to interact with hydrophobic amino acids [116–118] and, in general, with hydrophobic protein domains. The hydrophobicity of MβCD makes it an extensive object of research for use as a drug delivery system [119, 120]. Indeed, MβCD was found to interact with such proteins as ubiquitin, chymotrypsin inhibitor 2 (CI2), S6 and insulin [121]. Studies with other CD forms demonstrated binding of βCD to glucoamylase 1 and its variants [122], interaction of βCD with glycosyltransferase [123] and maltodextrin-binding protein [124, 125], and formation of complexes of αCD with pig pancreatic amylase [126] and β-amylase from Soybean [127]. CD-protein interaction was identified early on as one of the causes of CD induced hemolysis. Thus, Irie et al (1982) [11] reported that CDs differed in their capacity to induce hemolysis in the order of β>γ>α. Prominent hemolysis was observed at 3 mM βCD. Similarly, the potency of CD induced extraction of proteins from erythrocytes found to be in the order of: β≫γ>α [4]. CDs were shown to release a number of proteins from the murine T cell line P1798, including Thy-1, and induced partial removal of cell surface proteins CD46, CD26, MHC class I, and intracellular proteins Lck and Fyn [128]. A significant amount of GPI-anchored CD59 was released by MβCD from the human ECV304 cells [128]. The mechanism of this extraction, however, is unclear. Thus, Ilangumaran and Hoessli (1998) [128] noted that glycerophospholipids and glycoproteins are also released after treatment with MβCD, although no direct binding of MβCD to glycerophospholipids has been reported. Sheets et al (1999) [14] showed that MβCD-induced inhibition of tyrosine phosphorylation is associated with the loss of 70% of receptor-bound IgE, and 64% of ganglioside GD1b. The loss of FcεRI was attributed to vesicle shedding caused by MβCD treatment. It was also shown that CD can insert into the connexin pore [129], and bacterial α-hemolysin pore [130]. Moreover, further modifications of the pore functions can be made by small guest molecules, residing in CD cavities, leading to CDs being termed as “molecular adapters” [130]. The affinity of CDs for proteins was also employed for the creation of artificial chaperones [131].
In summary, there is no sufficient body of data available with systematic studies of the interaction of CDs with cell surface proteins to predict the effect of these compounds on cells in specific situations. Since these effects have a potential to affect cellular functions, more studies are needed to address this phenomenon.
4. CONTROL STRATEGIES
In light of all the evidence that exposure of cells to βCDs may have multiple non-specific effects in addition to cholesterol depletion, it becomes increasingly clear that these treatments require a rigorous set of control conditions. Several studies compared the effects of MβCD-induced cholesterol depletion with other methods of reducing cellular cholesterol, such as decreasing cellular cholesterol level by serum starvation (e.g. [77, 132]), sequestering cholesterol with filipin or nystatin (e.g. [12, 133, 134], or modification of membrane cholesterol by its enzymatic degradation with cholesterol oxidase (e.g. [62, 133]).
These methods, however, introduce additional factors that are difficult to quantify, such as changes in the local concentration of cholesterol or a build-up of products of cholesterol degradation. We are going to discuss, therefore, two quantitative control strategies that have been suggested in recent studies: “cholesterol-matched” controls and substitution of cholesterol by its structural analogues.
4.1. Cholesterol-matched control
One of the frequently used controls for the specificity of MβCD function is exposing cells to MβCD saturated with cholesterol. The weakness of this approach is that, as described above, exposure to MβCD saturated with cholesterol may not maintain cellular cholesterol at a normal level but is instead likely to significantly increase cellular cholesterol. This is a problem because both cholesterol depletion and cholesterol enrichment may affect cellular functions. We propose, therefore, that a stronger control is exposing cells to βCD-cholesterol complexes that have no effect on the level of cellular cholesterol. This can be achieved by varying the molar ratio between a βCD and cholesterol in the complex. Indeed, Christian et al (1997) [8] has shown that for both MβCD and 2OHPβCD, decreasing the degree of cholesterol saturation results in reduced ability of the complex to donate cholesterol until it reaches the “equilibrium point” after which the process is reversed, so that unsaturated βCD-cholesterol complex starts acting as cholesterol acceptor rather than cholesterol donor. The strength of this approach is that cells that are cholesterol-depleted, cholesterol-enriched and “cholesterol clamped”, are all exposed to the same concentration of βCD. We have used this approach to determine the effect of cholesterol on the biomechanical properties of aortic endothelial cells [135], and activities of K+ channels expressed in Chinese Hamster Ovary cells [17]. This approach has also been used to determine the role of cholesterol in membrane ruffling and pinocytosis in A431 cells [18]. Specific conditions for the “equilibrium point”, however, may be different for the different cell types. For example, while for BAEC the equilibrium point was achieved when the cells were exposed to1:1 MβCD: MβCD:cholesterol ratio [120]; under the same experimental conditions the equilibrium point for Chinese hamster ovary cells was achieved at 1:4 MβCD:MβCD:cholesterol ratio [15] (Figure 2). Therefore these conditions should be verified empirically.
Figure 2.
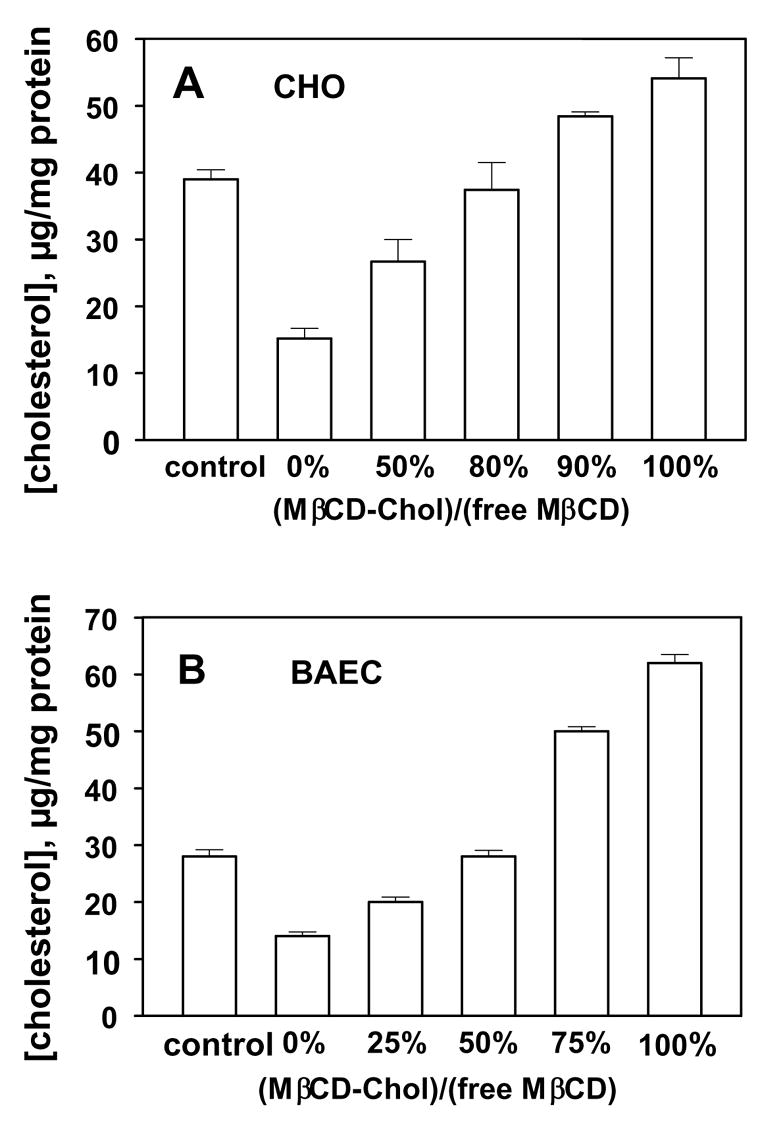
Modulation of cellular cholesterol by MβCD:MβCD-cholesterol mixtures in Bovine Aortic Endothelial cells (A) and in Chinese Hamster Ovary Cells (B). Modified from Romanenko et al. (2002) [74] and Byfield et al., [135]. MβCD saturated with cholesterol using the standard protocol. Briefly, small volume of a cholesterol stock solution in chloroform:methanol (1:1, vol:vol) was added to a glass tube and the solvent was evaporated. Then, 2.5 mM or 5 mM MβCD solution in DMEM medium without serum was added to the dried cholesterol. The tube was vortexed, sonicated, and incubated overnight in a shaking bath at 37°C. The mixture of MβCD and MβCD -cholesterol was prepared just by mixing the two solutions at different molar ratios.
It is important to note that while “cholesterol-matched” controls are designed to have no effect on the total amount of cellular cholesterol, they may still have an effect on cholesterol distribution between different membrane fractions. This problem applies to the same extent to depletion/repletion experimental protocols. This issue can be tested by loading matched βCD with hot cholesterol and monitoring the kinetics of exchange of cholesterol between the cell and βCD, as well as the exchange between the different membrane fractions. It is also possible that matched βCD-cholesterol complexes may have significant effects on the distributions of membrane phospholipids, which may be tested by analyzing the phospholipid compositions of the different fractions. With these possible caveats, “cholesterol-matched” conditions are expected to be a better control than exposing the cells to MβCD saturated with cholesterol that is likely to significantly increase cellular cholesterol level.
4.2 Substitution of cholesterol by sterol structural analogues
An alternative strategy is to remove membrane cholesterol and substitute it with a different sterol. The basic rationale for this approach is that removing membrane cholesterol and substituting it with an equivalent amount of another sterol allows identifying structural determinants of cholesterol function, thus providing an insight for the mechanism responsible for cholesterol sensitivity of a particular process. Earlier studies have used structural sterol analysis in artificial liposomes to investigate the mechanisms by which cholesterol interacts with phospholipids [136–138], regulates membrane permeability (e.g. [139–141]), affects the physical properties of the lipid bilayer [142, 143], and induces formation of ordered membrane domains [143]. Introduction of βCDs allows extending this approach to perform structural sterol analysis in cellular membranes in a quantitative and reproducible way.
This strategy has been applied in several studies in order to discriminate between cholesterol effects that are due to specific sterol-protein interactions or due to changes in the physical properties of membrane lipid bilayer. Specifically, performing multiple sterol substitutions, Gimple et al (1997) [133] showed that whereas the effects of sterols on cholecystokinin receptor correlated with changes in membrane fluidity, oxytocin receptor had a unique requirement for cholesterol suggesting that this receptor is regulated by specific cholesterol-protein interactions. More recently, it was also shown that an optical isomer of cholesterol, epicholesterol, substitutes functionally for cholesterol in the regulation of GABAA receptor [144]. A similar approach was undertaken in our recent studies to elucidate molecular mechanisms responsible for cholesterol sensitivity of K+ and Cl− ion channels in vascular endothelial cells [74, 78]. Our studies have shown that while endothelial inwardly-rectifying K+ channels are very sensitive to the substitution of cholesterol with epicholesterol [74], volume-regulated Cl− channels were not sensitive to this substitution at all [78]. Since the only difference between the two cholesterol analogues is the rotational angle of a hydroxyl group and since the two sterols have similar effects on physical properties of the membrane [133, 143], the sensitivity of K+ channels to this substitution suggests that these channels are regulated by specific sterol-protein interactions. In contrast, sensitivity of volume-regulated Cl− channels to different sterols correlated with the known effects of sterols on membrane fluidity. In both cases, repleting cells with cholesterol brought the currents back to control levels. These observations provide an additional example for distinct mechanisms underlying cholesterol sensitivity of different membrane proteins in the same cell. This study, however, did not address the question of how different sterols are distributed between raft and non-raft domains.
What are the possible pitfalls and limitations of this approach? One important condition to perform these experiments in a well-controlled manner is to optimize the conditions of substitution in such a way that the total sterol level of a cell is not changed. If this condition is fulfilled, then the only variable is the ratio between native cholesterol and a given sterol. It is also important to note that the substitution is likely not to be complete. In our experience, depending on MβCD concentration and the time of exposure, typically we could achieve ~50–70% of substitution but the degree of the substitution varied for different sterols even under the same experimental conditions (Figure 3, [74, 78]). It is important, therefore, to quantify the amount of sterols incorporated under each set of experimental conditions. The more critical limitation of this approach is the uncertainty about whether or not the sterols are incorporated into the rafts and a paucity of information regarding the impacts of different sterols on the physical properties of the membrane. One approach to partially alleviate this concern is to compare sterol-substitution experiments with cholesterol repletion experiments as controls.
Figure 3.
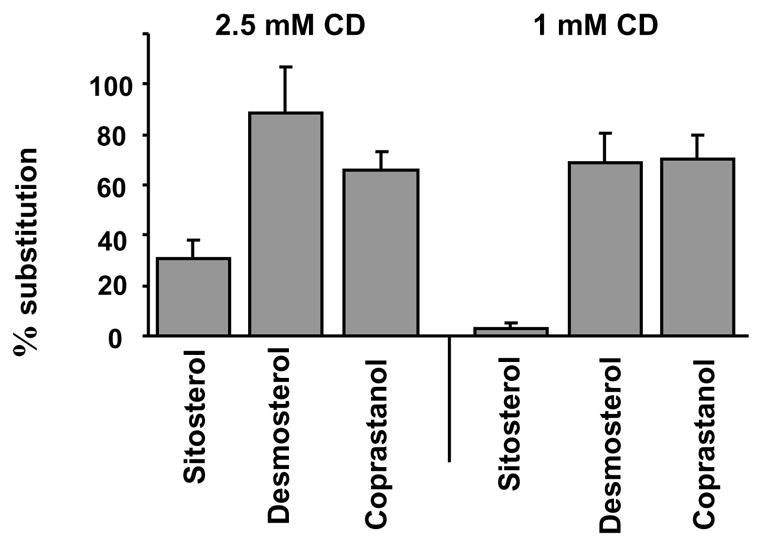
Efficiencies of different sterol-cholesterol substitutions in Bovine Aortic Endothelial Cells. Modified from Romanenko et al., (2004) [78]. Briefly, the substitution experiments were performed by exposing the cells to MβCD solution saturated with a specific analogue. Different MβCD-sterol solutions were prepared as described above for MβCD-cholesterol.
In summary, we suggest that this is a promising approach that will become increasingly valuable as more information becomes available about the contributions of different sterols on various membrane properties.
Conclusions
β-cyclodextrins (βCD) provide a unique tool to modulate cellular cholesterol in living cells, however it needs to be recognized that βCDs have pleiotropic effects on the level and distribution of different membrane components, as summarized in Figure 4. Furthermore, since under commonly used conditions βCDs extract cholesterol not only from the putative cholesterol-rich membrane rafts but also from the rest of the membrane (see Fig. 4A), a common interpretation of βCD-sensitivity as direct evidence for the involvement of membrane rafts is not justified. However, exposing the cells to low (≤1 mM) cyclodextrin concentration for the short (2–10 min) period of time may have preferential effect on membrane fractions proposed to contain membrane rafts. Based on the analysis of all the literature described in this review, we suggest that the “best practices” for the use of cyclodextrins include: (1) To limit the exposure to the conditions specified above and to verify the degree of cholesterol depletion in total and plasma membranes because the efficiency of βCD in extracting cholesterol may vary significantly depending on βCD concentration, duration of the exposure and the cell type. (2) It is important to recognize that saturated βCD-cholesterol complex, a commonly used control for the specificity of βCD effects to cholesterol, may actually bring cellular cholesterol significantly above the control level (Fig. 4B) and it may be beneficial to expose the cells to an “equilibrium” MβCD:cholesterol mixture. a “cholesterol-matched control”. (3) Finally, we suggest that substituting cholesterol with other sterols using βCDs as carriers, will provide additional insights into the role of cholesterol in cellular function.
Figure 4.
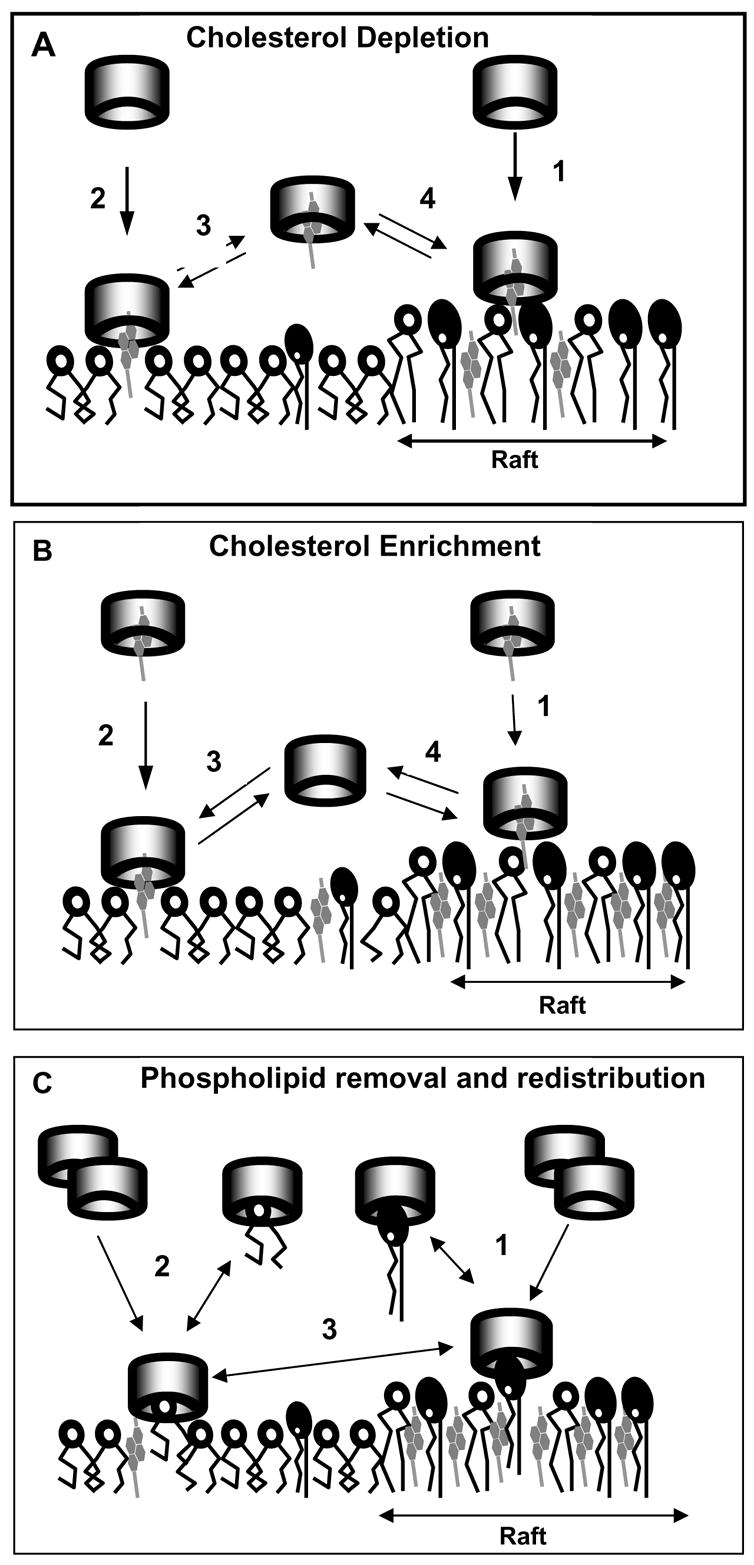
A simplified scheme of effects of MβCD on lipid membranes The scheme illustrates complexity of MβCD interactions when used, as in a majority of studies, to disrupt membrane “rafts”. Cylinders represent MβCD, dark-headgroup phospholipids sphingomyelin, open circle-headroup lipids represent “other” phospholipids, e.g., PC. No 1:1 stoichiometry is implied (see text). A. MβCD approaches the membranes and captures cholesterol from both raft (1) and nonraft (2) regions. The mechanism of capture likely involves a membrane fluctuation-induced partial protrusion of cholesterol molecules into the aqueous phase. The preponderance of evidence indicates that the removal of cholesterol from raft domains is more efficient; thus short (2–10 min) exposures and/or low (≤ 1 mM) concentrations of MβCD may preferentially remove cholesterol from the raft regions. Longer exposures and higher MβCD concentrations may also lead to redistribution of cholesterol between the raft and nonraft regions (3,4). B. Cholesterol enrichment of membranes is originated by incubation of membranes with MβCD, previously loaded with cholesterol. Both raft (1) and nonraft (2) regions may increase their cholesterol content, and the relative degrees of enrichment are unclear. MβCD molecules upon delivering cholesterol to the membrane may bring on redistribution of cholesterol between the membrane regions (3,4), as well as redistribution of phospholipids (panel C). C. Prolonged exposures and high concentrations of MβCD may lead to removal of phospholipids from both raft (1) and nonraft (2) regions, as well as redistribution of different phospholipids between the regions (3).
Acknowledgments
The authors wish to thank the sponsors and organizers of the Biophysical Society Discussions Meeting in Asilomar, 2004 for arranging the program that initiated the writing of this review. We are also grateful for encouragement, critical comments and suggestions made by Drs. M. Saxton, S. Pierce, G. Rothblat, M. Edidin and K. Jacobson. We also thank Mr. Gregory Kowalsky for the graphic design of Figure 4, summarizing the main effects of βCDs on membrane lipids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–35. doi: 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- 2.Uekama K. Design and evaluation of cyclodextrin-based drug formulation. Chem Pharm Bull (Tokyo) 2004;52:900–15. doi: 10.1248/cpb.52.900. [DOI] [PubMed] [Google Scholar]
- 3.Irie T, Fukunaga K, Pitha J. Hydroxypropylcyclodextrins in parenteral use. I: Lipid dissolution and effects on lipid transfers in vitro. J Pharm Sci. 1992;81:521–3. doi: 10.1002/jps.2600810609. [DOI] [PubMed] [Google Scholar]
- 4.Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- 5.Ohvo H, Slotte JP. Cyclodextrin-mediated removal of sterols from monolayers: effects of sterol structure and phospholipids on desorption rate. Biochemistry. 1996;35:8018–24. doi: 10.1021/bi9528816. [DOI] [PubMed] [Google Scholar]
- 6.Tsamaloukas A, Szadkowska H, Slotte PJ, Heerklotz H. Interactions of cholesterol with lipid membranes and cyclodextrin characterized by calorimetry. Biophys J. 2005;89:1109–19. doi: 10.1529/biophysj.105.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–6. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 8.Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. Journal of Lipid Research. 1997;38:2264–2272. [PubMed] [Google Scholar]
- 9.Atger VM, de la Llera Moya M, Stoudt GW, Rodrigueza WV, Phillips MC, Rothblat GH. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. Journal of Clinical Investigation. 1997;99:773–80. doi: 10.1172/JCI119223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levitan I, Christian AE, Tulenko TN, Rothblat GH. Membrane cholesterol content modulates activation of volume-regulated anion current (VRAC) in bovine endothelial cells. Journal of General Physiology. 2000;115:405–416. doi: 10.1085/jgp.115.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irie T, Otagiri M, Sunada M, Uekama K, Ohtani Y, Yamada Y, Sugiyama Y. Cyclodextrin-induced hemolysis and shape changes of human erythrocytes in vitro. J Pharmacobiodyn. 1982;5:741–4. doi: 10.1248/bpb1978.5.741. [DOI] [PubMed] [Google Scholar]
- 12.Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, Rose-John S. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE) J Biol Chem. 2003;278:38829–39. doi: 10.1074/jbc.M210584200. [DOI] [PubMed] [Google Scholar]
- 13.Niu SL, Mitchell DC, Litman BJ. Manipulation of cholesterol levels in rod disk membranes by methyl-beta-cyclodextrin: effects on receptor activation. J Biol Chem. 2002;277:20139–45. doi: 10.1074/jbc.M200594200. [DOI] [PubMed] [Google Scholar]
- 14.Sheets ED, Holowka D, Baird B. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcepsilonRI and their association with detergent-resistant membranes. J Cell Biol. 1999;145:877–87. doi: 10.1083/jcb.145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreja K, Voldstedlund M, Vinten J, Tranum-Jensen J, Hellstrand P, Sward K. Cholesterol depletion disrupts caveolae and differentially impairs agonist-induced arterial contraction. Arterioscler Thromb Vasc Biol. 2002;22:1267–72. doi: 10.1161/01.atv.0000023438.32585.a1. [DOI] [PubMed] [Google Scholar]
- 16.Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol. 1998;140:1357–67. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–61. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimmer S, van Deurs B, Sandvig K. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. Jornal of Cell Science. 2002;115:2953–2962. doi: 10.1242/jcs.115.14.2953. [DOI] [PubMed] [Google Scholar]
- 19.Fulop T, Jr, Douziech N, Goulet AC, Desgeorges S, Linteau A, Lacombe G, Dupuis G. Cyclodextrin modulation of T lymphocyte signal transduction with aging. Mech Ageing Dev. 2001;122:1413–30. doi: 10.1016/s0047-6374(01)00274-3. [DOI] [PubMed] [Google Scholar]
- 20.Davis PJ, Poznansky MJ. Modulation of 3-hydroxy-3-methylglutaryl-CoA reductase by changes in microsomal cholesterol content or phospholipid composition. Proc Natl Acad Sci U S A. 1987;84:118–21. doi: 10.1073/pnas.84.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slotte JP, Bierman EL. Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem J. 1988;250:653–8. doi: 10.1042/bj2500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange Y. Disposition of intracellular cholesterol in human fibroblasts. J Lipid Res. 1991;32:329–39. [PubMed] [Google Scholar]
- 23.Lange Y, Swaisgood MH, Ramos BV, Steck TL. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J Biol Chem. 1989;264:3786–93. [PubMed] [Google Scholar]
- 24.Kim JA, Maxwell K, Hajjar DP, Berliner JA. b-VLDL increases endothelial cell plasma membrane cholesterol. Journal of Lipid Research. 1991;32:1125–1131. [PubMed] [Google Scholar]
- 25.van Meer G. Lipid traffic in animal cells. Annu Rev Cell Biol. 1989;5:247–75. doi: 10.1146/annurev.cb.05.110189.001335. [DOI] [PubMed] [Google Scholar]
- 26.Hao M, Lin SX, Karylowski OJ, Wustner D, McGraw TE, Maxfield FR. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J Biol Chem. 2002;277:609–17. doi: 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- 27.Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–2270. [PubMed] [Google Scholar]
- 28.Lange Y, Ye J, Steck TL. How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proc Natl Acad Sci U S A. 2004;101:11664–7. doi: 10.1073/pnas.0404766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pike LJ. Rafts defined: a report on the Keystone symposium on lipid rafts and cell function. J Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Sowa G, Pypaert M, Sessa WC. Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc Natl Acad Sci U S A. 2001;98:14072–7. doi: 10.1073/pnas.241409998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh P, Schnitzer JE. Segregation of heterotrimeric G proteins in cell surface microdomains. G(q) binds caveolin to concentrate in caveolae, whereas G(i) and G(s) target lipid rafts by default. Mol Biol Cell. 2001;12:685–98. doi: 10.1091/mbc.12.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pike LJ, Han X, Chung KN, Gross RW. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry. 2002;41:2075–88. doi: 10.1021/bi0156557. [DOI] [PubMed] [Google Scholar]
- 33.Anderson RGW, Jacobson K. A Role for Lipid Shells in Targeting Proteins to Caveolae, Rafts, and Other Lipid Domains. Science. 2002;296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 34.Kiyokawa E, Baba T, Otsuka N, Makino A, Ohno S, Kobayashi T. Spatial and Functional Heterogeneity of Sphingolipid-rich Membrane Domains. J Biol Chem. 2005;280:24072–24084. doi: 10.1074/jbc.M502244200. [DOI] [PubMed] [Google Scholar]
- 35.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 36.Simons K, Toomre D. Lipid rafts and signal transduction. Nature reviews Molecular cell biology. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 37.Edidin M. Membrane cholesterol, protein phosphorylation and lipid rafts. Science’s STKE. 2001;67:PE1. doi: 10.1126/stke.2001.67.pe1. [DOI] [PubMed] [Google Scholar]
- 38.Edidin M. The state of lipid rafts: From model membranes to cells. Annual Review Biophysical Biomolecular Structure. 2003;32:257083. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 39.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–88. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- 41.McConnell HM, Vrljic M. LIQUID-LIQUID IMMISCIBILITY IN MEMBRANES. Annual Review of Biophysics and Biomolecular Structure. 2003;32:469–492. doi: 10.1146/annurev.biophys.32.110601.141704. [DOI] [PubMed] [Google Scholar]
- 42.Lommerse PHM, Spaink HP, Schmidt T. In vivo plasma membrane organization: results of biophysical approaches. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2004;1664:119. doi: 10.1016/j.bbamem.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 43.London E. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2005;1746:203. doi: 10.1016/j.bbamcr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Lagerholm BC, Weinreb GE, Jacobson K, Thompson NL. DETECTING MICRODOMAINS IN INTACT CELL MEMBRANES. Annual Review of Physical Chemistry. 2005;56:309–336. doi: 10.1146/annurev.physchem.56.092503.141211. [DOI] [PubMed] [Google Scholar]
- 45.Pike LJ. Growth factor receptors, lipid rafts and caveolae: An evolving story. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2005;1746:260. doi: 10.1016/j.bbamcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 46.Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silvius J. Lipid microdomains in model and biological membranes: how strong are the connections? Q Rev Biophys. 2005;38:373–83. doi: 10.1017/S003358350600415X. [DOI] [PubMed] [Google Scholar]
- 48.Heerklotz H. Triton promotes domain formation in lipid raft mixtures. Biophys J. 2002;83:2693–701. doi: 10.1016/S0006-3495(02)75278-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lichtenberg D, Goni FM, Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem Sci. 2005;30:430–6. doi: 10.1016/j.tibs.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 50.Schuck S, Honsho M, Ekroos K, Shevchenko A, Simons K. Resistance of cell membranes to different detergents. Proc Natl Acad Sci U S A. 2003;100:5795–800. doi: 10.1073/pnas.0631579100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Babiychuk EB, Draeger A. Biochemical characterization of detergent-resistant membranes: a systematic approach. Biochem J. 2006;397:407–16. doi: 10.1042/BJ20060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheiffele P, Roth MG, Simons K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. Embo J. 1997;16:5501–8. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng PC, Dykstra ML, Mitchell RN, Pierce SK. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J Exp Med. 1999;190:1549–60. doi: 10.1084/jem.190.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. European Journal of Immunology. 2000;30:954–963. doi: 10.1002/1521-4141(200003)30:3<954::AID-IMMU954>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 55.Predescu SA, Predescu DN, Shimizu K, Klein IK, Malik AB. Cholesterol-dependent syntaxin-4 and SNAP-23 clustering regulates caveolar fusion with the endothelial plasma membrane. J Biol Chem. 2005;280:37130–8. doi: 10.1074/jbc.M505659200. [DOI] [PubMed] [Google Scholar]
- 56.Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pralle A, Keller P, Florin EL, Simons K, Horber JK. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J Cell Biol. 2000;148:997–1008. doi: 10.1083/jcb.148.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gidwani A, Holowka D, Baird B. Fluorescence anisotropy measurements of lipid order in plasma membranes and lipid rafts from rbl-2h3 mast cells. Biochemistry. 2001;40:12422–9. doi: 10.1021/bi010496c. [DOI] [PubMed] [Google Scholar]
- 59.Gaus K, Gratton E, Kable EP, Jones AS, Gelissen I, Kritharides L, Jessup W. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc Natl Acad Sci U S A. 2003;100:15554–9. doi: 10.1073/pnas.2534386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ushio-Fukai MU, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling K, Alexander RW. Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells. The Journal of Biological Chemistry. 2001;276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- 61.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–44. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 62.Rouquette-Jazdanian AK, Pelassy C, Breittmayer JP, Aussel C. Revaluation of the role of cholesterol in stabilizing rafts implicated in T cell receptor signaling. Cellular Signalling. 2006;18:105. doi: 10.1016/j.cellsig.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 63.Larbi A, Douziech N, Khalil A, Dupuis G, Gherairi S, Guerard KP, Fulop T., Jr Effects of methyl-beta-cyclodextrin on T lymphocytes lipid rafts with aging. Exp Gerontol. 2004;39:551–8. doi: 10.1016/j.exger.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 64.Gaus K, Kritharides L, Schmitz G, Boettcher A, Drobnik W, Langmann T, Quinn CM, Death A, Dean RT, Jessup W. Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J. 2004:03-0486fje. doi: 10.1096/fj.03-0486fje. [DOI] [PubMed] [Google Scholar]
- 65.Gaus K, Rodriguez M, Ruberu KR, Gelissen I, Sloane TM, Kritharides L, Jessup W. Domain-specific lipid distribution in macrophage plasma membranes. J Lipid Res. 2005;46:1526–1538. doi: 10.1194/jlr.M500103-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Smart EJ, Ying Y, Mineo C, Anderson RGW. A Detergent-Free Method for Purifying Caveolae Membrane from Tissue Culture Cells. PNAS. 1995;92:10104–10108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sampson LJ, Hayabuchi Y, Standen NB, Dart C. Caveolae localize protein kinase A signaling to arterial ATP-sensitive potassium channels. Circ Res. 2004;95:1012–8. doi: 10.1161/01.RES.0000148634.47095.ab. [DOI] [PubMed] [Google Scholar]
- 68.Giurisato E, McIntosh DP, Tassi M, Gamberucci A, Benedetti A. T cell receptor can be recruited to a subset of plasma membrane rafts, independently of cell signaling and attendantly to raft clustering. J Biol Chem. 2003;278:6771–8. doi: 10.1074/jbc.M210758200. [DOI] [PubMed] [Google Scholar]
- 69.Schutz GJ, Kada G, Pastushenko VP, Schindler H. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. Embo J. 2000;19:892–901. doi: 10.1093/emboj/19.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swamy MJ, Ciani L, Ge M, Smith AK, Holowka D, Baird B, Freed JH. Coexisting Domains in the Plasma Membranes of Live Cells Characterized by Spin-Label ESR Spectroscopy. Biophys J. 2006;90:4452–4465. doi: 10.1529/biophysj.105.070839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Almeida PFF, Pokorny A, Hinderliter A. Thermodynamics of membrane domains. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2005;1720:1. doi: 10.1016/j.bbamem.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 72.Ottico E, Prinetti A, Prioni S, Giannotta C, Basso L, Chigorno V, Sonnino S. Dynamics of membrane lipid domains in neuronal cells differentiated in culture. J Lipid Res. 2003;44:2142–51. doi: 10.1194/jlr.M300247-JLR200. [DOI] [PubMed] [Google Scholar]
- 73.Klein U, Gimple G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol with b-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–93. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 74.Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–3222. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gimpl G, Klein U, Reilander H, Fahrenholz F. Expression of the human oxytocin receptor in baculovirus-infected insect cells: high-affinity binding is induced by a cholesterol-cyclodextrin complex. Biochemistry. 1995;34:13794–801. doi: 10.1021/bi00042a010. [DOI] [PubMed] [Google Scholar]
- 76.Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration Of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–34. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- 77.Breusegem SY, Halaihel N, Inoue M, Zajicek H, Lederer E, Barry NP, Sorribas V, Levi M. Acute and chronic changes in cholesterol modulate Na-Pi cotransport activity in OK cells. Am J Physiol Renal Physiol. 2005;289:F154–65. doi: 10.1152/ajprenal.00331.2004. [DOI] [PubMed] [Google Scholar]
- 78.Romanenko VG, Rothblat GH, Levitan I. Sensitivity of volume-regulated anion current to cholesterol structural analogues. J Gen Physiol. 2004;123:77–88. doi: 10.1085/jgp.200308882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T. Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-beta peptide. Faseb J. 2004;18:836–8. doi: 10.1096/fj.03-0372fje. [DOI] [PubMed] [Google Scholar]
- 80.Lange Y, Ramos BV. Analysis of the distribution of cholesterol in the intact cell. J Biol Chem. 1983;258:15130–4. [PubMed] [Google Scholar]
- 81.Bar LK, Barenholz Y, Thompson TE. Fraction of cholesterol undergoing spontaneous exchange between small unilamellar phosphatidylcholine vesicles. Biochemistry. 1986;25:6701–5. doi: 10.1021/bi00369a056. [DOI] [PubMed] [Google Scholar]
- 82.Haynes MP, Phillips MC, Rothblat GH. Efflux of cholesterol from different cellular pools. Biochemistry. 2000;39:4508–17. doi: 10.1021/bi992125q. [DOI] [PubMed] [Google Scholar]
- 83.Steck TL, Ye J, Lange Y. Probing red cell membrane cholesterol movement with cyclodextrin. Biophys J. 2002;83:2118–25. doi: 10.1016/S0006-3495(02)73972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones JD, Thompson TE. Spontaneous phosphatidylcholine transfer by collision between vesicles at high lipid concentration. Biochemistry. 1989;28:129–134. doi: 10.1021/bi00427a019. [DOI] [PubMed] [Google Scholar]
- 85.Wimley WC, Thompson TE. Phosphatidylethanolamine enhances the concentration-dependent exchange of phospholipids between bilayers. Biochemistry. 1991;30:4200–4204. doi: 10.1021/bi00231a014. [DOI] [PubMed] [Google Scholar]
- 86.Thurnhofer H, Hauser H. Uptake of cholesterol by small intestinal brush border membrane is protein-mediated. Biochemistry. 1990;29:2142–2148. doi: 10.1021/bi00460a026. [DOI] [PubMed] [Google Scholar]
- 87.Nichols JW, Pagano RE. Kinetics of soluble lipid monomer diffusion between vesicles. Biochemistry. 1981;20:2783–2789. doi: 10.1021/bi00513a012. [DOI] [PubMed] [Google Scholar]
- 88.Steck TL, Kezdy FJ, Lange Y. An activation-collision mechanism for cholesterol transfer between membranes. J Biol Chem. 1988;263:13023–31. [PubMed] [Google Scholar]
- 89.Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–76. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 90.Backer JM, Dawidowicz EA. Mechanism of cholesterol exchange between phospholipid vesicles. Biochemistry. 1981;20:3805–10. doi: 10.1021/bi00516a021. [DOI] [PubMed] [Google Scholar]
- 91.Butko P, Hapala I, Scallen TJ, Schroeder F. Acidic phospholipids strikingly potentiate sterol carrier protein 2 mediated intermembrane sterol transfer. Biochemistry. 1990;29:4070–7. doi: 10.1021/bi00469a007. [DOI] [PubMed] [Google Scholar]
- 92.Rodrigueza WV, Wheeler JJ, Klimuk SK, Kitson CN, Hope MJ. Transbilayer movement and net flux of cholesterol and cholesterol sulfate between liposomal membranes. Biochemistry. 1995;34:6208–17. doi: 10.1021/bi00018a025. [DOI] [PubMed] [Google Scholar]
- 93.Bojesen IN, Bojesen E. Oleic acid binding and transport capacity of human red cell membrane. Acta Physiol Scand. 1996;156:501–16. doi: 10.1046/j.1365-201X.1996.456173000.x. [DOI] [PubMed] [Google Scholar]
- 94.Jonas A, Maine GT. Kinetics and mechanism of phosphatidylcholine and cholesterol exchange between single bilayer vesicles and bovine serum high-density lipoprotein. Biochemistry. 1979;18:1722–8. doi: 10.1021/bi00576a014. [DOI] [PubMed] [Google Scholar]
- 95.Poznansky MJ, Czekanski S. Cholesterol exchange as a function of cholesterol/phospholipid mole ratios. Biochem J. 1979;177:989–91. doi: 10.1042/bj1770989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gottlieb MH. Rates of cholesterol exchange between human erythrocytes and plasma lipoproteins. Biochim Biophys Acta. 1980;600:530–41. doi: 10.1016/0005-2736(80)90454-x. [DOI] [PubMed] [Google Scholar]
- 97.McLean LR, Phillips MC. Cholesterol desorption from clusters of phosphatidylcholine and cholesterol in unilamellar vesicle bilayers during lipid transfer or exchange. Biochemistry. 1982;21:4053–9. doi: 10.1021/bi00260a022. [DOI] [PubMed] [Google Scholar]
- 98.McLean LR, Phillips MC. Mechanism of cholesterol and phosphatidylcholine exchange or transfer between unilamellar vesicles. Biochemistry. 1981;20:2893–900. doi: 10.1021/bi00513a028. [DOI] [PubMed] [Google Scholar]
- 99.Slotte JP, Lundberg B. Transfer of [3H]cholesterol between lipid vesicles and rat arterial smooth muscle cells in vitro. Biochim Biophys Acta. 1983;750:434–9. doi: 10.1016/0005-2760(83)90182-0. [DOI] [PubMed] [Google Scholar]
- 100.McLean LR, Phillips MC. Kinetics of phosphatidylcholine and lysophosphatidylcholine exchange between unilamellar vesicles. Biochemistry. 1984;23:4624–30. doi: 10.1021/bi00315a017. [DOI] [PubMed] [Google Scholar]
- 101.Nichols JW. Thermodynamics and kinetics of phospholipid monomer-vesicle interaction. Biochemistry. 1985;24:6390–8. doi: 10.1021/bi00344a011. [DOI] [PubMed] [Google Scholar]
- 102.Puglisi G, Fresta M, Ventura CA. Interaction of Natural and Modified β-Cyclodextrins with a Biological Membrane Model of Dipalmitoylphosphatidylcholine. Journal of Colloid and Interface Science. 1996;180:542–547. [Google Scholar]
- 103.Leventis R, Silvius JR. Use of cyclodextrins to monitor transbilayer movement and differential lipid affinities of cholesterol. Biophys J. 2001;81:2257–67. doi: 10.1016/S0006-3495(01)75873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Giocondi MC, Milhiet PE, Dosset P, Le Grimellec C. Use of cyclodextrin for AFM monitoring of model raft formation. Biophys J. 2004;86:861–9. doi: 10.1016/s0006-3495(04)74161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fukasawa M, Nishijima M, Itabe H, Takano T, Hanada K. Reduction of sphingomyelin level without accumulation of ceramide in Chinese hamster ovary cells affects detergent-resistant membrane domains and enhances cellular cholesterol efflux to methyl-beta -cyclodextrin. J Biol Chem. 2000;275:34028–34. doi: 10.1074/jbc.M005151200. [DOI] [PubMed] [Google Scholar]
- 106.Hansen GH, Immerdal L, Thorsen E, Niels-Christiansen LL, Nystrom BT, Demant EJ, Danielsen EM. Lipid rafts exist as stable cholesterol-independent microdomains in the brush border membrane of enterocytes. J Biol Chem. 2001;276:32338–44. doi: 10.1074/jbc.M102667200. [DOI] [PubMed] [Google Scholar]
- 107.Liu SM, Cogny A, Kockx M, Dean RT, Gaus K, Jessup W, Kritharides L. Cyclodextrins differentially mobilize free and esterified cholesterol from primary human foam cell macrophages. J Lipid Res. 2003;44:1156–66. doi: 10.1194/jlr.M200464-JLR200. [DOI] [PubMed] [Google Scholar]
- 108.Nakanishi K, Nadai T, Masada M, Miyajima K. Effect of cyclodextrins on biological membrane. II. Mechanism of enhancement on the intestinal absorption of non-absorbable drug by cyclodextrins. Chem Pharm Bull (Tokyo) 1992;40:1252–6. doi: 10.1248/cpb.40.1252. [DOI] [PubMed] [Google Scholar]
- 109.Singh I, Kishimoto Y. Effect of cyclodextrins on the solubilization of lignoceric acid, ceramide, and cerebroside, and on the enzymatic reactions involving these compounds. J Lipid Res. 1983;24:662–665. [PubMed] [Google Scholar]
- 110.Casu B, Grenni A, Naggi A, Torri G, Virtuani M, Focher B. Interaction of cyclodextrinscyclomalto-oligosaccharides with glycolipids: N.M.R. studies of aqueous system of cyclomaltohexaose and alkyl glycosides. Carbohydr Res. 1990;200:101–109. [Google Scholar]
- 111.Shiraishi T, Hiraiwa M, Uda Y. Effects of cyclodextrins on the hydrolysis of ganglioside GM1 by acid beta-galactosidases. Glycoconj J. 1993:170–174. doi: 10.1007/BF00737714. [DOI] [PubMed] [Google Scholar]
- 112.Ahmed SM, Casu B, Cedro A, Guerrini M, Lanzarotti E, Moltrasio D, Naggi A, Torri G. Disruption of micellar aggregates of ganglioside GM-1 by complexation with -cyclodextrin. Int J Pharm. 1994;109:99–106. [Google Scholar]
- 113.Monnaert V, Tilloy S, Bricout H, Fenart L, Cecchelli R, Monflier E. Behavior of alpha-, beta-, and gamma-cyclodextrins and their derivatives on an in vitro model of blood-brain barrier. J Pharmacol Exp Ther. 2004;310:745–51. doi: 10.1124/jpet.104.067512. [DOI] [PubMed] [Google Scholar]
- 114.Rawyler A, Siegenthaler PA. Cyclodextrins: a new tool for the controlled lipid depletion of thylakoid membranes. Biochim Biophys Acta. 1996;1278:89–97. doi: 10.1016/0005-2736(95)00190-5. [DOI] [PubMed] [Google Scholar]
- 115.Anderson TG, Tan A, Ganz P, Seelig J. Calorimetric measurement of phospholipid interaction with methyl-beta-cyclodextrin. Biochemistry. 2004;43:2251–61. doi: 10.1021/bi0358869. [DOI] [PubMed] [Google Scholar]
- 116.Cooper A. Effect of cyclodextrins on the thermal stability of globular proteins. J Am Chem Soc. 1992;114:9208–9209. [Google Scholar]
- 117.Horský J, Horsky J, Pitha J. Inclusion complexes of proteins: Interaction of cyclodextrins with peptides containing aromatic amino acids studied by competitive spectrophotometry. Journal of Inclusion Phenomena and Molecular Recognition in Chemistry. 1994;18:291–300. [Google Scholar]
- 118.Breslow R, Yang Z, Ching R, Trojandt G, Odobel F. Sequence Selective Binding of Peptides by Artificial Receptors in Aqueous Solution. J Am Chem Soc. 1998;120:3536–3537. [Google Scholar]
- 119.Uekama K. Cyclodextrins in drug delivery system. Adv Drug Delivery Rev. 1999;36:1–2. doi: 10.1016/s0169-409x(98)00057-x. [DOI] [PubMed] [Google Scholar]
- 120.Pitha J, Irie T, Sklar PB, Nye JS. Drug solubilizers to aid pharmacologists: amorphous cyclodextrin derivatives. Life Sci. 1988;43:493–502. doi: 10.1016/0024-3205(88)90150-6. [DOI] [PubMed] [Google Scholar]
- 121.Aachmann FL, Otzen DE, Larsen KL, Wimmer R. Structural background of cyclodextrin-protein interactions. Protein Eng. 2003;16:905–12. doi: 10.1093/protein/gzg137. [DOI] [PubMed] [Google Scholar]
- 122.Williamson MP, Le Gal-Coeffet MF, Sorimachi K, Furniss CS, Archer DB, Williamson G. Function of conserved tryptophans in the Aspergillus niger glucoamylase 1 starch binding domain. Biochemistry. 1997;36:7535–9. doi: 10.1021/bi9702896. [DOI] [PubMed] [Google Scholar]
- 123.Schmidt AK, Cottaz S, Driguez H, Schulz GE. Structure of cyclodextrin glycosyltransferase complexed with a derivative of its main product beta-cyclodextrin. Biochemistry. 1998;37:5909–15. doi: 10.1021/bi9729918. [DOI] [PubMed] [Google Scholar]
- 124.Evenas J, Tugarinov V, Skrynnikov NR, Goto NK, Muhandiram R, Kay LE. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J Mol Biol. 2001;309:961–74. doi: 10.1006/jmbi.2001.4695. [DOI] [PubMed] [Google Scholar]
- 125.Duan X, Hall JA, Nikaido H, Quiocho FA. Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: flexibility of tertiary structure and ligand binding. J Mol Biol. 2001;306:1115–26. doi: 10.1006/jmbi.2001.4456. [DOI] [PubMed] [Google Scholar]
- 126.Larson SB, Greenwood A, Cascio D, Day J, McPherson A. Refined molecular structure of pig pancreatic alpha-amylase at 2.1 A resolution. J Mol Biol. 1994;235:1560–84. doi: 10.1006/jmbi.1994.1107. [DOI] [PubMed] [Google Scholar]
- 127.Mikami B, Hehre EJ, Sato M, Katsube Y, Hirose M, Morita Y, Sacchettini JC. The 2.0-A resolution structure of soybean beta-amylase complexed with alpha-cyclodextrin. Biochemistry. 1993;32:6836–45. doi: 10.1021/bi00078a006. [DOI] [PubMed] [Google Scholar]
- 128.Ilangumaran S, Hoessli DC. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem J. 1998;335(Pt 2):433–40. doi: 10.1042/bj3350433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Locke D, Koreen IV, Liu JY, Harris AL. Reversible pore block of connexin channels by cyclodextrins. J Biol Chem. 2004;279:22883–92. doi: 10.1074/jbc.M401980200. [DOI] [PubMed] [Google Scholar]
- 130.Gu LQ, Braha O, Conlan S, Cheley S, Bayley H. Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature. 1999;398:686–90. doi: 10.1038/19491. [DOI] [PubMed] [Google Scholar]
- 131.Rozema D, Gellman SH. Artificial chaperone-assisted refolding of carbonic anhydrase B. J Biol Chem. 1996;271:3478–87. doi: 10.1074/jbc.271.7.3478. [DOI] [PubMed] [Google Scholar]
- 132.Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci U S A. 2003;100:13964–9. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gimpl G, Burger K, Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry. 1997;36:10959–74. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- 134.Ferraro JT, Daneshmand M, Bizios R, Rizzo V. Depletion of plasma membrane cholesterol dampens hydrostatic pressure and shear stress-induced mechanotransduction pathways in osteoblast cultures. Am J Physiol Cell Physiol. 2004;286:C831–9. doi: 10.1152/ajpcell.00224.2003. [DOI] [PubMed] [Google Scholar]
- 135.Byfield F, Aranda-Aspinoza H, Romanenko VG, Rothblat GH, Levitan I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys J. 2004;87:3336–43. doi: 10.1529/biophysj.104.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Slotte JP, Jungner M, Vilcheze C, Bittman R. Effect of sterol side-chain structure on sterol-phosphatidylcholine interactions in monolayers and small unilamellar vesicles. Biochim Biophys Acta. 1994;1190:435–43. doi: 10.1016/0005-2736(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 137.Mattjus P, Bittman R, Vilcheze C, Slotte JP. Lateral domain formation in cholesterol/phospholipid monolayers as affected by the sterol side chain conformation. Biochim Biophys Acta. 1995;1240:237–47. doi: 10.1016/0005-2736(95)00179-4. [DOI] [PubMed] [Google Scholar]
- 138.Slotte JP. Effect of sterol structure on molecular interactions and lateral domain formation in monolayers containing dipalmitoyl phosphatidylcholine. Biochim Biophys Acta. 1995;1237:127–34. doi: 10.1016/0005-2736(95)00096-l. [DOI] [PubMed] [Google Scholar]
- 139.Bittman R, Blau L. The phospholipid-cholesterol interaction. Kinetics of water permeability in liposomes. Biochemistry. 1972;11:4821–4839. doi: 10.1021/bi00775a029. [DOI] [PubMed] [Google Scholar]
- 140.Demel RA, Bruckdorfer KR, van Deenen LLM. The effect of sterol structure on the permeability of liposomes to glucose glycerol and Rb+ Biochem et Biophys Acta. 1972;255:321–330. doi: 10.1016/0005-2736(72)90031-4. [DOI] [PubMed] [Google Scholar]
- 141.De Kruyff B, Demel RA, van Deenen LLM. The effects of cholesterol and epicholesterol incorporation on the permeability and the phase transition of intact Acholeplasma Laidlawii cell emmbranes and derived liposomes. Biochem et Biophys Acta. 1972;255:331–347. doi: 10.1016/0005-2736(72)90032-6. [DOI] [PubMed] [Google Scholar]
- 142.Halling KK, Slotte JP. Membrane properties of plant sterols in phospholipid bilayers as determined by differential scanning calorimetry, resonance energy transfer and detergent-induced solubilization. Biochim Biophys Acta. 2004;1664:161–71. doi: 10.1016/j.bbamem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 143.Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry. 2000;39:843–849. doi: 10.1021/bi992543v. [DOI] [PubMed] [Google Scholar]
- 144.Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40:178–84. doi: 10.1016/s0028-3908(00)00159-3. [DOI] [PubMed] [Google Scholar]